
Abstract
There is a need to profile microorganisms which exist pre-and-post-production of umqombothi, to understand its microbial diversity and the interactions which subsequently influence the final product. Thus, this study sought to determine the relative microbial abundance in umqombothi and predict the functional pathways of bacterial and fungal microbiota present. Full-length bacterial 16S rRNA and internal transcribed spacer (ITS) gene sequencing using PacBio single-molecule, real-time (SMRT) technology was used to assess the microbial compositions. PICRUSt2 was adopted to infer microbial functional differences. A mixture of harmful and beneficial microorganisms was observed in all samples. The microbial diversity differed significantly between the mixed raw ingredients (MRI), customary beer brew (CB), and optimised beer brew (OPB). The highest bacterial species diversity was observed in the MRI, while the highest fungal species diversity was observed in the OPB. The dominant bacterial species in the MRI, CB, and OPB were Kosakonia cowanii, Apilactobacillus pseudoficulneus, and Vibrio alginolyticus, respectively, while the dominant fungal species was Apiotrichum laibachii. The predicted functional annotations revealed significant (p < 0.05) differences in the microbial pathways of the fermented and unfermented samples. The most abundant pathways in the MRI were the branched-chain amino acid biosynthesis super pathway and the pentose phosphate pathway. The CB sample was characterised by folate (vitamin B9) transformations III, and mixed acid fermentation. Biotin (vitamin B7) biosynthesis I and l-valine biosynthesis characterised the OPB sample. These findings can assist in identifying potential starter cultures for the commercial production of umqombothi. Specifically, A. pseudoficulneus can be used for controlled fermentation during the production of umqombothi. Likewise, the use of A. laibachii can allow for better control over the fermentation kinetics such as carbohydrate conversion and end-product characteristics, especially esters and aroma compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Umqombothi is a sorghum-based beer with a creamy constituency, an opaque pinkish colour, and a sour taste (Adekoya et al. 2018a). Due to its availability, affordability, and cultural importance, umqombothi is enjoyed by low-income populations in the rural and semi-urban areas of South Africa (Hlangwani et al. 2020) and is of significant economic importance. The beer is generally considered safe for human consumption when prepared under standard fermentation conditions, although microbial contamination could pose potential safety challenges and poor-quality characteristics (Ikalafeng 2008). The opaque beer is often prepared in-home or market settings, where poor handling conditions can lead to contamination. The source of contamination can be the raw materials or utensils used in the brewing process (Lues et al. 2009). Pathogenic and spoilage microbes belonging to the genera Bacillus, Escherichia, Proteus, Staphylococcus, Streptococcus, Aspergillus, Fusarium, Penicillium, Rhizopus have been reported in umqombothi (Katongole 2008). These microbial contaminants in traditional African beverages often lead to turbidity problems, flavour changes, reduced fermentation and yeast performance, aroma defects, and batch contamination, subsequently decreasing consumer confidence and leading to potential financial losses (Obi 2017).
Thus, the determination of the microbial profile of a product such as umqombothi can be an important step in ensuring the product’s quality and safety meet the minimum regulatory requirements for consumer products. Traditionally, the profiling of microbial communities has been based on culture-dependent methods, although these have drawbacks, as not all microorganisms are culturable on standard laboratory growth media. This inefficacy of culture-dependent techniques has led to the use of next-generation sequencing (NGS), a technique that provides the possibility of determining mixed microbial communities in a sample (Yap et al. 2022). To date, several NGS platforms have been developed, and are used depending on the application. Commonly used NGS platforms include Oxford Nanopore, Illumina, 454, PacBio, and Ion Torrent (Meera Krishna et al. 2019). Illumina is currently the most widely used NGS platform (Hodzic et al. 2017). However, the platform possesses limitations such as the potential for base-composition bias, loss of some information from original full-length transcripts, a short read length (25 to 300 bases), and the requirement for polymerase chain reaction (PCR) amplification of multiple DNA templates before sequencing (Cui et al. 2020).
