
Abstract
Resistance to European canker (Neonectria ditissima) in apple is currently one of the most important breeding targets for commercial production in Sweden. Previous research has identified significant genetic variation in susceptibility to the disease, with the local Swedish cultivar ‘Aroma’ considered as one of the most resistant cultivars. Identification of genetic regions underlying the resistance of this cultivar would be a valuable tool for future breeding. Thus, we performed Bayesian quantitative trait loci (QTL) mapping for resistance to European canker in a full-sib family of ‘Aroma’ × ‘Discovery’. Mapping was performed with the area under the disease progression curves (AUDPCs) from all seven (AUDPC_All7) and the first four assessments (AUDPC_First4), and three parameters of a sigmoid growth model for lesion length. As a scale for the effect of the different parameters, historic phenotypic data from screenings of a genetically diverse germplasm was compiled and re-analyzed. The parametrization of the data on lesion growth increased the number of QTL that could be identified with high statistical power, and provided some insight into their roles during different stages of disease development in the current experimental setup. Five QTL regions with strong or decisive evidence were identified on linkage groups 1, 8, 15, and 16. The QTL regions could be assigned to either of the parameters lesion length at the first assessment (‘LL_A1’), the maximal lesion growth rate (lesion length doubling time, ‘t_gen’), and the lesion length at girdling (‘LL_G’). Three of these QTL were traced along the pedigrees of some known relatives of the FS family, and discussed in relation to future crosses for breeding and genetic research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
European canker, caused by the fungus Neonectria ditissima, is currently one of the most serious threats to Swedish commercial apple (Malus domestica Borkh.) production. The fungus can infect trees through natural or artificial wounds throughout the year and is favored by cool and wet climates, such as North-West Europe, New Zealand, and Chile (Weber 2014). Infected wounds develop into necrotic lesions that may girdle branches and the stem, thereby killing the distal parts of the shoot or the entire tree. Apart from yield losses caused by reduced bearing capacity, the fungus can also infect fruits and thereby cause storage losses (Brown et al. 1994; Swinburne 1975).
N. ditissima commonly colonizes the phloem and xylem of the apple stem and then continues to spread until it reaches a defensive mature suberized cell layer (Krähmer 1980). However, beyond the importance of suberization, little is known about the defense mechanisms against N. ditissima in apple. The disease is managed by the removal of infected tissues, cultivation of resistant cultivars, and application of fungicides (Weber 2014). As applications of fungicides are limited in Sweden and pruning of cankered wounds is time consuming, there is a strong need for locally adapted cultivars with improved resistance to N. ditissima.
Despite the importance of the disease, there is currently no consensus on how to screen for resistance. Reported approaches differed in type of plant material (trees, detached shoots), cultivation (planted, potted, outdoors, or in a glasshouse), sources of inoculum (e.g., wild collected vs single-spore isolate), type of isolates (wild collected and in vitro propagated single-spore isolates), and type of spores (ascospores, conidia) as well as wounding techniques (e.g., naturally occurring leaf scars or manually inflicted by rasp or scalpel) (Bus et al. 2019; Delgado et al. 2022; Garkava-Gustavsson et al. 2016; Gómez-Cortecero et al. 2016; Karlström et al. 2022; van de Weg 1989; Wenneker et al. 2017). Also, various parameters to quantify resistance have been used, including area under the disease progression curve (AUDPC), infection rate, the slope of the lesion growth regression line, lesion length at a given time-point, and indexes quantifying the establishment of secondary infections (Bus et al. 2019; Garkava-Gustavsson et al. 2016; Gómez-Cortecero et al. 2016; Karlström et al. 2022; Wenneker et al. 2017). Different parameters might capture the effect of different resistance mechanisms depending on the timing of assessment in relation to the disease development of the experiment, due to variations in, e.g., isolate virulence, the conductivity of the experimental procedure to infection, physiological stage of the trees, and environmental conditions affecting disease progression (e.g., temperature). This diversity might account for variation in cultivar rankings in screening experiments as well as different outcomes in quantitative trait loci (QTL) mapping and validation studies. While likely to be of relevance for field conditions, assessment of colonization and infection rates might not be feasible for QTL mapping studies with large populations as it requires a high number of inoculation points (trees). In cases when repeated measurements have been made, the AUDPC can be used to average out the data on disease progression over time. This can help reduce the noise in the phenotypic data, as compared to using data from individual assessments. However, AUDPC does not provide information on the temporal dynamics of lesion growth. In the M. domestica – N. ditissima pathosystem, the lesion growth rate based on a linear regression over a defined time period has been proposed as a reproducible parameter of disease resistance (Wenneker et al. 2017). Parameters of non-linear models such as exponential-, logistic-, and Gompertz curves have been used successfully for functional QTL identification of growth in, e.g., Populus (Wu et al. 2003) and Arabidopsis (Bac-Molenaar et al. 2015), and provide information that has direct implications for the understanding of a trait’s genetic architecture. Employing such models to disease progression in the N. ditissima – M. domestica pathosystem might improve our understanding of the functional characteristics of different mapped QTL, thereby enabling a better understanding of underlying candidate genes, as well as facilitating better integration of results from different experiments.
Previous studies have shown that cultivars such as ‘Aroma’, ‘Santana’, and ‘Golden Delicious’ are partially resistant, while cultivars such as ‘Discovery’, ‘Elise’, and ‘Katja’ are highly susceptible to N. ditissima (Garkava-Gustavsson et al. 2016; van de Weg 1989; Wenneker et al. 2017). QTL for resistance to N. ditissima in apple have been mapped in a number of studies, e.g., ‘Scired’ x A045R14T055, ‘M9’ × ‘Robusta 5’, ‘Jonathan’ × ‘Discovery’, and ‘Golden Delicious’ × ‘Discovery’ (Bus et al. 2021; Bus et al. 2019; van de Weg et al. 2020) and recently in a set of five families including ‘Aroma’ as a parent of one full-sib (FS) family (Karlström et al. 2022). The cultivars ‘Aroma’ and ‘Discovery’ are both important parents and founders in the Nordic apple breeding programs and mapping of QTL for resistance to N. ditissima in these varieties is of high priority for the Swedish breeding program. Previous studies have indicated a complex inheritance of resistance to N. ditissima in M. domestica (Gómez-Cortecero et al. 2016; Karlström et al. 2022). Therefore, a Bayesian approach is likely better suited to QTL analysis than conventional interval-mapping, as it allows for simultaneous estimation not only for the position of the largest QTL, but for the number of QTL, the additive effect sizes, and credible interval of all QTL (Bink et al. 2014, 2008; van de Weg et al. 2018). Once QTL regions have been mapped, tracing the inheritance of the mapped loci through pedigrees can provide valuable information for further breeding and genetic research. This can be facilitated through calculation of Identity-by-Decent (IBD) probabilities of the founder haplotypes in a pedigreed germplasm. This can provide information on the presence of QTL regions in specific individuals and recombination events in the founder haplotypes, which can be valuable for increasing the applicability of the results.
The purpose of this study was to gain insight into the genetic control of partial resistance to N. ditissima in apple. To this end, an FS family from ‘Aroma’ × ‘Discovery’ was phenotyped, genotyped, and analyzed through Bayesian QTL mapping using two AUDPC parameters as well as parameters of a sigmoid growth model of lesion length. Finally, the use of the most relevant QTL and related parameters were evaluated through Identity-by-State (IBS) and IBD approaches, using historic phenotypic data compiled from previous cultivar screening experiments.
