
Abstract
In pomegranate (Punica granatum), seed hardness is an important trait directly affecting fruit marketability. However, seed formation in pomegranate has not been well studied. We investigated the genetic mechanism underlying pomegranate seed hardness by comparing protein expression profiles between soft- and hard-seeded varieties 60 and 120 days after flowering. We identified 1940 proteins, of which 399 were differentially expressed. Most of the differentially expressed proteins were involved in posttranslational modification and carbohydrate metabolism. Cell wall biosynthesis, which showed positive correlations with seed hardness, was selected as the candidate pathway. The mRNA levels of 14 proteins involved in cell wall biosynthesis were further analyzed by qPCR. Lignin biosynthesis-related differentially expressed proteins showed lower expression at protein and gene levels in a soft-seeded variety at the early stages. Moreover, cellulose biosynthesis-related differentially expressed proteins showed higher expression levels in the soft-seeded variety at 60 days after flowering. Thus, the soft-seeded variety showed lower lignin but higher cellulose biosynthesis at the early fruit developmental stage, suggesting that lignin and cellulose play opposing roles in cell wall formation in pomegranate seeds. Moreover, differentially expressed proteins involved in cell wall degradation showed higher expression levels in the soft-seeded variety at both developmental stages. These results suggested that differences in seed hardness between soft- and hard-seeded pomegranates might result from cell wall biosynthesis and also be affected by cell wall degradation. The present proteome-wide profiling of pomegranate genotypes with contrasting seed hardness adds to the current knowledge base of the molecular basis of seed hardness development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pomegranate (Punica granatum L.) has been cultivated for more than 2000 years, and it possesses high economic, nutritional, medicinal, and ornamental value (Cao et al. 2015). Pomegranate varieties can be classified into four types on the basis of seed hardness: soft-seeded, semi-soft seeded, semi-hard seeded, and hard-seeded (Khadivi et al. 2015). The soft seed trait in pomegranate is an important factor affecting fruit marketability because it is preferred and more acceptable by customers. Pomegranate has a complex genetic background, and only a few studies have investigated the genetic mechanisms underlying seed hardness in pomegranate. Cao et al. (2015) found that the upregulation of pyruvate dehydrogenase might play a role in the process of pomegranate seed hardness development. Dalimov et al. (2003) indicated that pomegranate seeds are rich in lignin and cellulose, suggesting their involvement in seed hardness. Lignin biosynthesis genes, including WRKY, MYB, and NAC, have higher expression levels in hard-seeded pomegranate variety than in soft-seeded variety and are believed to have key roles in seed formation (Xue et al. 2017). Two cellulose-related genes, cellulose synthase and sucrose synthase (SUS), have demonstrated higher expression levels in a soft-seeded pomegranate variety compared with a hard-seeded one, and their expression correlated with the cellulose content in pomegranate seeds (Zarei et al. 2016). However, the genetic mechanism underlying seed hardness in pomegranate is still not fully understood owing to the limited current knowledge on this aspect.
The cell wall, which is mainly composed of lignin and cellulose, is a key factor affecting seed hardness (Cosgrove 2005). Several studies have identified that cell wall biosynthesis-related genes are differentially expressed between soft- and hard-seeded varieties in fruit trees. Genes associated with lignin biosynthesis, such as caffeic acid 3-O-methyltransferase (COMT), ferulate-5-hydroxylase (F5H), and shikimate O-hydroxycinnamoyl transferase (HCT), were found to be significantly downregulated in soft-seeded hawthorn compared with hard-seeded hawthorn (Dai et al. 2013). Dang suggested that high phenylalanine ammonia lyase and peroxidase (PRX) activity and gene expression were associated with pericarp hardening of mangosteen (Dang et al. 2008). High accumulation of UDP-glucose pyrophosphorylase (UGP) and sucrose phosphate synthase were found to be involved in cell wall synthesis in seeded fruit than in parthenocarpic fruit during fruit development (Rounis et al. 2015).
In addition to cell wall biosynthesis, the cell wall degradation also affects the hardness of fruit. During fruit ripening, various enzymes involved in cell wall degradation show high expression, which is related to the softening of fruit. An α-l-arabinofuranosidase (ARF) gene implicated in cell wall degradation in Japanese pear was specifically expressed during the fruit ripening stage (Tateishi et al. 2005). Furthermore, the cell wall degradation-related genes β-d-xylosidase (XYL) and ARF showed high expression levels and enzymatic activity during tomato ripening (Tateishi et al. 2003). β-Galactosidase has been shown to markedly hydrolyze pectin and hemicellulose, resulting in fruit ripening (Lazan et al. 2004). However, little is known about the cell wall degradation-related gene functions in seed hardness formation. Breeding new soft-seeded pomegranate varieties is essential to meet the increasing demand for good-quality pomegranates. To explore the genetic mechanisms underlying seed hardness in pomegranate, proteomics analysis was performed using two pomegranate varieties: the soft-seeded variety Tunisia and the hard-seeded variety Sanbai. Our findings may provide a useful genetic resource for further analyses on the molecular mechanism of the pomegranate seed hardness.
Materials and methods
Pomegranate varieties: growth and harvest
Two varieties of pomegranates were selected based on their seed hardness and commercially relevant traits. The soft-seeded variety Tunisia, with a seed hardness of 2.12 kg (measured by a texture analyzer), is the most highly consumed soft-seeded pomegranate variety in China. This was chosen as the soft-seeded pomegranate for the study. The variety Sanbai, with a seed hardness of 7.56 kg, was chosen as the hard-seeded variety (Xue et al. 2017). Nine-year-old trees of both varieties were growing in the nursery of the Zhengzhou Fruit Research Institute, Chinese Academy of Agriculture Sciences.
Pomegranate fruit samples were harvested at 60 and 120 days after flowering (DAF). Twenty-four fruits (six replicates per variety and period) were collected, and three replicates were mixed into one biological replicate for proteomics analysis. The fruit seeds were rapidly collected and immediately snap-frozen in liquid nitrogen. The aril was peeled off for seed protein and RNA extraction. All frozen samples were stored at − 80 °C until experimental use.