Due to technical constraints, researchers must choose the most effective target regions to identify taxa from full-length 16S rRNA gene sequences containing nine hypervariable regions (V1–V9) as phylogenetically informative markers (Kim et al. 2011; Yang et al. 2016). Therefore, long-read sequencing of the 16S rRNA gene is a promising approach for providing species-level analysis of bacterial communities (Johnson et al. 2019). A similar approach is applicable in targeting the internal transcribed spacer (ITS) region which has the highest probability of successful identification for the broadest range of fungi (Bradshaw et al. 2023). This region has the most clearly defined barcode gap between inter-and-intraspecific variation (Yang et al. 2016). PacBio can generate reads with a median length of 8–10 kb, up to as long as 100 kb, to circumvent these limitations (Loman et al. 2015). For this study, PacBio SMRT (Single Molecule, Real-Time) technology was used to characterise both bacterial and fungal compositions in umqombothi. In addition, functional annotations of the opaque beer samples were assessed using PICRUSt2.
Methodology
Traditional beer (umqombothi) brewing process
The brewing process followed a method described by Hlangwani et al. (2021a), whereby 500 g of King Korn malted sorghum (Tiger Brands, Johannesburg, South Africa) was mixed with 1000 g of White Star maize meal (Pioneer Foods, Paarl, South Africa) in a sterile 10 L bucket filled with 7 L sterile tap water. The mixture was gently stirred, covered, and incubated (Labcon, Chamdor, South Africa) at 25 °C for 24 h to sour. The optimised beer brew (OPB) was prepared following the optimal production parameters generated by the optimisation model in a preceding study (Hlangwani et al. (2021a). The mixed ingredients were then cooked for 1.1 h at 95 °C, and the mixture was allowed to cool to 25 °C. To the cooled porridge, 500 g of King Korn malted sorghum (Tiger Brands, Johannesburg, South Africa) was added and homogenised with gentle stirring. The mixture was then fermented at 29.3 °C for 25.9 h to obtain the OPB. In contrast, the customary beer brew (CB) was prepared following the traditional production method highlighted in Hlangwani et al. (2021b). The mixed ingredients were cooked for 30 min at 95 °C and the mixture was allowed to cool to 25 °C. To the cooled porridge, 500 g of King Korn malted sorghum (Tiger Brands, Johannesburg, South Africa) was added and homogenised with gentle stirring and the subsequent mixture was then fermented at 25 °C for 24 h. The mixed raw ingredients (MRI) were prepared by mixing 500 g of King Korn malted sorghum (Tiger Brands, Johannesburg, South Africa), 1000 g of White Star maize meal (Pioneer Foods, Paarl, South Africa), and 7 L of sterile tap water. The physicochemical properties of the respective samples were previously investigated (Hlangwani et al. 2021b) and are presented in Table 1.
Microbial community profiling of umqombothi
DNA isolation
A modified cetyltrimethylammonium bromide (CTAB) extraction method was used to isolate the DNA (Tamari et al. 2013). Three sterile conical tubes were each used to suspend 1 g of freshly prepared optimised beer brew (OPB), customary beer brew (CB), and mixed raw ingredients (MRI) in 800 µL prewarmed (60 °C) CTAB extraction buffer and then incubated for 1 h. Thereafter, 800 µL chloroform:isoamyl alcohol (24:1) was added and the tubes were gently inverted 4 times to mix. The samples were centrifuged (Eppendorf, Hamburg, Germany) at 3 000 × g for 10 min. The aqueous phase was transferred to sterile microtubes, and 1 µL RNase (Sigma-Aldrich, Missouri, USA) was added to each microtube and incubated for 30 min at 37 °C. Subsequently, 600 µL of isopropanol was added to each microtube and the resulting mixture was gently mixed. The samples were left at room temperature for 4 h to precipitate. The samples were then centrifuged (Eppendorf, Hamburg, Germany) at 3000 × g for 20 min to form a pellet. Thereafter, the supernatant was removed, and the pellet was washed three times with cold (–4 °C) 70% ethanol and then centrifuged (Eppendorf, Hamburg, Germany) three times at 3000 × g for 10 min to remove excess ethanol. The microtubes were left open and allowed to air-dry under a laminar flow (Esco Micro Pte. Ltd., Changi, Singapore) at room temperature for 30 min to dry the pellets. Lastly, the pellets were resuspended in sterile distilled water and stored at –20 °C for subsequent analysis.
Deoxyribonucleic acid (DNA) quantification
The quality and quantity of the DNA were determined using a spectrophotometer at a wavelength of 200–400 nm (Implen GmbH, Munich, Germany). The instrument was calibrated by placing a drop of sterile distilled water on the instrument well before use.