Materials and methods
Plant material
For QTL discovery, an FS family consisting of 172 offspring from an ‘Aroma’ × ‘Discovery’ cross, was phenotyped in two independent experiments over two consecutive years (2019 and 2020), together with their parents and six reference cultivars ‘Cox’s Orange Pippin’, ‘Prima’, ‘Jonathan’, ‘Katja’, ‘Golden Delicious’, and ‘Santana’. In the second year of phenotyping, two grandparents (‘Filippa’ and ‘Ingrid Marie’) and three half-siblings (‘Fredrik’, ‘Frida’, and ‘Julia’) of the FS family were included. To trace QTL intervals detected in the FS family and to provide a scale for the disease resistance parameters used, we also compiled and reanalyzed phenotypic data from 98 genotypes that had been screened previously. These cultivar screening experiments were performed over 5 years (2011–2016), with seven reference cultivars being included in all experiments: ‘Aroma’, ‘Cox’s Orange Pippin’, ‘Discovery’, ‘Elise’, ‘Golden Delicious’, ‘Jonathan’, and ‘Santana’ (File S1). Results for a subset of 15 cultivars from the screening germplasm were published previously (Garkava-Gustavsson et al. 2016) using various parameters for estimation of resistance.
Phenotyping
Phenotyping of the FS family and a set of reference cultivars as controls was performed essentially as described previously (Garkava-Gustavsson et al. 2016). Each autumn, new trees grown in 3L pots were obtained, which had been grafted on rootstock ‘M9’ in the spring of the same year. A single-spore isolate of N. ditissima (‘SLU-E1’) was propagated, from a spore solution maintained at − 80 °C in 50% glycerol, on 20% Potato Glucose Agar (70,139, Sigma-Aldrich) supplemented with bactoagar (B1000, Saveen o Werner) for 4–6 weeks on a laboratory bench under ambient light and temperature. Three buds (typically the 11:th, 14:th, and 17:th axillary buds from the apex) were removed with a scalpel (File S1) and the wounds were inoculated with a mixture of micro- and macroconidia (mostly macroconidia with two septae or less) to a total of 1000 conidia in 10 μl water. Once the suspension was absorbed by the plant tissue, the wound was covered with white petrolatum (Special Care Vitt vaselin, ACO Sweden). The petrolatum was removed 5 days after inoculation, using tissue paper. The inoculations were performed block-wise in late autumn (November–December), with all trees of a block being inoculated on the same date. The inoculation dates of the blocks were separated by approximately 1 week, thus leaves were typically fully present at the first inoculation occasion and mostly shed at the fourth and final inoculation occasion. Throughout the trial period, the trees were kept in a glasshouse with minimal heating and no artificial cooling or light, with a temperature fluctuating between 9 and 11 °C until the end of February. From then on, changes in outdoor climate led to increased fluctuations in temperature, ranging between 5 °C (night) and occasionally above 20 °C (day). Lesions were measured using a digital caliper at seven time points, every second week, starting approximately 6 weeks after inoculation when the first visual symptoms had appeared in a meaningful proportion of the block (typically more than half of the wounds having developed lesions at least 5 mm long). Lesions shorter than 5 mm were recorded as ‘0’. In cases where two or three lesions merged during the trial period, their total length was measured and divided between the lesions according to the relative proportions of their last individual measurement. In case lesions could not be measured because the stems were girdled, thereby strangling the distal parts and preventing further upward growth of the lesion, the lesions were assigned the last true measurement for the remainder of the trial. For the experiments with the FS family, approximately half of the developed lesions girdled. The previous screening experiments had been performed similarly, but using a wild-collected spore solution (mostly with three septae) prepared as described previously (Garkava-Gustavsson et al. 2016) and one year using rootstock ‘B9’ (File S1). All current and previous experiments were performed using a randomized complete block design with three trees per individual in each block, except for the FS family where each individual was represented by a single tree in each of four blocks for practical reasons (File S1).
Parameters of disease resistance and heritability
For the AUDPC parameter, we initially considered all seven assessments (‘AUDPC_All7’) as the phenotypic parameter for QTL mapping. Next, to investigate how robust the detected QTL regions were to artifacts introduced by girdling of wounds, we considered the AUDPC from the first to the fourth (‘AUDPC_First4’) assessment, as most wounds that girdled typically did so after the fourth assessment. For the sigmoid growth curve model we used the parameters initial lesion length (‘LL_A1’), the shortest doubling time of the length of the lesion (‘t_gen’), and the lesion length at girdling (‘LL_G’).
For all parameters, wounds that had not developed any lesions at the 6th assessment were treated as missing values. The AUDPC parameters were calculated as:
where Yi is the lesion length at the ith assessment, Xi is the number of days at the ith observation and n is the total number of observations. To estimate the lesion growth rate, a logistic regression was performed over each pseudo-replicate (wound) using the R (R Core Team 2020) package GrowthCurveR (Sprouffske and Wagner 2016), fitting the model:
where Nt is the lesion length at time t, K is the carrying capacity (maximum lesion length reached at girdling of the wound), N0 is the lesion length at the beginning of the growth curve, and r is the intrinsic growth rate of the lesion, which would occur if there were no restrictions. The generation time (t_gen) is the shortest time it could take for the lesion to double in length under the model:
where r is the intrinsic growth rate obtained from (2). To improve the ability of the model to fit the growth rate parameter, an artificial time point was added to the assessment data for the logistic regression, corresponding to the time of inoculation, which was given the phenotypic value ‘0’ for all wounds. The regression curves of all wounds with outlier residual standard errors were inspected manually, and wounds with poorly fitting regression curves were excluded from further analysis. The lesion length at the last assessment for all wounds that had girdled was used as the parameter for the maximal lesion length (LL_G), i.e., the plateau of the sigmoid growth curve.
Best linear unbiased estimates (BLUEs) combined over the experiments were used as phenotypes and BLUEs were calculated using the R package ‘lme4’ (Bates et al. 2015) with Genotype treated as a fixed effect and with Tree nested within Block, and Block in turn nested within Year, as random effects. Wounds with outlier residuals, identified visually from QQ-plots, were investigated manually and excluded from the analysis if the assessment data were problematic.
Broad sense heritability estimates (Ĥ2) were calculated using the regression method previously described (Schmidt et al. 2019). In short, the model described above was fitted to calculate BLUEs from each data set, and a similar model but with the Genotype as a random effect was used to calculate Best Linear Unbiased Predictors (BLUPs). By regressing BLUPs on BLUEs, the regression coefficient can be used as an approximation of Ĥ2. A Genotype x Year (‘G x Y’) interaction term was not included in the models, as this was not considered biologically significant for the current experimental protocol.
The residuals of all parameters (AUDPC_All7, AUDPC_First4, LL_A1, t_gen, and LL_G) showed slight deviations from a normal distribution in both the FS family and the screening population (File S2). Initially, various transformation methods were considered, where Box-Cox transformation gave distributions that were closest to normal. However, preliminary QTL-mapping analysis indicated that Box-Cox-transformed and untransformed data for the AUDPC parameters gave very similar results. In addition, the application of Box-Cox transformation to mixed models is problematic and makes biologically relevant interpretations of BLUEs and, e.g., QTL effects complicated. Thus, untransformed data were used for all QTL analyses.