Quantitative proteomics
Chemicals and regents used for iTRAQ analysis were purchased from various suppliers, and all solvents were of LC-MS grade (Supplementary Table 1).
Protein extraction, trypsin digestion, and iTRAQ labelling
Proteins from pomegranate seeds were extracted using the trichloroacetic acid (TCA)/acetone method with minor modifications (Mechin et al. 2003). The samples were ground with liquid nitrogen, and the powders were transferred to 5 mL centrifuge tubes and sonicated with lysis buffer (containing 8 M urea, 1% Triton-100, 65 mM dithiothreitol, and 0.1% protease inhibitor cocktail) three times on ice by using a high-intensity ultrasonic processor (Scientz, Ningbo, China). The remaining debris was removed by centrifugation at 20,000×g at 4 °C for 10 min. The protein was precipitated with cold 15% TCA for 2 h at − 20 °C. After centrifugation, the precipitate was redissolved in a buffer (8 M urea, 100 mM tetraethylammonium bromide, pH 8.0), and the protein concentration was determined using a 2-D Quant kit (GE Healthcare, Piscataway, NJ, USA). After the proteins were detected using SDS-PAGE, the iTRAQ experiment was performed as previously described (Jin et al. 2016).
The protein solution was reduced with 10 mM dithiothreitol for 1 h at 37 °C and alkylated with 20 mM indole acetic acid for 45 min at room temperature in the dark. It was then diluted by adding 100 mM tetraethylammonium bromide to a urea concentration of less than 2 M. Subsequently, each sample (approximately 100 μg protein) was digested with trypsin at a 1:50 trypsin-to-protein mass ratio for the first digestion overnight, and a 1:100 trypsin-to-protein mass ratio for the second 4 h digestion.
After trypsin digestion, the peptides were desalted using a Strata X-C18 SPE column (Phenomenex, Los Angeles, USA) and vacuum-dried. The peptides were reconstituted in 0.5 M tetraethylammonium bromide and processed according to the manufacturer’s protocol for the 8plex iTRAQ kit (AB Sciex, Foster City, CA, USA). Briefly, 1 U of iTRAQ reagent (defined as the amount of reagent required to label 100 μg of protein) was thawed and reconstituted in 24 μL acetonitrile. The peptide mixtures were then incubated for 2 h at room temperature, pooled, desalted, and dried by vacuum centrifugation.
The peptide sample was fractionated using high-pH reverse-phase high-performance liquid chromatography by using the Agilent 300 Extend C18 column (5 μm, 4.6 mm × 250 mm). Briefly, peptides were first separated with a gradient of 2 to 60% acetonitrile in 10 mM ammonium bicarbonate (pH 10) over 80 min into 80 fractions. Next, they were combined into 18 fractions and dried by vacuum centrifugation.
Liquid chromatography–tandem mass spectrometry analysis
The peptides were dissolved in 0.1% formic acid and loaded onto a reversed-phase pre-column (Acclaim PepMap 100; Thermo Scientific, Waltham, MA, USA). They were separated using a reverse-phase analytical column (Acclaim PepMap RSLC; Thermo Scientific). The gradient included an increase from 7 to 20% solvent B (0.1% formic acid in 98% acetonitrile) over 22 min, 20 to 35% for 6 min, and a gradual increase to 80% for 3 min, followed by holding at 80% for the last 4 min. The column was run at 300 nL min−1 on an EASY-nLC 1000 UPLC system. All peptide samples were subjected to a nanoelectrospray ionization source, followed by tandem mass spectrometry (MS/MS) by using the Q Exactive™ Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to an ultra-performance liquid chromatography system. For MS scans, intact peptides were detected in the Orbitrap with the m/z scan range of 350–1800 at a resolution of 70,000. Peptides were selected for MS/MS by using the normalized collision energy setting as 35. Ion fragments were detected using Orbitrap at a resolution of 17,500. The fixed first mass was set as 100 m/z. The MS spectra were acquired using a data-dependent procedure that alternated between MS1 and MS2, followed by the acquisition of 20 MS/MS scans with up to 20 precursor ions above a threshold ion count of 2E4 in the MS survey scan. The dynamic exclusion duration was 15.0 s, and the electrospray voltage applied was 2.0 kV. Automatic gain control was used to prevent overfilling of the Orbitrap; 5E4 ions were accumulated for the generation of MS/MS spectra.
Real-time quantitative reverse transcription PCR
Total RNA was extracted from pomegranate seeds using the CTAB-LiCl method. The actin gene, which is expressed similarly in soft- and hard-seeded pomegranates, was used for the normalization of gene expression (Chen et al. 2017). Total RNA of each sample was treated with DNase I (Thermo Scientific) to exclude genomic DNA contamination. Then, the RNA samples were analyzed at an absorbance ratio of A260/A280 with a Nanodrop ND-1000 system (Thermo Scientific). First-strand complementary DNA (cDNA) was synthesized from 2 μg of total RNA using PrimeScript RT reagent kit with gDNA Eraser (Takara, Shiga, Japan) following the instruction manual. Reactions were performed in triplicate for each sample, and the Roche LightCycler 480/480II (Roche, Basel, Switzerland) with SYBR Green PCR master mix (Roche Diagnostics, Penzberg, Germany) were used. In this study, the primers were designed on the basis of conserved sequences detected by de novo transcriptome sequencing of Punica granatum (Xue et al. 2017). Gene sequences were blasted against the NCBI database, and primers were designed using Primer 5.0 software for PCR experiments (Supplementary Table 2). The amplification reactions contained 10 μL SYBR Green PCR master mix, 2 μL cDNA, and 0.5 μL of each primer in a final reaction volume of 20 μL. The thermal cycling program began with 5 min at 95 °C, followed by 45 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 10 s.