Data sequencing and analysis
PCR amplification of the 16S rRNA gene
The polymerase chain reaction (PCR) was performed using a procedure by Klindworth et al. (2013). Primer pairs used were: 5\({\prime}\)-CCTACGGGNGGCWGCAG-3′ and 5′-GACTACHVGGGTATCTAATCC-3′ (Schloss et al. 2016). The primers targeted V1–V9 hypervariable regions. In 50 μL volumes, the reaction mixture contained 0.3 mg/mL bovine serum albumin (BSA), 250 mM dTNPs, 0.5 mM of each primer, 0.02 U Phusion High-Fidelity DNA Polymerase (Finnzymes OY, Espoo, Finland) and 5× Phusion HF Buffer containing 1.5 mM MgCl2. The PCR conditions used were initial denaturation at 95 °C for 5 min, followed by 25 cycles of denaturation (95 °C for 40 s), annealing (2 min), and extension (72 °C for 1 min), with a final extension step at 72 °C for 7 min. For the primer pair, the annealing temperature was adjusted to 55 °C. A QiaQuick PCR purification kit was used to purify the PCR products (QIAGEN, Hilden, Germany). The PCR products were stored at –20 °C until sequencing.
PCR amplification of the ITS gene
The ITS gene amplification followed the procedure by Akwa et al. (2020). Primer pairs used were: 5′-TCCGTAGGTGAACCTGCGG-3′ and 5′-TCCGTAGGTGAACCTGCGG-3′. In 50 µL volumes, the reaction mixture contained 1 µL of DNA (final concentration of 10 µM), 250 mM dTNPs, 2 µM of each primer, 0.02 U Phusion High-Fidelity DNA Polymerase (Finnzymes OY, Espoo, Finland) and 5X Phusion HF Buffer containing 1.5 mM MgCl2. PCR conditions used were as follows: 34 cycles initial denaturation (98 °C for 1 min), annealing (58 °C for 45 s), and extension (72 °C for 1.5 min), with a final extension step at 72 °C for 10 min. The PCR products were stored at –20 °C until sequencing.
Library preparation for sequencing
The extracted DNA was sent to Inqaba Biotechnical Industries (Pty) Ltd., (Pretoria, South Africa) for sequencing. Reads were generated using PacBio single-molecule, real-time (SMRT) technology. To create highly accurate readings (> QV40), raw subreads were processed using the SMRTlink (v7.0.1) circular consensus sequences (CCS) method. The reads were demultiplexed at the sequencing facility prior to analysis.
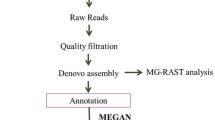
Bioinformatics analyses
The R package DADA2 was used to denoise the reads and identify the relative abundance of each amplicon sequence variant (ASV). Briefly, filtering and removal of low-quality reads were done using the standard parameters. The chimera was removed with the removeBimeraDenovo function. The assignTaxonomy and addspecies functions were used for taxonomic assignment. Amplicon sequencing variants (ASVs) were assigned species-level taxonomy with the full-length trained SILVA138 database for bacterial classification, while the fungal ASVs classification was done using UNITE databases.
Exploratory analyses and prediction analyses
Graphical taxa visualisations were performed in R v.3.5.1 and Bioconductor v.3.0 with Phyloseq, and Microbial R packages were used. The diversity () function in vegan was used to calculate alpha diversity measures. The alpha diversity indices were used to calculate diversity by accounting for dominance and richness. PICRUSt2 was used to predict the microbial function in the microbiota, while subsequent results were visualized and tested for statistical significance in STAMP.
Results
Measures of diversity
The richness and evenness of bacterial and fungal species in each sample were quantified using Chao1, ACE, Shannon, Simpson, InvSimpson, and Fisher alpha diversity indices. The Chao1 (> 200), ACE (> 200), Shannon (> 1.68), Simpson (< 0.5), and Fisher (> 30) alpha diversity indices showed the highest bacterial species diversity for the MRI sample (Fig. 1). Conversely, these alpha diversity indices showed the lowest bacterial species diversity for the CB. Only the Shannon (< 1.6) index showed the lowest bacterial species diversity for the OPB (Fig. 1). The Chao1 (> 200), ACE (> 200), Shannon (> 1.68), Simpson (< 0.5), and Fisher (> 30) alpha diversity indices showed the highest bacterial species diversity for the MRI sample (Fig. 1). The Chao1 (> 30), ACE (> 30), and Fisher (> 6.0) alpha diversity indices showed the highest fungal species diversity for the OPB sample (Fig. 2). Only the Shannon (> 2.7) index showed the highest fungal species diversity for the MRI samples (Fig. 2). The Chao1 (< 25), ACE (< 25), Shannon (< 2.4), and Fisher (< 4.0) alpha diversity indices showed the lowest fungal species diversity for the CB sample (Fig. 2). Conversely, the highest fungal species diversity was observed for the CB sample in the Simpson index (< 0.87) (Fig. 2).