Genotyping and marker curation
DNA from the FS family and part of the screening population was extracted from lyophilized leaf tissue using the DNeasy 96 Plant Kit (Qiagen) and was used for genotyping with the 20 K apple Infinium® single nucleotide polymorphism (SNP) array (Bianco et al. 2014). Marker data were initially curated at the level of individual SNPs, as described by Vanderzande and Howard et al. (2019), with the modification that a subset of 10 K SNPs retained in a previous study on marker integration (Howard et al. 2021) were called in Genome Studio (GS) v 2.0 (Illumina Inc.), using cluster definition files kindly made available by the authors. For computational efficiency of the QTL analysis in the FS family, the marker set was pruned for SNPs with pairwise r2 above 0.2 in 2 Mb sliding windows with a 200-kb shift using PLINK 1.9 (Chang et al. 2015) and physical positions on the HFTH1 whole genome sequence (WGS) (Eric van de Weg, unpubl. data). This was done separately for SNPs exhibiting maternal monohybrid segregation, paternal monohybrid segregation, and dihybrid segregation, resulting in a set of 4506 SNPs in approximate linkage equilibrium. Marker data for linkage groups (LGs) with QTL loci in the FS family were further curated as haploblocks (HBs) as described by Vanderzande and Howard et al. (2019) in the screening germplasm, resulting in 370 HBs representing 2348 SNPs on linkage groups (LGs) 1, 8, 15, and 16. Genotypic 20 K SNP array data on some of the accessions in the screening population, and close relatives, was made available from other previous and ongoing projects (Hjeltnes et al. 2019; Skytte af Sätra et al. 2020). For curation as well as subsequent QTL analysis, genetic positions were taken from an advanced draft of a virtual linkage map (the 15K-iGW-map, Eric van de Weg, unpubl. data) resulting from integration of the iGLMap (Di Pierro et al. 2016) and the HFTH1 WGS (Zhang et al. 2019).
QTL analysis
QTL analysis was conducted using a Bayesian approach as embedded in FlexQTL™ (www.flexqtl.nl), which uses Markov chain Monte Carlo (MCMC) simulations and bi-allelic QTL models to evaluate possible combinations of the number of QTL, QTL positions, QTL effects, and their transmission through pedigrees (Bink et al. 2014, 2008). In this study, each parameter was analyzed four times using different seeds and priors for the number of QTL (1 or 3). The maximum number of QTL was set to 15 for all runs, and all runs consisted of at least 100,000 iterations with a thinning of 100 using additive genetic models with normal prior distributions and random (Co) variance matrix diagonals. While the phenotypes of parents are usually not included in QTL analyses, initial runs consistently identified ‘Aroma’ as the susceptible parent and ‘Discovery’ as the resistant parent. By adding the phenotypes of the parents to the model, they were correctly identified as resistant/susceptible, and as the same QTL regions were identified with or without the phenotypes of the parents in preliminary FlexQTL runs, these were included for further analysis.
All analyses were run until the effective chain samples exceeded 100, i.e., until convergence. The level of evidence provided for the presence of a QTL was indicated by two times the natural logarithm of the Bayes factors (‘2lnBF’) for an incremental number of QTL per LG through a pair-wise comparison. A 2lnBF value above 0, 2, 5, and 10 is considered to indicate hardly any, positive, strong, and decisive evidence for the presence of a QTL, respectively. Further analysis was limited to the five QTL with a distinct peaks in posterior position for the AUDPC_All7 parameter. QTL regions were defined as successive 2-cM bins with 2lnBF above 2. For individual QTL regions, the Bayesian probability (B-Prob.) of a QTL is based on the posterior QTL intensity, i.e., the frequency at which any of the 2-cM bins within the QTL region were included in the model. Given the probability of more than one QTL per LG, the Bayesian probability of a QTL region might exceed 1.0 in case multiple genetic bins within the QTL region are frequently included in the model. The proportion of phenotypic variance explained (PVE) by a QTL was calculated using the formula:
where VP is the total phenotypic variance and AVt is the additive variance explained by the QTL region (from PostQTL analysis). For all QTL regions (Table 1), trace plots of QTL positions for all runs were investigated to confirm convergence for the QTL position, i.e., that the QTL regions were not included in the model alternatingly. In FlexQTL outputs Q and q denotes alleles with increasing and decreasing effects on the phenotype, respectively, and this notation is used throughout this paper. In addition to the model with additive effects only, FlexQTL also supports models with dominance and additive effects. Tests of these models with the AUPDC parameters indicated similar QTL regions as in the additive effects only model, but with very low statistical power and very small estimated dominance effects. Thus, dominance was not considered further for QTL analysis. While there is little documentation accompanying FlexQTL, some previous publications include informative material and methods sections and supplementary material that has been very valuable to the current QTL analysis (Mangandi et al. 2017; van de Weg et al. 2018).
Compound QTL genotypes
QTL intervals were identified as two or more consecutive 2-cM genetic bins with a 2lnBF above five, in all four runs performed for the parameter analyzed. Individuals with recombination events within these bins were excluded from the analyses of compound genotype effects. Segregation and effects of the QTL regions were investigated by fitting a series of linear models using the non-recombinant genotypes in base R with the ‘car’ an ‘lmtest’ packages (Fox and Weisberg 2019; R Core Team 2020; Zeileis and Hothorn 2002; File S2). Nested models were evaluated by likelihood ratio tests and non-nested models were compared based on their Akaike information criterion (AIC). The models with the lowest AIC were also evaluated by their Bayesian information criterion (BIC).
Search for epistatic loci
As the Q alleles at the LG1 and LG8 as well as the q alleles at the LG15 and LG16b loci did not have significantly different additive effects, they were grouped into subsets based on their total number of q alleles for AUDPC_First4 across both respective loci. These subset of individuals with 0, 1, 2, or 3 q alleles for LG1/LG8 and LG15/LG16b, respectively, were used to identify potentially epistatic loci segregating in the population. Only individuals without recombination events in consensus QTL regions were used, resulting in subsets consisting of 17, 49, 35, and 10 individuals for the LG1/LG8 loci, and 12, 42, 49, and 12 individuals for the LG15/LG16b loci. A single FlexQTL run was performed for each subset and parameter, using AUDPC_All7, AUDPC_First4, and t_gen for the LG1/LG8 loci, and AUDPC_All7, AUDPC_First4, and LL_A1 for the LG15/LG16b loci. Additionally, subsets of 89 and 71 individuals carrying the q and Q alleles from ‘Discovery’ at the LG16a locus, respectively, were also used to search for additional epistatic loci for the LL_G parameter.
Prevalence of the segregating haplotypes in the screening germplasm
Genotypes that were IBS to the haplotypes segregating in the FS family were identified from HB genotypes, and individuals having identical haplotype calls along the entire consensus QTL intervals were considered IBS. Next, IBD genotypes for estimation of allele effects were identified as IBS genotypes where the haplotype could be traced through their known pedigree in cultivars with phased genotypic data to the segregating haplotype from three of the FS-family’s grandparents ‘Filippa’, ‘Worcester Pearmain’, or ‘Beauty of Bath’ and two of its grand-grandparents, ‘Cox’s Orange Pippin’ and ‘Cox’s Pomona’. For visualization of QTL haplotype inheritance through the pedigree, the probability of IBD was calculated in FlexQTL and used as input together with AUDPC_First4 BLUEs for visualization in PediMap (Voorrips et al. 2012).
Results
Phenotypic data
The infection rate was above 95% of inoculated wounds in all experiments, except in 2012 (89.4%). All parameters (AUDPC_All7, AUDPC_First4, LL_A1, t_gen, and LL_G) were heteroscedastic and had residual distributions that deviated from normality (File S2). Ĥ2 was moderate to high for the FS family across both years (0.78, 0.78, 0.67, 0.73, and 0.52 for AUDPC_All7, AUDPC_First4, LL_A1, t_gen, and LL_G, respectively). Similarly, moderate to high Ĥ2 point estimates were observed for the screening germplasm across all years (0.87, 0.87, 0.62, 0.74, and 0.75 for AUDPC_All7, AUDPC_First4, LL_A1, t_gen, and LL_G, respectively). The two AUDPC parameters, AUDPC_All7 and AUDPC_First4, were highly correlated, while the three parameters relating to the sigmoid growth model were very weakly correlated with each other (Fig. 1a). The year-year correlation for the FS family (represented by 1 tree/block) was low for all parameters (Pearson corr. = 0.43, 0.42, 0.34, 0.54, 0.12, and 0.35 for AUDPC_All7, AUDPC_First4, LL_A1, t_gen, and LL_G, respectively, Fig. 1b), but the correlation between years was moderate to high for the control cultivars included both those years (represented by 3 trees/block) (Pearson Corr. = 0.83, 0.87, 0.90, 0.74, and 0.75 for AUDPC_All7, AUDPC_First4, LL_A1, t_gen, and LL_G, respectively, Fig. 1c). The low correlation between years for the FS-family is likely caused by extensive experimental noise in single-year data when using only one tree per block. As a consequence, differences between years cannot be reliably estimated in the FS family, and the BLUEs over both years were used as phenotypes for QTL mapping.