MS data analysis
The resulting MS/MS data were processed using the Mascot search engine (v.2.3.0). Tandem mass spectra were searched against the Punica granatum transcriptome sequence database (Xue et al. 2017). Trypsin/P was specified as a cleavage enzyme, which allows up to two missing cleavages. Cysteine carbamidomethylation, iTRAQ-8plex (N-term), and iTRAQ-8plex (K) were specified as fixed modifications, and methionine oxidation was specified as the variable modification. The false discovery rate was adjusted to < 1%, and the peptide ion score was set as > 20. The DEPs were the proteins with more than 1.2-fold change (p < 0.05) between Tunisia and Sanbai. The mass spectrometry proteomics data have been uploaded to the ProteomeXchange database. Functional categorization was performed using MapMan (http://mapman.gabipd.org/) (Thimm et al. 2004). The heat map was visualized using heatmap 2.0 in the gplot R package. The protein functional network was analyzed by STRING 9.0 (http://string-db.org). Expression stability of the target genes was analyzed using the Delta Ct method. The analysis of statistically significant differences from gene expression was performed by the independent samples t test analysis at p < 0.05 using SPSS 17.0 software. Graphs were constructed using Excel software. Correlation analysis between cell wall-related gene and protein expression was carried out with Pearson’s correlation coefficient analysis (Sato et al. 2015).
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD007659.
Results
Quantitative proteomics of pomegranate seeds
Quantitative proteomics was used to identify the DEPs between soft- and hard-seeded pomegranate varieties at 60 and 120 DAF. The mass error of all identified peptides was checked. The distribution of the mass error was near 0, and most of the errors were less than 0.02 Da, indicating that the identified MS data conformed to the experimental requirement. Furthermore, the length of most peptides was distributed between 8 and 16, which fitted with the property of tryptic peptides. A total of 166,169 spectra were generated from the iTRAQ experiment by using soft- and hard-seeded pomegranate tissue. Of these, 13,005 spectra were matched to known spectra, and 15,272 unique spectra matched to 19,015 unique peptides. Of these, 7604 unique peptides corresponded to 1940 unique proteins, and 1889 proteins were quantified (Fig. 1a). In terms of protein mass distribution, a good coverage was obtained for a wide molecular weight range for proteins larger than 7 kDa (Fig. 1b). In all, 399 proteins were differentially expressed between Tunisia and Sanbai (Supplementary Table 3). Of these, 117 were upregulated and 95 were downregulated in Tunisia compared with Sanbai at 60 DAF; 50 were upregulated and 137 were downregulated in Tunisia compared with Sanbai at 120 DAF; and 64 were co-expressed at 60 and 120 DAF.

Peptides and proteins that were identified from iTRAQ proteomics by searching against the database. (a) Number of peptides that match proteins using MASCOT. (b) Distribution of the proteins that were identified among different molecular weights
Functional classification of the identified DEPs
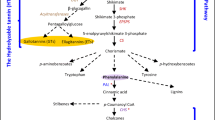
Bioinformatics analysis was performed to annotate and classify the DEPs on the basis of protein functional classification and hierarchical cluster analysis. MapMan analysis revealed that most of the DEPs were involved in carbohydrate metabolism and posttranslational modification (Fig. 2). Among these, most of the regulated proteins were upregulated at 60 DAF and downregulated at 120 DAF in Tunisia compared with Sanbai. Notably, only cell wall-related DEPs showed consistent downregulation during both growth periods in Tunisia. Moreover, about 30% of the DEPs were involved in translation and posttranslational modification. To further explore the expression change in cell wall-related DEPs, hierarchical cluster analysis was used (Fig. 3). Compared with Sanbai, in Tunisia, 38 cell wall-related DEPs were clustered closely at 60 and 120 DAF. Most cell wall-related DEPs were involved in lignin biosynthesis (8; Fig. 3a), glycoside hydrolase function (7; Fig. 3b), sucrose metabolism (11; Fig. 3c), and sugar metabolism (12; Fig. 3d).

MapMan classification analysis of pomegranate differentially expressed proteins (DEPs) in Tunisia compared with Sanbai at 60 and 120 DAF. The log2-fold change in the proteomic levels was used in the analysis. Green and red indicate down- and up-regulated proteins, respectively

Heat map of different expression levels of 38 proteins in Tunisia compared with Sanbai at 60 and 120 DAF, which are associated with cell wall metabolic processes. Red indicates significantly upregulated proteins, and green indicates significantly downregulated proteins. Black indicates proteins with no significant changes. (a) Heat map of phenylpropanoid biosynthesis-associated protein expression. (b) Heat map of galactose metabolism-associated protein expression. (c) Heat map of starch and sucrose metabolism-associated protein expression. (d) Heat map of amino sugar and nucleotide sugar metabolism-associated protein expression. c16669_g2: cinnamyl-alcohol dehydrogenase; c4938_g1: cinnamyl-alcohol dehydrogenase 2; c7211_g1: peroxidase; c1747_g1: ferulate-5-hydroxylase; c13334_g1: shikimate O-hydroxycinnamoyl transferase; c20820_g2: peroxiredoxin; c30491_g1: cinnamyl-alcohol dehydrogenase 9; c26596_g1: β-glucosidase; c32663_g1: trehalose 6-phosphate synthase/phosphatase; c14647_g1: beta-fructofuranosidase1; c15956_g1: UDP-glucose 6-dehydrogenase; c13244_g2: sucrose synthase 1; c23454_g1: glucuronosyl transferase; c1196_g1: β-fructofuranosidase 4; c13860_g1: β-d-xylosidase 1; c17832_g1: UTP-glucose-1-phosphate uridylyltransferase; c13244_g1: sucrose synthase 3; c26596_g1: β-glucosidase; g.117645: β-d-xylosidase 4; c13860_g1: β-d-xylosidase 1; c17832_g1: UTP-glucose-1-phosphate uridylyltransferase; c15956_g1: UDP glucose 6-dehydrogenase; c11819_g1: alpha-N-arabinofuranosidase; c12832_g1: reversibly glycosylated polypeptide/UDP-arabinopyranose mutase; c16913_g4: glucose-6-phosphate isomerase; c6593_g1: chitinase; g.240956: GDP-d-mannose 3′,5′-epimerase; c20858_g1: chitinase; c34241_g1: glucuronokinase; c8135_g1: cytochrome-b5 reductase; g.11830: raffinose synthase; c12404_g1: alpha-1,3-glucosidase; c23470_g1: raffinose synthase; c14647_ g1: beta-fructofuranosidase1; c6366_g1: alpha-mannosidase; c1196_g1: beta-fructofuranosidase 4; c11906_g1: alpha-glucosidase
Interaction analysis of DEPs
Seed maturation is a complicated physiological and biochemical process in plants. Development from normal ovules into mature seeds is regulated by the co-expression of many proteins in the short term. In the present study, the signal transmission underlying the development of seed hardness in pomegranate was studied by analysing the protein–protein interactions using the STRING database. We identified 505 and 549 proteins to be involved in protein–protein interaction at 60 and 120 DAF, respectively (Supplementary Fig. 1 and Supplementary Fig. 2). Among these, 7 proteins were involved in phenylpropanoid biosynthesis and 15 proteins were involved in starch and sucrose metabolism (Fig. 4). In the interaction network, cell wall-related DEPs, such as cinnamyl-alcohol dehydrogenase (CAD), PRX, HCT, F5H (Fig. 4a), UTP-glucose-1-phosphate uridylyltransferase (UGP2), beta-d-xylosidase 4 (XYL4), beta-d-xylosidase 1 (XYL1), beta-1,3-glucosidase (β-GLU), and sucrose synthase 3 (SUS3) (Fig. 4b) interacted with most of the other DEPs, indicating that they are the most suitable candidates for further studies on seed hardness development.