Bacterial alpha diversity metrics based on Chao1, ACE, Shannon, Simpson, InvSimpson, and Fisher’s diversity indices

Fungal alpha diversity metrics based on Chao1, ACE, Shannon, Simpson, InvSimpson, and Fisher’s diversity indices
Microbial community
At the phylum level, Proteobacteria were more abundant in the MRI and OPB, while Firmicutes were dominant in the CB (Fig. 3). The dominant classes in the CB, MRI, and OPB samples were Bacilli, γ-proteobacteria, and α-proteobacteria, respectively (Appendix 1). The dominant order greatly varied in the CB (Lactobacillales), MRI (Acetobacterales), and OPB (Rhodobacterales) samples (Appendix 2). At the family level, the dominant groups were Lactobacillaceae, Acetobacteraceae, and Rhodobacteraceae in the CB, MRI, and OPB respectively (Appendix 3). Only the OPB sample contained Vibrionaceae at the family level (Appendix 3). Diversity in the dominant genus was observed in all three samples (Appendix 4). Apilactobacillus, Kosakonia, and Nautella were the dominant classes in the CB, MRI, and OPB respectively (Appendix 4). In addition, each sample contained at least one of the essential lactic acid-producing genera such as Apilactobacillus, Ligilactobacillus, Lactiplantibacillus, Lactobacillus, Lactococcus, and Paenibacillus (Appendix 4) (Quilodrán-Vega et al. 2020). At the species level, A. pseudoficulneus, Staphylococcus pseudoficulneus, and Fructobacillus pseudoficulneus were the dominant species in the CB sample (Fig. 4). Conversely, Kosakonia cowanii was the dominant species in the MRI sample (Fig. 4). Vibrio alginolyticus was the dominant species in the OPB (Fig. 4).

Relative abundance of bacterial communities in the customary beer brew (CB), mixed raw ingredients (MRI), and optimised beer brew (OPB) at phylum level

Relative abundance of bacterial communities in the customary beer brew (CB), mixed raw ingredients (MRI), and optimised beer brew (OPB) at species level
The total fungal read count was 1233, 1940, and 2037 for MRI, CB, and OPB, respectively. The dominant phylum in all three samples was Rozellomycota, followed by Basidiomycota (Fig. 5). Tremellomycetes were the dominant class in all three samples (Appendix 5). However, this dominance was lower in the MRI compared to the fermented samples, CB, and OPB (Appendix 5). The dominant order in all three samples was Trichosporonales (Appendix 6). At the family level, the MRI and OPB had a greater abundance of fungal communities than the CB (Appendix 7). Trichosporonaceae was the dominant family in all samples, with Trichocomaceae being the second most dominant family group in the fermented samples. The genus Apiotrichum was dominant in the MRI, CB, and OPB samples (Appendix 8). The genera Leucosporidium and Penicillium were also present in the OPB (Appendix 8). At the species level, A. laibachii was the dominant group in all three samples (Fig. 6). A. tropicalis and A. orientalis, were only present in the MRI sample, while A. porosum was only present in the OPB sample (Fig. 6).

Relative abundance of fungal communities of the customary beer brew (CB), mixed raw ingredients (MRI), and optimised beer brew (OPB) at phylum level

Relative abundance of fungal communities of the customary beer brew (CB), mixed raw ingredients (MRI), and optimised beer brew (OPB) at species level
Amplicon sequencing variants (ASVs)
The analyses showed that the bacterial community had higher amplicon sequencing variants (ASVs) (215) (Fig. 7) than the fungal community (29) (Fig. 8). Unique ASVs were highly associated with the MRI group in both bacteria (209) and fungi (27). The high unique ASVs in the MRI in both bacteria (209) and fungi (27) suggest a higher variability (mutations) in the microorganisms within the sample. This result corresponded with the high variability observed in bacterial and fungal communities at the species level (Fig. 4 and Fig. 6). Two bacterial ASVs were shared between CB and MRI, while no shared ASVs were found in the fungal ASVs distribution. The two shared ASVs suggested common genetic features or traits shared by a group of individuals.