Distributions of, and correlations between, parameters and years. a Correlogram from the screening population with histograms for each parameter in the diagonal, Pearson correlation coefficient (r) in the upper off-diagonal, and pairwise scatter plots in the lower left off-diagonal. b Correlations between the two years of phenotyping for the QTL mapping in the FS family and c the reference cultivars that were common between the 2 years. d–h Distributions in the FS family for the different parameters where phenotypic values of the parents are indicated by red (‘Aroma’) and blue (‘Discovery’) arrows
QTL mapping
First, we considered the AUDPC over all seven assessments (AUDPC_All7), and subsequently we also assessed the AUDPC over only the first four assessments (AUDPC_First4), when most wounds had not started to girdle. Next, QTL mapping was performed on three parameters of a sigmoid growth curve: initial lesion length (LL_A1), growth rate (t_gen), and maximum lesion length before the wounds girdle (LL_G).
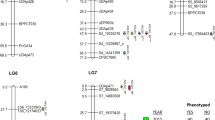
For AUDPC_All7 there was strong statistical evidence for three QTL, one each on LG1, LG8, and LG15. In addition, there was positive evidence for one QTL on LG16, although this evidence was the joined result of two peaks in the posterior probability far apart, while there was hardly any evidence for 2 QTL (2lnBF: 0.6 – 1.3) (Table 1, Fig. 2, File S2).

Posterior QTL positions from FlexQTL for each parameter, from a run with 3 prior QTL, a AUDPC_All7, b AUDPC_First4, c LL_A1, d t_gen, and e LL_G. Note that statistical evidence is based on the area under a peak rather than the height of a peak
AUDPC_First4 provided decisive evidence for one QTL each on LG8 and LG15, and strong evidence for one QTL each on LG1 and LG16. In contrast to AUDPC_All7, there was a single consistent peak in posterior intensity for LG16, coinciding with one of the peaks for AUDPC_All7 on that LG, denoted LG16b (Table 1, Fig. 1, File S2).
The LL_A1 parameter provided strong evidence for one QTL, and positive evidence for a second QTL on LG15, one of which overlapped with the QTL detected for AUDPC_All7 and AUDPC_First4 on this LG. There was also strong evidence for a QTL for LL_A1 on LG16, which overlapped with the LG16b locus detected for AUDPC_First4, and positive evidence for a QTL on LG8, overlapping with the region identified for other parameters. Additionally, there was strong evidence for a QTL for LL_A1 on LG13, which was not detected for any of the other parameters analyzed and was not considered further (Table 1, Fig. 2, File S2).
The parameter t_gen provided strong evidence for one QTL on LG8, overlapping with the QTL on LG8 detected for AUDPC_All7 and AUDPC_First4. In addition, one QTL with only positive evidence was detected on LG1, overlapping with the QTL for AUDPC_All7 and AUDPC_First4 on this LG (Table 1, Fig. 2, File S2).
LL_G provided strong evidence for one QTL on LG16, which coincided with one of the QTL for AUDPC_All7 on LG16, but not with the LG16b locus. Thus, we denote this locus LG16a (Table 1, Fig. 2, File S2).
All identified QTL regions explained a low to moderate proportion of the variance of the respective parameter (Table 1). None of the parameters showed strong evidence (2lnBF > 5) for more than one QTL on any LG, in any replicate FlexQTL run. All FlexQTL runs converged (ECS > 100 for all parameters) and QTL intervals were reproducible between runs (File S2). The LG15 locus was the only one with strong evidence for segregation in ‘Aroma’, while there was strong evidence for segregation in ‘Discovery’ for all five loci (Table 1). The two peaks for the LL_A1 parameter on LG15 (Fig. 2c) seemed to be fitted alternatingly according to the trace plot and were thus interpreted as representing a single QTL with an uncertain position. All other identified QTL regions showed independence in their discovery according to the trace plots (File S2). The consensus regions of all 5 QTL intervals were investigated (i.e., the outer boundaries of the range across parameters), and the presence of segregating markers and recombination events could be confirmed in all five cases.
Compound QTL genotypes
Thus, five QTL regions related to different parameters of a sigmoid growth curve for lesion length were identified and considered further. Next, we analyzed the segregation patterns (monohybrid vs dihybrid) and potential interaction effects within and between loci, considering the pairs of loci affecting the same parameter. To evaluate the behavior of the compound QTL genotypes, individuals without recombination in the consensus QTL regions for the relevant loci were selected. For LG1 and LG8, the t_gen parameter was used as phenotype as there were no other QTL regions with LG-wide positive evidence for that parameter. For the LG15 and LG16b loci, the LL_A1 parameter was used and the LL_G parameter was used for the LG16a locus, despite LG-wide strong and positive evidence for a locus segregating at LG13, respectively. The genotypes for the QTL regions were then analyzed by a series of nested linear models to investigate segregation patterns and interaction effects (File S2). None of the loci exhibited skewed segregation.
For the t_gen parameter QTL at LG1 and LG8, 111 individuals were identified without recombination events in the QTL regions. Considering the possible compound genotypes across both loci, the model with paternal segregation at LG1 and dihybrid segregation at LG8 was significantly better than the alternative models. The desirable Q alleles for t_gen came from ‘Beauty of Bath’ at LG1, and from ‘Ingrid Marie’ and ‘Worcester Pearmain’ at LG8. Adding terms for dominance or allele-specific epistasis between the two loci did not cause any significant improvements over the additive effects only model. Indeed, a simple linear model accounting for the total number of Q alleles across both loci had lower AIC (62.0) and BIC (70.0) than a model accounting for the segregation at each locus separately (65.0 and 78.5, respectively). Adding an extra Q allele increased the average t_gen by 0.07, 0.21, and 0.25 days when going from zero to one, from one to two, and from two to three Q alleles, respectively, corresponding to an average decrease in AUDPC_All7 of 149, 335, and 339 mm*days (Fig. 3a). As the average increase in t_gen is about three times larger when going from one to two, or from two to three Q alleles than when going from zero to one Q allele, there might be some synergistic epistasis for the number of Q alleles at the LG1 and LG8 loci. Indeed, alternative models accounting for epistasis but otherwise similar to the simple model (accounting for the total number of Q alleles across both loci) all had AICs in the same range (within 2 units of the minimum). However, there were only two alternative full models with a BIC that was less than 3 units above the simple model, indicating that they might be equally competitive. These models were ones that had an additional log10-transformed term for the number of Q alleles across both loci, and one which instead of the total number of Q alleles, had one term for going from zero to one Q allele, and one term for additional Q alleles beyond one.