Analysis of the functional network by STRING 9.0 of the cell-wall related proteins in Tunisia compared with Sanbai at 60 and 120 DAF. A confidence level of 0.4 was applied as the analysis parameter. Different line colours represent the types of evidence used in predicting the associations: gene fusion (red), neighbourhood (green), co-occurrence across genomes (blue), co-expression (black), experimental (purple), association in curated databases (light blue), or co-mentioned in PubMed abstracts (yellow). Two clusters of highly interacting protein nodes are marked with circles and include proteins involved in (a) phenylpropanoid biosynthesis metabolism, (b) starch and sucrose metabolism and amino sugar and nucleotide sugar metabolism. c30491_g1: cinnamyl-alcohol dehydrogenase 9 (CAD9); c16669_g2: cinnamyl-alcohol dehydrogenase (CAD); c7211_g1: peroxidase (PRX); c1747_g1: ferulate-5-hydroxylase (F5H); c13334_g1: shikimate O-hydroxycinnamoyl transferase (HCT); c20820_g2: peroxiredoxin 6 (PRXII6); c10292_g1: peroxiredoxin 2 (PRXII2); c32663_g1: trehalose 6-phosphate synthase/phosphatase (TPS); c14647_g1: beta-fructofuranosidase (FFase1); c15956_g1: UDP-glucose 6-dehydrogenase (UGD); c13244_g2: sucrose synthase 1 (SUS1); c1196_g1: β-fructofuranosidase 4 (FFase4); c17832_g1: UTP-glucose-1-phosphate uridylyl-transferase (UGP2); c13244_g1: sucrose synthase 3 (SUS3); c26596_g1: β-glucosidase (β-GLU); g.117645: β-d-xylosidase 4 (XYL4); c6593_g1: chitinase (CHI), c12832_g1: reversibly glycosylated polypeptide/UDP-arabinopyranose mutase (RGP); c16913_g4: glucose-6-phosphate isomerase (GPI); g.240956: GDP-d-mannose 3′,5′-epimerase (GME); c11819_g1: alpha-N-arabinofuranosidase (ARF); c23454_g1: glucuronosyltransferase (UGT)
Network analysis between proteomics and transcriptomics
To compare protein abundance changes with transcript-level alterations during seed development, the correlation of proteome data with transcriptome data was determined. The transcript data were obtained from our previous research with the same varieties and fruit maturation stages (Xue et al. 2017). The results showed that 328 and 1456 proteins were related to known transcripts at 60 and 120 DAF, respectively, by using RNA-seq (Fig. 5). Among them, 27 and 35, respectively, had corresponding changes in the RNA-seq data (Supplementary Table 4).

Venn diagram intersection of differentially expression protein and mRNA in Tunisa compared with Sanbai at 60 and 120 DAF
Gene expression and correlation analysis
To investigate the molecular mechanism underlying pomegranate seed hardness at the transcription level, 14 candidate proteins involved in cell wall biosynthesis—CAD, CAD2, CAD9, PRX, HCT, F5H, SUS3, UGP2, GST, XYL4, XYL1, α-MAN, FFase1, and β-GLU—were chosen from the DEPs based on the present proteomics analysis and previous transcriptional-level analyses by quantitative RT-PCR (Dai et al. 2013; Dang et al. 2008; Rounis et al. 2015; Tateishi et al. 2003). At 60 and 120 DAF, most of the candidate genes were expressed differently between Tunisia and Sanbai (Fig. 5). Specifically, at 60 DAF, lignin-related candidate genes, CAD, CAD2, CAD9, PRX, and HCT, were significantly upregulated in Tunisia compared with Sanbai; GST also showed upregulation, but the difference was not significant; and F5H was significantly downregulated. Among cellulose-related genes, SUS3 and UGP2 were significantly upregulated in Tunisia compared with Sanbai, and the cell wall degradation-related gene α-MAN was significantly upregulated and FFase1 was significantly downregulated. At 120 DAF, the lignin biosynthesis genes CAD, PRX, and F5H were significantly downregulated, while GST and CAD2 were significantly upregulated in Tunisia compared with Sanbai, and HCT and CAD9 did not show significant expression difference between the varieties. Among cellulose-related genes, SUS3 was significantly downregulated, and the expression difference of UGP2 was not significant. The cell wall degradation-related genes XYL4, β-GLU, FFase1, and α-MAN were significantly upregulated, while XYL1 was significantly downregulated. We further investigated the correlations between protein and mRNA expression profiles to compare Tunisia and Sanbai. The expression of seven candidate genes showed strong correlations with their encoding proteins, including F5H (r = 0.873, p < 0.01), PRX (r = 0.835, p < 0.01), GST (r = 0.815, p < 0.05), XYL4 (r = 0.720, p < 0.05), CAD (r = 0.711, p < 0.05), FFase1 (r = 0.7839, p < 0.05), and CAD2 (r = 0.983, p < 0.01) (Supplementary Table 5). Seven genes, HCT, SUS3, UGP2, XYL1, β-GLU, CAD9, and α-MAN, showed poor correlation with their corresponding protein expression.