An upset plot showing shared and unique bacterial ASV(s) across different umqombothi samples

An upset plot showing shared and unique fungal ASV(s) across different umqombothi samples
Predictive functional profiles of microbial communities
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) was used to computationally predict the functional composition of metagenomes from marker gene data and a database of reference genomes. These predictions establish connections between metabolic profiles and the phylogenetic structure of microbes. Overall, a total of 120 MetaCyc metabolic pathways were analysed based on the 16S rRNA amplicon data to predict the microbial functional differences between the samples (FDR p < 0.05) (Figs. 9 and 10). MetaCyc metabolic pathways for bacteria are shown in Figs. 9, 10 and 11. No significant differences in the metabolic pathways between samples were observed for fungi. The MetaCyc metabolic pathways for fungi are shown in Table 2. The pathways associated with carbohydrate metabolism, amino acid biosynthesis and vitamin biosynthesis were the major enrichments for both bacteria (Figs. 9, 10 and 11) and fungi (Table 2).

Bar plots showing the mean proportion (%) of significantly different predicted functional compositions between CB and OPB samples

Bar plots showing the mean proportion (%) of significantly different predicted functional compositions between MRI and CB samples

Bar plots showing the mean proportion (%) of significantly different predicted functional compositions between MRI and OPB samples
Predictive functional profiles of bacterial communities
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway predictions revealed a high abundance of functional metagenomes in the CB sample (Figs. 9, 10 and 11). The MRI was found to be enriched in predicted superclasses related to amino acid biosynthesis, nicotinamide adenine dinucleotide (NAD) biosynthesis, carbohydrate metabolism, and coenzyme A biosynthesis (Figs. 10 and 11). The most abundant corresponding pathways in the MRI were the branched-chain amino acid (BCAA) biosynthesis super pathway (p = 1 × 10–15), NAD de novo biosynthesis I pathway (p = 8.99 × 10–15), pentose phosphate pathway (p = 1 × 10–15), and coenzyme A biosynthesis I pathway (p = 3.26 × 10–3) (Figs. 10 and 11). In contrast, the CB sample was characterized by B-group vitamin biosynthesis, amino acid biosynthesis, and carbohydrate metabolism-related pathways (Figs. 9 and 10). The most abundant pathways in the CB were folate (vitamin B9) transformations III (p = 1 × 10–5), L-threonine biosynthesis super pathway (p = 1.03 × 10–4), and mixed acid fermentation (p = 1 × 10–5) (Figs. 9 and 10). Similarly, B-group vitamin biosynthesis, amino acid biosynthesis, and fatty acid and lipid metabolism-related pathways were characteristic of the OPB sample (Figs. 9 and 11). The most abundant pathways in the OPB were biotin (vitamin B7) biosynthesis I (p = 1 × 10–15), L-valine biosynthesis (p = 1 × 10–15), and fatty acid elongation (p = 1 × 10–15) (Fig. 9 and 11).
Predictive functional profiles of fungal communities
Fungal communities showed a high abundance of functional metagenomes in the MRI sample (p-adjusted FDR = 0.00) (Table 2). The MRI was found to be enriched in predicted superclasses related to amino acid biosynthesis, cofactor, carrier, vitamin biosynthesis, and carbohydrate metabolism (Table 2). The corresponding pathways in the MRI were heme b biosynthesis II (p-adjusted FDR = 0.00), and coenzyme A biosynthesis I (p-adjusted FDR = 0.00) (Table 2). The CB sample was enriched in a predicted superclass related to amino acid degradation, specifically the L-histidine degradation I pathway (p-adjusted FDR = 0.00). The OPB sample was enriched in a predicted superclass related to carbohydrate metabolism, amino acid degradation, and carboxylate degradation (Table 2). The corresponding pathways in the OPB were hexitol degradation (p-adjusted FDR = 0.00), l-arginine degradation super pathway (p-adjusted FDR = 0.00), and d-galactarate/d-glucarate degradation I (p-adjusted FDR = 0.00) (Table 2).
Discussion
Bacterial and fungal alpha diversity
Chao1 and ACE estimate species richness, while the Shannon and Simpson indices consider both species richness and evenness, with the Simpson also measuring dominance (Kumar et al. 2022). On the other hand, Fisher’s alpha measures comparable diversity between communities (Chen and Shen 2017). The microbial diversity differed significantly between the MRI, CB, and OPB samples. The highest bacterial species diversity was observed in the MRI, which could be associated with differing endogenous microbiota of the raw materials, which was subsequently reduced significantly after fermentation. This finding is consistent with other studies that have found a reduction in bacterial diversity during the fermentation process (Deng et al. 2021; Huang et al. 2022). As a fermentation process proceeds, certain species from dominant bacterial phyla such as Proteobacteria outcompete species from other phyla, as shown by the relative abundance of bacterial communities in this study (Fig. 3 and Fig. 4).