Phenotype distributions of compound genotypes within the ArDi FS family, grouped by a number of Q alleles for t_gen at the LG1 and LG8 loci, b number of q alleles for LL_A1 at the LG15 and LG16b loci, and c number of q alleles for LL_G at the LG16a locus. Individuals with recombination events within the QTL regions of the respective QTL loci are excluded. Note that for LG1, LG16a, and LG16b the homozygous state of ‘Aroma’ cannot be determined, thus those alleles are not counted
For the LL_A1 parameter QTL at LG15 and LG16b, 115 individuals were identified without recombination events in the LG15 or LG16b QTL regions. Considering the additive effects of the four possible alleles at each locus, the best model was for dihybrid segregation at LG15 and paternal monohybrid segregation at LG16b. The desirable q alleles for LL_A1 came from ‘Filippa' and ‘Beauty of Bath’ at LG15, and from ‘Worcester Pearmain’ at LG16b. There were, however, no significant effects of dominance or allele-specific epistasis. As the three q alleles appeared to have similar effects, different linear models with the total number of q alleles regardless of locus were fitted and compared to the alternative models with individual alleles. Adding an extra q allele decreased LL_A1 by 0.85, 2.84, and 1.18 mm when going from zero to one, from one to two, and from two to three alleles, respectively (Fig. 3), corresponding to an average decrease in AUDPC_All7 of 2, 423, and 523 mm*days. The model with the lowest AIC accounted only for linear additive effects from the total number of q alleles across both loci, although a number of models which had additional terms for non-linear or allele-specific effects, had AICs that were less than 2 units larger than the simple model. The latter had a BIC value 3.7 units lower than the alternative full model with the second lowest BIC. Similar to the case for the LG1 and LG8 loci, the second-best model was one that instead of the total number of Q alleles, had one term for going from zero to one Q allele and one term for additional Q alleles beyond one. While this might indicate the presence of some degree of synergistic epistasis for the LG1 and LG8 loci, and the LG15 and LG16b loci with regard to the number of q alleles, it could also be an artefact from the data as the distribution is skewed, particularly LL_A1 for which there are ‘zero’ observations (Fig. 1 and File S2).
There were 159 individuals without recombination events within the LG16a QTL region. Considering either maternal or paternal alleles separately, or compound genotypes, provided statistical evidence only for maternal segregation, with the decreasing q allele coming from ‘Worcester Pearmain’. Individuals carrying the q allele had lesions that were on average 9.18 mm shorter when they girdled, and a 325 mm*days lower AUDPC_All7.
Search for epistatic loci
As the phenotypic distributions of some of the subgroups of the FS family seemed to indicate some residual segregation (e.g., the two Q alleles subgroup in Fig. 3a and the one q allele subgroup in Fig. 3b), we considered the possibility of epistatic interactions specific to each subgroup. To search for potential epistatic loci, we divided the FS population into four subsets with individuals having zero to three desirable alleles at the LG1 and LG8 loci, four subsets with individuals having zero to three desirable alleles at the LG15 and LG16b loci, and two groups carrying the q or Q allele from ‘Discovery’ at the LG16a locus. However, as this resulted in very small subsets, this must be considered very preliminary results. For individuals with one Q allele for t_gen at LG1 or LG8, there was strong LG-wide evidence for a QTL on LG3 for the t_gen parameter (2lnBF = 5.6), which was not found in the full FS family for any of the parameters. In the LG16a subgroups there was LG-wide positive evidence for one QTL on LG3 (2lnBF = 3.4) and on LG10 (2lnBF = 2.6), in the subgroups with one and zero q allele for LL_G, respectively.
In the original mapping using the full FS-family, strong LG-wide evidence for a QTL on LG13 was found for the LL_A1 parameter (2lnBF = 5.5–6.1). Investigation of the genotype probabilities assigned by FlexQTL for one of the replicate runs with the full mapping population and LL_A1 as parameter indicated that individuals with a probability above 0.5 for having a Qq genotype at the LG13 locus occurred at a frequency close to 0.5 for both the 1q and 2q subgroups (0.5 and 0.43, respectively). Thus, the LG13 locus is expected to segregate in both subgroups, but positive evidence was found for one QTL on LG13 for the AUDPC_All7 and AUDPC_First4 (2lnBF = 4.2 and 3.1, respectively) parameters only in the subgroup carrying a single q allele for AUDPC or LL_A1 at either the LG15 or the LG16b locus. That the locus on LG13 is only detected in the subgroup with one q allele might indicate that there is a locus on LG13 that is partially recessive to the other loci mapped in this study, although the absence of detectable segregation in the 2q subgroup could be due to scale effects. However, this is expected to be less of an issue with the AUDPC_All7 parameter than with LL_A1 (File S2). Furthermore, positive LG-wide evidence was found for the LL_G parameter (2lnBF = 2.8–3.6) on the same LG, although there was barely any evidence using the AUDPC or t_gen parameters (Fig. 2, File S2). The peak for the LL_A1 parameter appears on the lower end of LG13 together with the LG16b locus on the upper end of LG16, and the peak for the LL_G parameter appear of the upper end of LG13 together with the LG16a locus on the lower end of LG16 (Fig. 2). Chromosome 13 is known to be homoeologous to chromosome 16 (Daccord et al. 2017), although the QTL mapping was done based on genetic distances, which do not follow a linear relationship to the physical position across the entire LG. Due to the low power for the LG13 loci and the large QTL intervals, the potential homoeology between the two LG13 peaks and the LG16a and LG16b loci was not investigated further, although it might indicate a complex interaction between these regions.
Prevalence of the segregating haplotypes in the screening germplasm
Several of the phenotyped individuals in the screening germplasm were related to either of the parents of the FS family, with phased marker data available. Thus, transmission of IBD haplotypes can be traced and haplotypes being IBS to those segregating in the FS family can be identified. While validation of QTL with moderate effects on a polygenic trait can be difficult in a genetically diverse germplasm, it can still provide information on the effect of a QTL and guide future crosses for breeding and genetic research. Among the identified QTL regions, we focused on LG8, LG15, and LG16b for further analysis due to the low prevalence of the desirable allele from ‘Beauty of Bath’ at LG1 and the uncertain biological effect of the q allele at LG16a. The LG1 locus was not considered further, as the donor of the q allele, ‘Beauty of Bath’, had very few descendants in the germplasm studied. Also, the LG1 locus had lower Bayesian probability across parameters (Table 1). The LG16a locus was omitted due to its small effect, uncertainties in its segregation (low Bayesian probability, relatively few segregating SNP markers in the QTL region, and q allele of LG16a being in coupling phase with the q allele at the LG16b locus) and ambiguity in which allele should be considered desirable (see the “Discussion” section). Thus, haplotypes of the LG8, LG15, and LG16b loci that segregated in the FS family were traced along the known pedigrees of some cultivars related to ‘Aroma’ and ‘Discovery’ (Fig. 4), and individuals with IBS haplotypes were identified in the germplasm (File S1).

Transmission of QTL haplotypes of the QTL a LG8, b LG15, and LG16b along the pedigrees of some cultivars, which have inherited parts of the haplotypes of the consensus QTL regions segregating in ‘Aroma’ and ‘Discovery’. The color of the frames indicates the cultivars BLUE for AUDPC_First4, ranging from purple (high value, susceptible) to green (low value, resistant). The boundaries of the consensus QTL regions are indicated by white lines. The founder haplotypes segregating in ‘Aroma’ × ‘Discovery’ are indicated as dark/light green (q) and dark/light purple (Q), all other haplotypes are grey. Red lines indicate maternal inheritance and blue lines indicate paternal inheritance. Note that both haplotypes of ‘Cox’s Orange Pippin’ are traced for LG15, as ‘Ingrid Marie’ seems to have a recombination event within the consensus QTL region, such that the first 3 cM comes from one haplotype (light purple) and the upper 7 cM from the other haplotype (dark purple) of ‘Cox’s Orange Pippin’. Additionally, ‘Ingrid Marie’ seems to have two subsequent double recombination events on LG15 from ‘Cox’s Orange Pippin’, including a 10-cM region upstream of the QTL regions where ‘Cox’s Orange Pippin’ is homozygous for the markers included in this study
Concerning LG8, ‘Lillarosaröda’ (an unreleased selection from the Swedish breeding program, ‘Lillarosarod’ in Fig. 4) and the highly susceptible ‘Julia’ are the only individuals, apart from ‘Discovery’, to have inherited the q allele for AUDPC from ‘Worcester Pearmain’. However, the q allele for AUDPC coming from ‘Cox’s Orange Pippin’ has been transmitted to several descendants, and there are 15 haplotypes that are IBS to either of the q alleles (Fig. 4a, File S1).