Discussion
Proteomics analysis for DEPs in soft- and hard-seeded pomegranate varieties
Seed hardness in pomegranate is of economic relevance, and it affects the market value of the fruit. However, the molecular mechanism of seed hardness is still not fully understood in pomegranate. In recent years, with the continuous development of proteomics, researchers have focused on investigating seed hardness development using protein profiling techniques. Previous studies have evaluated the proteome-level differences in seeds between Zhongnonghong (soft-seeded) and Sanbai (hard-seeded) pomegranate varieties (Cao et al. 2015). However, the 2-DE technique has many limitations, such as relative quantitation only, low resolution and proteome coverage, and low data throughput (Li et al. 2013). An iTRAQ-based quantitative proteomics analysis was conducted in the present study, which provides whole-proteome profiling with more accurate and sensitive protein quantification. Compared with 2-DE results (Cao et al. 2015), more than twice the total number of proteins (1940 versus 892) and 4 times the DEPs (399 versus 76) were identified in pomegranate seeds in the current study.
Most of the DEPs were involved in translational and posttranslational modification. Notably, DEPs involved in lipid metabolism and defence mechanisms showed significant differences between soft- and hard-seeded pomegranate varieties. However, these differences might be influenced by genetic variability between the two varieties, and they are not correlated with the development of seed hardness in pomegranate seeds (Eikani et al. 2012; Kalaycıoğlu and Erim 2017). Only cell wall-related DEPs showed positive correlations with seed hardness changes in pomegranates at 60 DAF and 120 DAF in the current study. The cell wall has also been known to play important roles in seed hardness development in other fruits (Dai et al. 2013; Rounis et al. 2015). Thus, cell wall-related DEPs were chosen as research candidates, and their gene expression was further investigated. Seven of them showed correlated expressions at gene and protein expression levels. The expression of 7 of 14 candidate genes showed poor correlation with their protein expression. The inconsistency between transcriptional and translational expression could be attributed to posttranscriptional and posttranslational regulation, reversible phosphorylation, and translation efficiency, which has also been found in many other studies (Battle et al. 2015; Weeda et al. 2010). This inconsistency indicated that the efficiency of gene translation is an important regulatory factor during pomegranate seed development.
DEPs involved in the cell wall biosynthesis pathway determine pomegranate seed hardness
Lignin plays an important role in the structural integrity of the secondary cell wall and provides stiffness and mechanical strength to the cell wall. Several genes, such as F5H, HCT, and CAD, have been found to regulate lignin biosynthesis. The lignin polymer generally consists of syringyl (S), guaiacyl (G), and p-hydroxyphenyl (H) units (Boerjan et al. 2003). Bonawitz indicated that F5H specifically participated in S lignin biosynthesis (Bonawitz and Chapple 2010). F5H overexpression resulted in a 97.5% increase in S lignin unit biosynthesis in poplar (Stewart et al. 2009). Moreover, F5H overexpression in Arabidopsis thaliana also increased the deposition of S lignin (Sibout et al. 2002). The silencing of HCT in Arabidopsis thaliana resulted in a distinct alteration in lignin composition, causing an increase in H units and a decrease in S units (Hoffmann et al. 2004). Zhao et al. (2013) found that a CAD1 mutant with lignification defects led to a major alteration in lignin structure in Medicago truncatula. In this study, eight DEPs involved in lignin biosynthesis were identified, among which F5H and HCT proteins were downregulated in Tunisia compared with Sanbai, and their corresponding genes were also downregulated consistently (Figs. 3 and 6). These results imply that low expression of lignin biosynthesis-related proteins leads to a reduced deposition of lignin in the soft-seeded variety Tunisia. Therefore, F5H and HCT can be regarded as candidate proteins for further functional characterization to understand the development of seed hardness. In addition, the gene and protein expression of CAD, CAD2, and CAD9 was upregulated at 60 DAF in Tunisia compared with Sanbai. However, this result was inconsistent with related studies in strawberry, peach, and loquat (Salentijn et al. 2003; Prinsi et al. 2011; Shan et al. 2008). Therefore, the role of CAD proteins in pomegranate seed hardness needs to be further studied.

The expression levels of cell wall-related genes quantified by qRT-PCR. c16669_g2: cinnamyl-alcohol dehydrogenase (CAD); c7211_g1: peroxidase (PRX); c1747_g1: ferulate-5-hydroxylase (F5H); c13244_g1: sucrose synthase (SUS3), g.117645: beta-d-xylosidase 4 (XYL4), c15749_g1: glutathione S-transferase (GST), c13334_g1: shikimate O-hydroxycinnamoyl transferase (HCT); c17832_g1: UTP-glucose-1-phosphate uridylyltransferase (UGP2); c13860_g1: beta-d-xylosidase 1 (XYL1); c4938_g1: cinnamyl-alcohol dehydrogenase 2 (CAD2); c30491_g1: cinnamyl-alcohol dehydrogenase 9 (CAD9); c26596_g1: β-glucosidase (β-GLU);c14647_g1: beta-fructofuranosidase (FFase1); c6366_g1: alpha-mannosidase (α-MAN)
Cellulose is also a central component in the plant cell wall, and it is highly abundant in the secondary cell wall. Recent studies have shown that UGP and SUS are closely associated with cellulose biosynthesis in plants (Andersson et al. 2006). For example, UGP overexpression was shown to increase the height and cellulose content in jute (Zhang et al. 2013) and the cellulose content in cotton (Wang et al. 2011) and poplar (Coleman et al. 2007). SUS overexpression in poplar resulted in increased cellulose deposition and thicker secondary cell walls in the wood (Coleman et al. 2009). Moreover, cellulose deposition has been found to be correlated with high SUS activity in wheat roots (Albrecht and Mustroph 2003). In the present study, we found that UGP2 and SUS3 were upregulated at 60 DAF and downregulated at 120 DAF at both gene and protein expression levels in Tunisia compared with Sanbai (Figs. 3 and 6). This is in accordance with results from an earlier study that indicated that soft-seeded pomegranate varieties had higher cellulose-related gene expression and cellulose content in contrast with the hard-seeded variety (Zarei et al. 2016). Thus, it is possible that the higher expression of these two genes in Tunisia at 60 DAF had a close correlation with cellulose biosynthesis and seed hardness development in pomegranate. The higher lignin-related protein expression and lower cellulose-related protein expression in Tunisia compared with Sanbai indicate that lignin and cellulose deposition might play contradictory roles in the cell wall structure in soft-seeded pomegranate.