In contrast, the highest fungal species diversity was observed in the OPB sample, suggesting that the fermentation process might have enhanced fungal diversity. The fungi’s relatively high mutation rate allows species to quickly adapt to new and changing microbial environments (Gambhir et al. 2022). Specifically, Candida spp. such as Candida tropicalis (Fig. 6), Saccharomyces spp. and Hanseniaspora spp. have been shown to grow well, co-dominate, adapt, and survive stressful alcoholic fermentation (Cureau et al. 2021). The low fungal diversity in the CB, as shown by the Chao1, ACE, Shannon, and Fisher alpha diversity indices, could be due to its limited fermentation time and lower fermentation temperature, factors that drive microbial proliferation (CB fermentation conditions = 24 h at 25 °C compared to OPB fermentation conditions = 25.9 h at 29.3 °C) (Hlangwani et al. 2021a). However, the Simpson index showed the highest fungal species diversity in the CB (< 0.87) (Fig. 2).
Composition of bacterial communities
The phylum Proteobacteria is the largest and most phenotypically diverse phylogenetic lineage within the domain Bacteria (Kersters et al. 2006a). Because Proteobacteria respond positively to labile carbon compounds, they are often the most prominent keystone taxa in the sorghum rhizosphere networks (Oberholster et al. 2018). The dominance of Firmicutes in the CB is not unusual, as a noticeable shift from Proteobacteria to Firmicutes occurs during fermentation (Mareque et al. 2018). This shift has been demonstrated in the fermentation of the traditional Mexican alcoholic beverage pulque and traditional Korean alcoholic beverages nuruk and makgeolli (Escalante et al. 2008; Jung et al. 2012). Certain, bacterial groups belonging to Proteobacteria and Firmicutes in Sesotho beer, a type of umqombothi produced in The Kingdom of Lesotho, have been reported in a study by Cason et al. (2020). Their study suggests that the Proteobacteria and Firmicutes present likely originated from the raw materials (maize, sorghum, or malted sorghum) used in the preparation of the Sesotho beer, as well as from the brewers themselves (Cason et al. 2020).
The proliferation of Bacilli reduces the growth of unwanted microbes in the beer’s souring mash while producing lactic acid, providing a unique flavour profile and improving the quality and shelf life of the final beer product (Cissé et al. 2019). The classes Acidimicrobiia, Desulfuromonadia, Planctomycetes, and Phycisphaerae were only present in the OPB. Acidimicrobiia are acidophilic chemotrophs with an optimal growth pH of around 2 which possess the ability to oxidize ferrous iron at relatively fast rates (Johnson 2009). Desulfuromonadia, on the other hand, are typical thermophiles that are capable of anaerobic respiration by using sulphur, manganese, iron, trichloroacetic acid, and cobalt as electron acceptors (Brenner et al. 2005). Planctomycetes are biotechnologically important bacteria capable of sterol biosynthesis, nitrogen-fixation, endocytosis, and phagocytosis-like process, anaerobic ammonia oxidation, and sulphate polysaccharides metabolism (Kaboré et al. 2020). The emergence of these classes in the OPB is not surprising, as the sample had a lower pH (Table 1), and an abundance of minerals (iron, nitrogen, sulphur, manganese), and sugar compounds (maltose, glucose, mannitol) (Hlangwani et al. 2021a, b).
Members of Lactobacillales produce proteinaceous bacteriocins, lactic acid and other metabolic products (Katongole 2008) which contribute to the organoleptic and textural profile of umqombothi. Acetobacterales are mainly found in sugary raw materials (Kersters et al. 2006b). Fermentation negatively affected the presence of Acetobacterales, as shown by the decrease in relative abundance in the OPB and CB samples (Appendix 2). Acetobacterales achieve optimum growth between pH 5–6.5 (Gomes et al. 2018), and, as a result, the low pH values in the CB (pH = 4.23) and OPB (pH = 3.27) (Table 1) limited their growth. The proliferation of Rhodobacterales in the OPB can be attributed to the sample’s high glutamine content (1.6 g/100 g) (Hlangwani et al. 2021b). The olsBA operon in Rhodobacterales contains a gene that codes a homologue of OlsB (olsB2), which catalyses glutamine-containing compounds (Geiger et al. 2010). In co-cultures and/or symbiotic systems, certain members of Rhodobacterales have the ability to shift from mutualism to parasitism, increasing the group’s dominance (Wang et al. 2014). To dominate and outcompete other groups for nutrients, Rhodobacterales produces antibiotics and other bactericidal compounds and then utilises the fixed carbon from the lysed cells (Cooper and Smith 2015).