Regarding the LG15 locus, ‘Gratia’ seems to be the only descendant of ‘Aroma’ to share the q allele by IBD, and the only individual that shares the q allele of ‘Beauty of Bath’ by IBD is the susceptible cultivar ‘Julia’. Notably, ‘Ingrid Marie’ seems to have a recombination event within the consensus QTL region, so it is unclear which of the descendants of the ‘Cox’s Orange Pippin’ inherited the Q allele segregating in the ‘Aroma’ × ‘Discovery’ family. However, 10 haplotypes in the screening germplasm are IBS to either of the Q alleles, while only 5 are IBS to either of the q alleles, all of which are IBD (Fig. 4b, File S1).
The q allele from ‘Worcester Pearmain’ at the LG16b locus has been transmitted to six individuals in the screening germplasm, and several other cultivars which were not phenotyped. Offspring of ‘Worcester Pearmain’ that have inherited the q allele include ‘Discovery’, ‘Katja’, and ‘Lord Lambourne’ (Fig. 4c, File S1). These have in turn transmitted the q allele to cultivars such as ‘Agnes’, ‘Lillarosaröda’, ‘Øye’, ‘Nanna’, ‘Estelle’, ‘Maj-Britt’, and ‘Skaistis’ (‘Estelle’ and ‘Maj-Britt’ are unreleased selections from the Swedish breeding program).
While not considered further, it should be noted that the LG16a q allele for AUDPC_All7 is in coupling phase with the q allele of the LG16b locus in ‘Worcester Pearmain’. Also, a relatively large number of haplotypes in the screening germplasm are IBS to either the Q or the q alleles (17 haplotypes of each allele, File S1).
Discussion
QTL related to resistance to European canker
We identified several QTL for resistance against N. ditissima segregating in ‘Aroma’ × ‘Discovery’, five of which were considered for further analysis. Two QTL on LG15 and LG16 (LG16b) were identified for the LL_A1 parameter as well as AUDPC_First4. Two QTL on LG1 and LG8 were identified for the t_gen parameter and both AUDPCs. Lastly, a second QTL on LG16 (LG16a) was identified for the LL_G parameter. The AUDPC_All7 parameter gave strong evidence for only three QTL and no evidence for presence of two QTL on LG16. However, further analysis by parametrization provided strong evidence for two QTL on LG16 with effects during different stages of lesion growth. Thus, parametrization of the phenotypic data allowed us to identify more QTL with better statistical power, and provided a first insight into their roles during different stages of disease development under these current experimental conditions. For example, ‘Discovery’ segregated for a q allele for LL_G at the LG16a locus. The q allele results in a shorter lesion length when the wound girdles, which implies a smaller lesion with less potential as a source for secondary infections. On the other hand, a shorter lesion length when the wound girdled implies that distal parts of the stem dies quicker. From an epidemiological point the q allele might be desirable while the effect of the Q allele might be preferred by the farmer as the distal parts of the branch can remain productive for a longer time before it dies off. Thus, the Q allele could be considered a tolerance allele rather than a susceptibility allele. In the current study, we found no statistically significant dominance effects for the five loci, although this could be due to a lack of statistical power in the available data and monohybrid segregation as for, e.g., the LG16b locus. Thus, further validation studies would benefit from being designed to enable the assessment of dominance effects at the QTL of interest. On the other hand, we identified potentially epistatic loci on LGs 3 and 13, with effects specific to individuals in the FS family carrying one q allele for AUDPC at the LG1 and LG8 or the LG15 and LG16b loci, respectively. We also identified a potential epistatic interaction between the LG3 and LG10 loci for subsets of the FS family that have one and zero q allele at the LG16a locus, respectively. However, these potentially epistatic loci were identified based on small subsets of the FS family, with the parameters of each subgroup suffering from scale effects to different degrees. Thus, their status as epistatic loci must be considered very preliminary, and interpretations should be made with great caution until their effects have been further verified. Putative epistatic QTL interactions have previously been identified in other pathosystems of M. domestica, e.g., resistance to fire blight (van de Weg et al. 2018).
In the current study, the LG8, LG15, and LG16b loci had the highest probabilities across parameters (Table 1). Bus et al. (2021) also identified QTL for resistance against European canker on LG8 and LG16 segregating in a bi-parental family. The SNP being most strongly linked to the LG8 locus in that study was within the consensus QTL peak of the LG8 locus in the current study and might thus represent the same locus. Similarly to the current study, the LG8 locus was segregating in the cultivar considered to be susceptible (‘Scired’). On the other hand, the SNP being reported by Bus et al. (2021) as most tightly linked to the LG16 locus is located in between the LG16a and LG16b loci identified in the current study, outside the consensus QTL regions of both loci. Additionally, Karlström et al. (2022) recently performed QTL mapping through Pedigree Based Analysis (PBA) using several disease parameters and found positive or strong evidence for QTL regions on several linkage groups, including LGs 8, 15 and 16. While the QTL regions on LG15 of that study barely coincided with the QTL at LG15 in the current study, the statistical power was much higher in the current study, with decisive evidence for the AUDPC_First4 parameter. This might in part be due to the much smaller families used by Karlström et al. (2022) (60–69 individuals). Considering a trait that is controlled by several QTL with moderate effect, large families will be required for both breeding and QTL mapping. For further efforts in pre-breeding or validation of QTL effects, one might for example consider the cross ‘Gratia’ × ‘Lillarosaröda’ where ‘Gratia’ is carrying the q allele from ‘Filippa’ at LG15 and ‘Lillarosaröda’ is carrying the q alleles for AUDPC from ‘Worcester Pearmain’ at the LG8 and LG16b loci (Fig. 4).
Experimental procedures
The experimental procedures of the current study provided phenotypic data of high quality, which facilitated high statistical power in QTL detection. By dissecting the phenotypic data further, in light of a sigmoid growth curve for lesion growth, these QTL regions were found to play key roles during specific stages of the infection. This indicates that the time points at which assessments are being made can have a crucial effect on which QTL regions are detected in a study. Wenneker et al. (2017) suggested LGR as the most robust metric for quantification of resistance to European canker. In the current study, we used the parameter t_gen, which is conceptually similar to LGR as both are measures of growth rate. In line with Wenneker et al. (2017), we found the t_gen parameter to provide high heritability and correlations between years in both the screening germplasm and the FS family. While the t_gen parameter provided strong evidence for the LG8 QTL, it did not provide any evidence for the LG15, LG16a, or LG16b QTL regions.
While it has been questioned whether screening tests in controlled conditions are relevant to field conditions (Delgado et al. 2022), there are large differences between experimental set-ups that might appear similar at a first glance, e.g., potted trees experiment. Karlström et al. (2022) also made use of a controlled conditions experiment with potted trees, which in some respects was similar to the experimental set-up of the current study. There were, however, several differences in the procedure, including physiological growth stage of the trees, climatic conditions, number of replicates and pseudo-replicates, number of conidia applied to each wound, and assessment intervals. They reported a broad sense heritability of 0.46 for their potted trees experiments, which is much lower than what is reported here (0.78 for AUDPC_All7). Consequently, they reported a much weaker statistical support for the QTL regions identified in their study.