DEPs involved in cell wall remodelling and degradation pathways in pomegranate seed hardness formation
Several glycosyl hydrolases (GHs) are involved in cell wall degradation, such as ARF, XYL, alpha 1,3-glucosidase (α-GLU), β-GLU, and alpha-mannosidase (α-MAN), and they play important roles in fruit development (Brummell et al. 2004). An ARF gene in peach showed increased expression during fruit ripening and has been identified as a fruit softening-related gene (Di et al. 2009; Hayama et al. 2006). In sweet cherry, β-GLU with high activity in the cell wall was found to participate in the degradation of cell wall components and lead to fruit softening (Gerardi et al. 2001). Decreased XYL activity in the siliques of Arabidopsis thaliana altered the composition of the secondary cell wall (Goujon et al. 2003). Moreover, XYL was found to be involved in hemicellulose degradation in Cellulosimicrobium cellulans (Yuan et al. 2016). The activity of α-MAN significantly and steadily increases during fruit ripening in tomato and may play a critical role in fruit softening (Jagadeesh et al. 2004). Consistent with these findings, in this study, XYL4 protein and gene expression were upregulated at 60 and 120 DAF in the soft-seeded variety compared with the hard-seeded one (Figs. 3 and 6). Moreover, α-GLU, FFase1, XYL1, β-GLU, α-MAN, and ARF also showed higher protein expression in the soft-seeded variety at seed developmental stages (Figs. 3 and 6). The significantly increased expression of these GH proteins suggests their dynamic roles in pomegranate seed softening. In addition, other cell wall degradation-related proteins also showed increased expression in soft-seeded pomegranate. GST played a critical role in beta-aryl ether cleavage, which is an indispensable step in lignin degradation (Otsuka et al. 2003; Masai et al. 2003). PRX can induce cell wall loosening, and it is generally considered a lignin-degrading enzyme (Passardi et al. 2004; Adav et al. 2012). In this study, PRX protein and gene expression were upregulated at 60 DAF (Figs. 3 and 6), and GST protein and gene expression were upregulated at 120 DAF in the soft-seeded variety compared with the hard-seeded one (Supplementary Table 3; Fig. 6). Thus, the GST and PRX proteins might also have certain roles in pomegranate seed softening.
Conclusions
The data presented herein provide new information on the proteomes in pomegranate seeds, identifying several candidate proteins determining seed hardness. The results showed that lignin biosynthesis-related proteins were significantly downregulated in the soft-seeded variety compared with the hard-seeded one. However, cellulose biosynthesis-related proteins and their corresponding genes showed significantly higher expression in the soft-seeded variety at 60 DAF but lower expression at 120 DAF. This indicates that the soft-seeded variety showed lower lignin but higher cellulose biosynthesis at the early fruit development stage, which suggests that lignin and cellulose play contradictory roles in cell wall formation in soft-seeded pomegranate. A similar result has also been identified in a previous study (Zarei et al. 2016). In addition, DEPs involved in cell wall degradation, including XYL4, α-GLU, β-GLU, ARF, FFase1, and α-MAN, showed higher expression levels in the soft-seeded variety at both developmental stages. Altogether, these findings provide important insights into pomegranate seed formation and indicate that cell wall biosynthesis and degradation play important roles in the development of seed hardness. Furthermore, the identified candidate proteins could form the basis for conducting further studies to analyze the molecular mechanisms underlying seed hardness in pomegranate and provide a theoretical basis for the cultivation of new varieties of soft seed fruit.
References
Adav SS, Ravindran A, Sze SK (2012) Quantitative proteomic analysis of lignocellulolytic enzymes by Phanerochaete chrysosporium on different lignocellulosic biomass. J Proteome 75(5):1493–1504. https://doi.org/10.1016/j.jprot.2011.11.020
Albrecht G, Mustroph A (2003) Localization of sucrose synthase in wheat roots: increased in situ activity of sucrose synthase correlates with cell wall thickening by cellulose deposition under hypoxia. Planta 217(2):252–260. https://doi.org/10.1007/s00425-003-0995-6
Andersson-Gunnerås S, Mellerowicz EJ, Love J et al (2006) Biosynthesis of cellulose-enriched tension wood in Populus: global analysis of transcripts and metabolites identifies biochemical and developmental regulators in secondary wall biosynthesis. Plant J 45:144–165. https://doi.org/10.1111/j.1365-313X.2005.02584, 2
Battle A, Khan Z, Wang SH (2015) Impact of regulatory variation from RNA to protein. Sci 347(6222):664–667. https://doi.org/10.1126/science.1260793
Boerjan W, Ralph J, Baucher M (2003) Lignin biosynthesis. Ann Rev Plant Biol 54(1):519–546. https://doi.org/10.1146/annurev.arplant.54.031902.134938
Bonawitz ND, Chapple C (2010) The genetics of lignin biosynthesis: connecting genotype to phenotype. Annu Rev Genet 44(1):337–363. https://doi.org/10.1146/annurev-genet-102209-163508
Brummell DA, Cin VD, Crisosto CH, Labavitch JM (2004) Cell wall metabolism during maturation, ripening and senescence of peach fruit. J Exp Bot 55(405):2029–2039. https://doi.org/10.1093/jxb/erh227
Cao SH, Niu J, Cao D et al (2015) Comparative proteomics analysis of pomegranate seeds on fruit maturation period (Punica granatum L.) JIA 14(12):60345–60352. https://doi.org/10.1016/S2095-3119(15)61029-2