Staphylococcaceae was significantly prevalent in the CB (9.36%) and moderately present in the MRI (0.13%) (Appendix 3). This result is not surprising, as Staphylococcaceae can contaminate raw maize and then rapidly multiply when lactic acid bacteria (LAB) are still in the lag phase and a significant amount of lactic acid is yet to be produced (Tchakounté et al. 2018). At the exponential growth phase of LAB, lactic acid concentration increased with a decrease in pH (Table 1), imparting bacteriostatic and bactericidal effects which subsequently led to a reduction in the prevalence of Staphylococcaceae (Appendix 3). The reduction of Staphylococcaceae during fermentation is a consequence of the longer fermentation period in the OPB sample (time = 25.9 h) compared to the CB sample (time = 24 h). As a result of a higher pH value and a lower lactic acid concentration in the CB compared to the OPB (Table 1), the relative abundance of Staphylococcaceae was higher in the CB sample (Appendix 3). Studies by Ikalafeng (2008) and Lues et al. (2011) have reported the presence of Staphylococcaceae in umqombothi. Vibrionaceae are metabolically diverse facultative anaerobes capable of D-glucose, D-fructose, maltose, and glycerol catabolism (Gomez-Gil et al. 2014). It is possible that the reduction in the concentration of the sugars arabitol, fructose, sorbitol, myo-inositol, and sucrose during the fermentation of umqombothi (i.e., lower in CB and OPB compared to MRI) was due to the growth of members of this family (Hlangwani et al. 2021b).
Using culture-dependent and culture-independent methods, Mukisa et al. (2012) showed the dominance of LAB in obushera, a collective name for popular traditional fermented cereal beverages obutoko, enturire, ekitiribita, and obuteire consumed in western, southwestern, and central Uganda. Similarly, a high-throughput sequencing study by Eltayeb et al. (2020) found that the bacterial community in hulumur (a Sudanese sorghum-based fermented beverage) was dominated by LAB at the genus level. Currently, not much is known about A. pseudoficulneus. However, the majority of Apilactobacillus species including A. pseudoficulneus, A. timberlakei, A. micheneri, A. quenuiae, and A. kunkeei have been described to be heterofermentative, facultatively anaerobic fructophilic lactic acid bacteria (FLAB) which produce lactic acid, and antimicrobial compounds, including bacteriocins (Maeno et al. 2021; Molina et al. 2023). It is possible some of these species were present in the CB but were not detected during sequencing, construction, and/or analysis. Such screening limitations exist in the species-level metagenomic analysis of fermented foods, and thus the use of two or more approaches is recommended (Walsh et al. 2017).
Composition of fungal communities
Several groups of Rozellomycota, observed in all samples, prefer low pH conditions (3–4) and ambient temperatures, which are simulated in the production of umqombothi (Table 1) (Ikalafeng 2008). Tremellomycetes are often isolated from cereal grains such as wheat, rice, maize, and sorghum, and may be found in the mucosal surfaces of approximately 1–10% of the healthy human population (Shoff and Perfect 2021). However, interestingly, they have been the dominant class in some wineries and breweries (Sohlberg et al. 2022). Groups within this class may be extremely harmful or beneficial to humans (Weiss et al. 2014). For example, pathogenic Tremellomycetes may cause white Piedra, an infection of the hair follicles (Shivaprakash et al. 2011), while Tremellomycetes are mycoparasitic fungi that produce a wide range of bioactive, antimicrobial metabolites, carbon-active and proteolytic enzymes, and yeast oils (Abe et al. 2006; Vujanovic 2021). Therefore, Tremellomycetes are useful in the biocontrol of plant-pathogenic fungi such as Smut diseases, Botrytis, common bunt, and Tilletia, as well as the biodegradation of anti-nutritional compounds such as phenolic compounds (Fonseca et al. 2011; Vujanovic 2021).