Future prospects
As the identified QTL all had low to moderate effects, and the estimates of broad-sense heritability were high, each of the identified QTL will likely have a low predictiveness by themselves. Thus, maximal genetic gain will likely be obtained by first identifying the most suitable parent-parent combination, followed by a two-step selection process, first by culling the worst genotypes using marker-assisted selection and subsequently by phenotypic screening of the remaining individuals (Vanderzande et al. 2018; Ru et al. 2016). However, this requires that the identified QTL haplotypes can be converted to a small number of markers that can be routinely analyzed on a relatively cheap platform. As the SNPs for the 20 K SNP array were selected from a narrow genetic base of only 14 individuals, mostly with high MAF, single SNPs that are diagnostic of a given QTL region are unlikely to be identified. Rather, subsets of SNPs that can distinguish a given haplotype in a given breeding germplasm could be searched for (Chagné et al. 2019). Additionally, QTL effects from desirable alleles that are rare in a germplasm, such as the q allele at LG15 from ‘Filippa’, are difficult to verify, and even more so in the presence of several additional QTL of moderate effect segregating in the germplasm. Thus, future efforts to validate the QTL regions identified in the current study, and potential subsets of SNPs for marker-assisted seedling selection, would require FS-families that are expected to segregate for the specific QTL alleles to be validated. Furthermore, a promising source of resistance for the Swedish breeding program, the cultivar ‘Santana’, was not IBS for the favorable alleles at any of the QTL loci. Thus, ‘Santana’ might pose a supplementary source of resistance, and further breeding efforts would benefit from the mapping of resistance QTL segregating in that cultivar.
Data archiving statement
BLUEs for phenotypic parameters for the screening germplasm and phased SNP calls of the parents of the FS family for each consensus QTL region are given in File S1. SNP calls and BLUEs for the different parameters of the FS-family can be made available upon request.
References
Bac-Molenaar JA, Vreugdenhil D, Granier C, Keurentjes JJB (2015) Genome-wide association mapping of growth dynamics detects time-specific and general quantitative trait loci. J Exp Bot 66:5567. https://doi.org/10.1093/JXB/ERV176
Bates D, Mächler M, Bolker BM, Walker SC (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48. https://doi.org/10.18637/JSS.V067.I01
Bianco L, Cestaro A, Sargent DJ, Banchi E, Derdak S, Di Guardo M, Salvi S, Jansen J, Viola R, Gut I, Laurens F, Chagné D, Velasco R, van de Weg E, Troggio M (2014) Development and validation of a 20K single nucleotide polymorphism (SNP) whole genome genotyping array for apple (Malus × domestica Borkh). Plos One 9:e110377. https://doi.org/10.1371/journal.pone.0110377
Bink MCAM, Uimari P, Sillanpää MJ, Janss LLG, Jansen RC (2002) Multiple QTL mapping in related plant populations via a pedigree-analysis approach. Theor Appl Genet 104:751–762. https://doi.org/10.1007/s00122-001-0796-x
Bink MCAM, Boer MP, Ter Braak CJF, Jansen J, Voorrips RE, van de Weg WE (2008) Bayesian analysis of complex traits in pedigreed plant populations. Euphytica 161:85–96. https://doi.org/10.1007/s10681-007-9516-1
Bink MC, Jansen J, Madduri M, Voorrips RE, Durel CE, Kouassi AB, Laurens F, Mathis F, Gessler C, Gobbin D, Rezzonico F, Patocchi A, Kellerhals M, Boudichevskaia A, Dunemann F, Peil A, Nowicka A, Lata B, Stankiewicz-Kosyl M, Jeziorek K, Pitera E, Soska A, Tomala K, Evans KM, Fernandez-Fernandez F, Guerra W, Korbin M, Keller S, Lewandowski M, Plocharski W, Rutkowski K, Zurawicz E, Costa F, Sansavini S, Tartarini S, Komjanc M, Mott D, Antofie A, Lateur M, Rondia A, Gianfranceschi L, van de Weg WE (2014) Bayesian QTL analyses using pedigreed families of an outcrossing species, with application to fruit firmness in apple. Theor Appl Genet 127:1073–1090. https://doi.org/10.1007/s00122-014-2281-3
Brown AE, Muthumeenakshi S, Swinburne TR, Li R (1994) Detection of the source of infection of apple trees by Cylindrocarpon heteronema using DNA polymorphisms. Plant Pathol 43:338–343. https://doi.org/10.1111/j.1365-3059.1994.tb02693.x
Bus VGM, Scheper RWA, Walter M, Campbell RE, Kitson B, Turner L, Fisher BM, Johnston SL, Wu C, Deng CH, Singla G, Bowatte D, Jesson LK, Hedderley DI, Volz RK, Chagné D, Gardiner SE (2019) Genetic mapping of the European canker (Neonectria ditissima) resistance locus Rnd1 from Malus ‘Robusta 5.’ Tree Genet Genomes 15:1–13
Bus V, Single G, Horner M, Jesson L, Walter M, Kitson B, López-Girona E, Kirk C, Gardiner S, Chagné D, Volz R (2021) Preliminary genetic mapping of fire blight and European canker resistances in two apple breeding families. Acta Hortic 1307:199–203
Chagné D, Vanderzande S, Kirk C, Profitt N, Weskett R, Gardiner SE, Peace CP, Volz RK, Bassil NV (2019) Validation of SNP markers for fruit quality and disease resistance loci in apple (Malus × domestica Borkh.) using the OpenArray® platform. Hortic Res 6:30. https://doi.org/10.1038/s41438-018-0114-2
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. https://doi.org/10.1186/s13742-015-0047-8
Daccord N, Celton JM, Linsmith G, Becker C, Choisne N, Schijlen E, van de Geest H, Bianco L, Micheletti D, Velasco R, Di Pierro EA, Gouzy J, Rees DJG, Guerif P, Muranty H, Durel CE, Laurens F, Lespinasse Y, Gaillard S, Aubourg S, Quesneville H, Weigel D, van de Weg E, Troggio M, Bucher E (2017) High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat Genet 49:1099–1106. https://doi.org/10.1038/ng.3886
Delgado Á, García-Fernández B, Gómez-Cortecero A, Dapena E (2022) Susceptibility of Cider Apple Accessions to European Canker - Comparison between Evaluations in Field Planted Trees and Rapid Screening Tests. Plants (Basel). https://doi.org/10.3390/plants11091145
Di Pierro E, Gianfranceschi L, Di Guardo MM, Koehorst-van H, Kruisselbrink J, Longhi S, Troggio M, Bianco L, Muranty H, Pagliarani G, Tartarini S, Letschka T, Luis LL, Garkava-Gustavsson L, Micheletti D, Bink M, Voorrips R, Aziz E, Velasco R, Laurens F, van de Weg E (2016) A high-density, multi-parental SNP genetic map on apple validates a new mapping approach for outcrossing species. Hortic Res 3:16057
Fox J, Weisberg S (2019) An R companion to applied regression, 3rd edn. Sage, Thousand Oaks CA. https://socialsciences.mcmaster.ca/jfox/Books/Companion/
Garkava-Gustavsson L, Ghasemkhani M, Zborowska A, Englund J-E, Lateur M, van de Weg E (2016) Approaches for evaluation of resistance to European canker (Neonectriaditissima) in apple. Acta Hortic 1127:75–82. https://doi.org/10.17660/ActaHortic.2016.1127.14
Gómez-Cortecero A, Saville RJ, Scheper RWA, Bowen JK, Agripino De Medeiros H, Kingsnorth J, Xu X, Harrison RJ (2016) Variation in host and pathogen in the Neonectria/Malus interaction; toward an understanding of the genetic basis of resistance to European canker. Front Plant Sci 7:1365. https://doi.org/10.3389/fpls.2016.01365