Chen L, Zhang J, Li H et al (2017) Transcriptomic analysis reveals candidate genes for female sterility in pomegranate flowers. Front Plant Sci 8:1430. https://doi.org/10.3389/fpls.2017.01430
Coleman HD, Canam T, Kang KY et al (2007) Over-expression of UDP-glucose pyrophosphorylase in hybrid poplar affects carbon allocation. J Exp Bot 58(15-16):4257–4268. https://doi.org/10.1093/jxb/erm287
Coleman HD, Yan J, Mansfield SD (2009) Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc Natl Acad Sci U S A 106(31):13118–13123. https://doi.org/10.1073/pnas.0900188106
Cosgrove DJ (2005) Growth of the plant cell wall. Nat Rev Mol Cell Bio 6(11):850–861. https://doi.org/10.1038/nrm1746
Dai H, Han G, Yan et al (2013) Transcript assembly and quantification by RNA-Seq reveals differentially expressed genes between soft-endocarp and hard-endocarp hawthorns. PLoS One 8(9):1–11. https://doi.org/10.1371/journal.pone.0072910
Dalimov DN, Dalimova GN, Bhatt M (2003) Chemical composition and lignins of tomato and pomegranate seeds. Chem Nat Compd 39(12):37–40. https://doi.org/10.1016/S2095-3119(15)61029-2
Dangcham S, Bowen J, Ferguson IB, Ketsa S (2008) Effect of temperature and low oxygen on pericarp hardening of mangosteen fruit stored at low temperature. Postharvest Biol Tec 50(1):37–44. https://doi.org/10.1016/j.postharvbio.2008.02.005
Di SMC, Pagano EA, Sozzi GO (2009) Differential expression of a-L-arabinofuranosidase and a-L-arabinofuranosidase/β-D-xylosidase genes during peach growth and ripening. Plant Physiol Bioch 47(7):562–569. https://doi.org/10.1016/j.plaphy.2009.02.007
Eikani MH, Golmohammad F, Homami SS (2012) Extraction of pomegranate (Punica granatum L.) seed oil using superheated hexane[J]. Food Bioprod Process 90(1):32–36. https://doi.org/10.1016/j.fbp.2011.01.002
Gerardi C, Blando F, Santino A (2001) Purification and characterisation of a β-glucosidase abundantly expressed in ripe sweet cherry (Prunus avium L.) fruit. Plant Sci 160(5):795–805. https://doi.org/10.1016/S0168-9452(00)00423-4
Goujon T, Minic Z, EIAmrani A et al (2003) AtBXL1, a novel higher plant (Arabidopsis thaliana) putative β-xylosidase gene, is involved in secondary cell wall metabolism and plant development. Plant J 33(4):677–690. https://doi.org/10.1046/j.1365-313X.2003.01654.x
Hayama H, Shimada T, Fujii H (2006) Ethylene-regulation of fruit softening and softening-related genes in peach. J Exp Bot 57(15):4071–4077. https://doi.org/10.1093/jxb/erl178
Hoffmann L, Besseau S, Geoffroy PC et al (2004) Silencing of hydroxycinnamoyl-coenzyme A shikimate/quinate hydroxycinnamoyl transferase affects phenylpropanoid biosynthesis. Plant Cell 16(6):1446–1465. https://doi.org/10.1105/tpc.020297
Jagadeesh BH, Prabha TN, Srinivasan K (2004) Activities of glycosidases during fruit development and ripening of tomato (Lycopersicum esculantum L.): implication in fruit ripening. Plant Sci 166(6):1451–1459. https://doi.org/10.1016/j.plantsci.2004.01.028
Jin W, Wu F (2016) Proteome-wide identification of lysine succinylation in the proteins of tomato (Solanum lycopersicum). PLoS One 11:1–12. https://doi.org/10.1371/journal.pone.0147586
Khadivi-Khub A, Kameli M, Moshfeghi N et al (2015) Phenotypic characterization and relatedness among some Iranian pomegranate (Punica granatum L.) accessions. Trees 293(3):893–901. https://doi.org/10.1007/s00468-015-1172-9
Kalaycıoğlu Z, Erim FB (2017) Total phenolic contents, antioxidant activities, and bioactive ingredients of juices from pomegranate cultivars worldwide. Food Chem 221:496–507. https://doi.org/10.1016/j.foodchem.2016.10.084
Lazan H, Ng SY, Goh LY (2004) Papaya β-galactosidase/galactanase isoforms in differential cell wall hydrolysis and fruit softening during ripening. Plant Physiol Biochem 42(11):748–853. https://doi.org/10.1016/j.plaphy.2004.10.007
Li H, Li G, Zhao X et al (2013) Complementary serum proteomic analysis of autoimmune hepatitis in mice and patients. J Transl Med 11:113. https://doi.org/10.1186/1479-5876-11-146
Masai E, Ichimura A, Sato Y et al (2003) Roles of the enantioselective glutathione S-transferases in cleavage of beta-aryl ether. J Bacteriol 185(6):1768–1775. https://doi.org/10.1128/JB.185.6.1768-1775.2003
Mechin V, Consoli L, Guilloux M, Damerval C (2003) An efficient solubilization buffer for plant proteins focused in immobilized pH gradients. Proteomics 3(7):1299–1302. https://doi.org/10.1002/pmic.200300450
Otsuka Y, Sonoki T, Ikeda S et al (2003) Detection and characterization of a novel extracellular fungal enzyme that catalyzes the specific and hydrolytic cleavage of lignin guaiacylglycerol beta-aryl ether linkages. Eur J Biochem 270(11):2353–2362. https://doi.org/10.1046/j.1432-1033.2003.03545.x
Passardi F, Penel C, Dunand C (2004) Performing the paradoxical: how plant peroxidases modify the cell wall. Trends Plant Sci 9(11):534–540. https://doi.org/10.1016/j.tplants.2004.09.002
Prinsi B, Negri AS, Fedeli C, Morgutti S, Negrini N, Cocucci M, Espen L (2011) Peach fruit ripening: aproteomic comparative analysis of the mesocarp of twocultivars with different flesh firmness at two ripening stages. Phytochemistry 72(10):1251–1262. https://doi.org/10.1016/j.phytochem.2011.01.012
Rounis V, Skarmoutsos K, Tsaniklidis G et al (2015) Seeded and parthenocarpic cherry tomato fruits exhibit similar sucrose, glucose, and fructose levels, despite dissimilarities in UGPase and SPS gene expression and enzyme activity. J Plant Growth Regul 34(1):47–56. https://doi.org/10.1007/s00344-014-9441-1
Salentijn EM, Aharoni A, Schaart JG et al (2003) Differential gene expression analysis ofstrawberry cultivars that differ in fruit firmness. Physiol Plant 118:571–578. https://doi.org/10.1034/j.1399-3054.2003.00138
Sato A, Zhang T, Yonekura L et al (2015) Antiallergic activities of eleven onions (Allium cepa) were attributed to quercetin 4′-glucoside using QuEChERS method and Pearson’s correlation coefficient. J Funct Foods 14:581–589. https://doi.org/10.1016/j.jff.2015.02.029
Shan L, Li X, Wang P et al (2008) Characterization of cDNAs associated with lignification and their expression profiles in loquat fruit with different lignin accumulation. Planta 227(6):1243–1254. https://doi.org/10.1007/s00425-008-0696-2
Sibout R, Baucher M, Gatineau M et al (2002) Expression of a poplar cDNA encoding a ferulate-5-hydroxylase/coniferaldehyde 5-hydroxylase increases S lignin deposition in Arabidopsis thaliana. Plant Physiol Biochem 40(12):1087–1096. https://doi.org/10.1016/S0981-9428(02)01474-2
Stewart JJ, Akiyama T, Chapple C et al (2009) The effects on lignin structure of overexpression of ferulate 5-hydroxylase in hybrid poplar. Plant Physiol 150:621–635. https://doi.org/10.1104/pp.109.137059
Tateishi A, Mori H, Watari J et al (2003) AtBXL1, a novel higher plant (Arabidopsis thaliana) putative β-xylosidase gene, is involved in secondary cell wall metabolism and plant development. Plant J 33(3):677–690. https://doi.org/10.1104/pp.104.056655
Tateishi A, Mori H, Watari J et al (2005) Isolation, characterization, and cloning of a-L-arabinofuranosidase expressed during fruit ripening of Japanese pear. Plant Physiol 138(3):1653–1664. https://doi.org/10.1104/pp.104.056655
Thimm O, Bläsing O, Gibon Y et al (2004) mapman: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37(6):914–939. https://doi.org/10.1111/j.1365-313X.2004.02016.x
Wang Q, Zhang X, Li F et al (2011) Identification of a UDP-glucose pyrophosphorylase from cotton (Gossypium hirsutum L.) involved in cellulose biosynthesis in Arabidopsis thaliana. Plant Cell Rep 30:1303–1312. https://doi.org/10.1007/s00299-011-1042-x
Weeda SM, Kumar GN, Knowles NR (2010) Correlative changes in proteases and protease inhibitors during mobilisation of protein from potato (Solanum tuberosum) seed tubers. Funct Plant Biol 37: 32–42. https://doi.org/10.1071/FP09188
Xue H, Cao S, Li H (2017) De novo transcriptome assembly and quantification reveal differentially expressed genes between soft-seed and hard-seed pomegranate (Punica granatum L.) PLoS One 7(6):1–15. https://doi.org/10.1371/journal.pone.0178809
Yuan Y, Hu Y, Zhang H, Leng J, Li F, Zhao X, Gao J, Zhou Y (2016) Characterization of a recombinant multifunctional glycoside hydrolase family 3 β-xylosidase/α-l-arabinofuranosidase/β-glucosidase from Cellulosimicrobium cellulans sp. 21. J Mol Catal B-Enzym 131:65–72. https://doi.org/10.1016/j.molcatb.2016.06.002
Zarei A, Zamani Z, Fatahi R et al (2016) Differential expression of cell wall related genes in the seeds of soft- and hard-seeded pomegranate genotypes. Sci Hortic 205:7–16. https://doi.org/10.1016/j.scienta.2016.03.043
Zhang G, Qi J, Xu J, Niu X, Zhang Y, Tao A, Zhang L, Fang P, Lin LH (2013) Overexpression of UDP-glucose pyrophosphorylase gene could increase cellulose content in jute (Corchorus capsularis L.) Biochem Bioph Res Co 442(3-4):153–158. https://doi.org/10.1016/j.bbrc.2013.11.053
Zhao Q, Tobimatsu YK, Zhou R (2013) Loss of function of cinnamyl alcohol dehydrogenase 1 leads to unconventional lignin and a temperature sensitive growth defect in Medicago truncatula. PNAS 110(33):13660–13665. https://doi.org/10.1073/pnas.131223-4110
Acknowledgements
This work was supported by the Key Project of the National Science and Technology Basic Work of China (2012FY110100) and the Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2015-ZFRI).
Author information
Authors and Affiliations
Contributions
Shangyin Cao and Juan Niu designed the study and experiment protocols. Juan Niu and Da Cao directed proteomic research. Shangyin Cao, Juan Niu, and Da Cao drafted of the manuscript. Juan Niu and Haoxian Li performed the experiments. Juan Niu, Da Cao, Lina Chen, and Beibei Liu participated in protein identification and bioinformatics analysis. Fuhong Zhang and Diguang Zhao contributed reagents or other essential material. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Table 1
Reagents used in the iTRAQ experiment. (DOC 18 kb)
Supplementary Table 2
Sequences of specific primers used for qRT-PCR experiments. (XLS 27 kb)
Supplementary Table 3
Differentially accumulated proteins and their assigned or assumed functions. (XLSX 52 kb)
Supplementary Table 4
Correlation analysis between differentially expression protein and mRNA in Tunisa compared with Sanbai at 60 and 120 DAF. (XLSX 28 kb)
Supplementary Table 5
Correlation analysis between cell wall-related gene and protein expression levels. (DOC 24 kb)
Supplementary Fig 1
Analysis of the functional network by STRING 9.0 of differentially expressed proteins in Tunisia compared with Sanbai at 60DAF. (PDF 592 kb)
Supplementary Fig 2
Analysis of the functional network by STRING 9.0 of differentially expressed proteins in Tunisia compared with Sanbai at 120DAF. (PDF 548 kb)
ESM 1
(RAR 1190 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Niu, J., Cao, D., Li, H. et al. Quantitative proteomics of pomegranate varieties with contrasting seed hardness during seed development stages. Tree Genetics & Genomes 14, 14 (2018). https://doi.org/10.1007/s11295-018-1229-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-018-1229-1