Hjeltnes S-H, Garkava-Gustavsson L, Alsheikh M, Borve J, Davik J, Haikonen T, Ikase L, Keldmaë H, Karhu S, Kjos B, Odilbekov F, Parikka P, Pedersen C, Rentanen M, Rumpunen K, Røen D, Sasnauskas A, Volens K, van de Weg E (2019) Public Private Partnership project NORDFRUIT: meeting the future challenges in Nordic apple and strawberry production [Poster presentations]. XV EUCARPIA Fruit Breeding and Genetics Symposium, Prague
Howard NP, Troggio M, Durel C-E, Muranty H, Denancé C, Bianco L, Tillman J, van de Weg E (2021) Integration of Infinium and Axiom SNP array data in the outcrossing species Malus × domestica and causes for seemingly incompatible calls. BMC Genomics 22:1–18. https://doi.org/10.1186/s12864-021-07565-7
Karlström A, Gómez-Cortecero A, Nellist CF, Ordidge M (2022) Dunwell JM, Harrison RJ (2022) Identification of novel genetic regions associated with resistance to European canker in apple. BMC Plant Biol 221(22):1–16. https://doi.org/10.1186/S12870-022-03833-0
Krähmer H (1980) Wound reactions of apple trees and their influence on infections with Nectria galligena. J Plant Dis Prot 87(2):97–112
Mangandi J, Verma S, Osorio L, Peres NA, van de Weg E, Whitaker VM (2017) Pedigree-based analysis in a multiparental population of octoploid strawberry reveals QTL alleles conferring resistance to Phytophthoracactorum. G3 Genes. Genomes Genet 7:1707–1719
R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Ru S, Hardner C, Carter PA, Main Evans K, D, Peace C, (2016) Modeling of genetic gain for single traits from marker-assisted seedling selection in clonally propagated crops. Hortic Res 3:16015. https://doi.org/10.1038/hortres.2016.15
Schmidt P, Hartung J, Rath J, Piepho HP (2019) Estimating broad-sense heritability with unbalanced data from agricultural cultivar trials. Crop Sci 59:525–536. https://doi.org/10.2135/CROPSCI2018.06.0376
Sprouffske K, Wagner A (2016) Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics 17:172. https://doi.org/10.1186/s12859-016-1016-7
Skytte af Sätra J, Troggio M, Odilbekov F, Sehic J, Mattisson H, Hjalmarsson I, Ingvarsson PK, Garkava-Gustavsson L, (2020) Genetic status of the Swedish central collection of heirloom apple cultivars. Sci Hortic (Amsterdam) 272:109599. https://doi.org/10.1016/j.scienta.2020.109599
Swinburne TR (1975) European canker of apple (Nectria galligena). Rev Plant Pathol 54:787–799
van de Weg E, Di Guardo M, Jänsch M, Socquet-Juglard D, Costa F, Baumgartner I, Broggini GAL, Kellerhals M, Troggio M, Laurens F, Durel C-EE, Patocchi A (2018) Epistatic fire blight resistance QTL alleles in the apple cultivar ‘Enterprise’ and selection X-6398 discovered and characterized through pedigree-informed analysis. Mol Breed 38:1–18. https://doi.org/10.1007/s11032-017-0755-0
Vanderzande S, Howard NP, Cai L, Da Silva Linge C, Antanaviciute L, Bink MCAM, Kruisselbrink JW, Bassil N, Gasic K, Iezzoni A, van de Weg E, Peace C (2019) High-quality, genome-wide SNP genotypic data for pedigreed germplasm of the diploid outbreeding species apple, peach, and sweet cherry through a common workflow. Plos One 14:e0210928. https://doi.org/10.1371/journal.pone.0210928
Voorrips RE, Bink MCAM, van ee Weg WE (2012) Pedimap: Software for the Visualization of Genetic and Phenotypic Data in Pedigrees. J. Hered. 103:903–907. https://doi.org/10.1093/JHERED/ESS060
Weber RWS (2014) Biology and control of the apple canker fungus Neonectria ditissima (syn. N. galligena) from a Northwestern European perspective. Erwerbs-Obstbau 56:95–107. https://doi.org/10.1007/s10341-014-0210-x
Wenneker M, Goedhart PW, van der Steeg P, van de Weg WE, Schouten HJ (2017) Methods for the quantification of resistance of apple genotypes to European fruit tree canker caused by Neonectria ditissima. Plant Dis 101:2012–2019. https://doi.org/10.1094/PDIS-04-17-0576-RE
Wu R, Ma CX, Zhao W, Casella G (2003) Functional mapping for quantitative trait loci governing growth rates: A parametric model. Physiol Genomics 14:241–249. https://doi.org/10.1152/physiolgenomics.00013.2003
van de Weg E, van der Steeg P, Wenneker M, Koerst-van Putten H, Smulders MJM, Schouten H (2020) Resistance to Neonectria ditissima in apple: what QTL discovery teaches us about plant resistance tests and resistance parameters [Conference presentation]. 4th International workshop on European fruit tree canker and resilient orchards, Online via Zoom
van de Weg EW (1989) Breeding for resistance to Nectria galligena in apple: differences in resistance between seedling populations. In Gessler C, Butt DJ, Koller B (eds) Integrated control of pome fruit diseases, vol II. Proceedings of a workshop of the Subgroup on integrated control of pome fruit diseases of the Working Group: “Integrated Plant Protection in Orchards” pp 137–145
Vanderzande S, Piaskowski JL, Luo F, Edge Garza DA, Klipfel J, Schaller A, Martin S, Peace C (2018) Crossing the finish line: How to develop diagnostic DNA tests as breeding tools after QTL discovery. J Hortic 5:1. https://doi.org/10.4172/2376-0354.1000228
Zeileis A, Hothorn T (2002) Diagnostic checking in regression relationships. R News 2(3):7–10. https://CRAN.R-project.org/doc/Rnews/
Zhang L, Hu J, Han X, Li J, Gao Y, Richards CM, Zhang C, Tian Y, Liu G, Gul H, Wang D, Tian Y, Yang C, Meng M, Yuan G, Kang G, Wu Y, Wang K, Zhang H, Wang D (2019) Cong P (2019) A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat Commun 10:1–13
Acknowledgements
We gratefully acknowledge access to germplasm for genotyping granted by Inger Hjalmarsson and the Programme for Diversity of Cultivated Plants (Swedish Central Collection, Alnarp). We are appreciative of Tuuli Haikonen (Luke, Finland), Dag Røen (Graminor, Norway), and Vidmantas Bendokas (Lithuanian Research Centre of Agriculture and Forestry, Lithuania) for providing material through the PPP Nordfruit project. Excellent genotyping service was provided by the Sequencing and Genotyping platform at Fondazione Edmund Mach (FEM, Italy). Thanks to Jasna Sehic, Marjan Ghasemkhani, Anna Zborowska, and Malin Dörre for technical assistance as well as Adam Flöhr and Jan-Eric Englund for statistical support. Thanks also to Stijn Vanderzande, Nicholas P. Howard, and Sujeet Verma for valuable comments on various aspects of data curation and the use of FlexQTL. Data analysis benefited from the Udda server at the Department of Plant Breeding, LTV Faculty, SLU.
Funding
Open access funding provided by Swedish University of Agricultural Sciences. This work was funded by the SLU GroGrund project “Accelerated and cost efficient cultivar development through genome informed apple breeding”, Formas (grant nr 2014–00924 and grant nr 2017–00496), Nordic Co-operation (“PPP Nordfruit”), and SLF (grant V1036010). SNP data were shared by the Fruitbreedomics Project No. 265582, which was funded by the EU seventh Framework Programme, and the PPP Nordfruit, which was funded by the Nordic Co-operation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by D. Chagné.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skytte af Sätra, J., Odilbekov, F., Ingvarsson, P.K. et al. Parametric mapping of QTL for resistance to European canker in apple in ‘Aroma’ × ‘Discovery’. Tree Genetics & Genomes 19, 12 (2023). https://doi.org/10.1007/s11295-023-01587-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-023-01587-w