Trichosporonales consists of a diverse group of members involved in fermentation processes and biotransformation of biarylic compounds (Takashima et al. 2019; Martínez-Herrera et al. 2021). Saccharomycetales, a common group in maize, were only present in the MRI sample (Sun et al. 2019). Members of Saccharomycetales such as Schwanniomyces occidentalis, Saccharomyces cerevisiae, and Candida are responsible for the fermentation of sugars (Johnson and Echavarri-Erasun 2011). Because Saccharomycetales synthesise butyric acid and caproic acid, they play a key role in beer brewing and flavour formulation (Liu et al. 2021). The mycoparasitic, dimorphic yeast, Leucosporidiales, were only present in the OPB samples (Appendix 6) (van der Klei et al. 2011).
Trichocomaceae are a relatively large family of fungi which are thermotolerant, psychrotolerant, osmotolerant, and xerotolerant, and can grow on diverse substrates (Houbraken and Samson 2011). As a result, Trichocomaceae commonly colonise sorghum grains and a wide range of fermented alcoholic beverages such as Chinese ‘Baiyunbian’ liquor, marcha, and thiat (Moretti and Sarrocco 2016; Hu et al. 2017; Sha et al. 2017). The secondary metabolites (extrolites) secreted by members of Trichocomaceae may be beneficial (e.g., the production of antibiotics such as penicillin and the cholesterol-lowering agent lovastatin) or harmful (e.g., the production of mycotoxins such as patulin, aflatoxins, ochratoxins, and fumonisins) (Moretti and Sarrocco 2016). In addition, members of Trichocomaceae produce organic acids and different enzymes that degrade a range variety of complex biomolecules (Houbraken and Samson 2011).
During fermentation, members of Leucosporidium may produce the therapeutic enzyme L-asparaginase (ASNase), coenzymes Q9 and Q10, lipases, and Rhodotorulic acid, which is effective against spore germination of the fungus Botrytis cinerea (Sansone et al. 2005; Moguel et al. 2020). In a study by Odhav and Naicker (2002) species of Penicillium were isolated from sorghum and sorghum malt grains but umqombothi was not studied. Adekoya et al. (2018b) reported the presence of 2.30 × 105 CFU/mL Penicillium species in umqombothi samples. Although members of Penicillium produce several mycotoxins, certain species can produce extracellular enzyme systems that alter the chemical composition of the sorghum grains, leading to changes in the flavour and colour of the beer (Odhav and Naicker 2002).
A. laibachii has the ability to utilise complex nitrogenous compounds (hydroxyproline, tyramine, uric acid, L-phenylamine and ethylamine), aliphatic lipids, cellobiose, aromatic compounds, ammonium sulphate, and amines as sole sources of carbon and energy (Johnson and Echavarri-Erasun 2011). During fermentation, A. laibachii produces the hydrolytic enzymes β-glucosidase, xylanase, and thermostable cellulases which break down insoluble and soluble substrates at elevated temperatures over a wide pH range (Touijer et al. 2019). The use of Candida tropicalis as a starter culture in the alcoholic fermentation of tchapalo, a traditional sorghum beer of Côte d’Ivoire, resulted in the higher production of organic acids and 2-butanone in pure culture (N’Guessan et al. 2010).
Fructophilic lactic acid bacteria (FLAB) such as A. porosum can only metabolize a limited number of carbohydrates (Endo et al. 2018). Generally, they show high metabolic activity in a D-fructose-rich substrate and limited metabolic activity in a D-glucose-rich substrate (Maeno et al. 2021). Furthermore, FLAB are metabolically characterised by a higher production of acetate and not alcohol unlike other groups of heterofermentative LAB (Maeno et al. 2021). This may explain the relatively lower alcohol production (i.e., approximately 4.5–5.5%) reported in a preceding study (Hlangwani et al. 2021a). A. porosum has useful applications in alcoholic fermentation, especially in the production of high-value products such as gluconic acid, single-cell oil, and low-alcohol beverages (Gorte et al. 2019). Furthermore, A. porosum (syn. T. porosum) is highly active against pathogenic basidiomycetous and ascomycetous fungi, while the fungicidal preparation extracted from the culture broth has been recorded to be effective against Filobasidiella neoformans and Candida albicans cells (Kulakovskaya et al. 2010). These antifungal and fungicidal cellobiose lipids produced by T. porosum have been isolated in Malaysian fermented foods such as tapai, tempeh, and miso (Lim et al. 2011).
Bacterial and fungal amplicon sequencing variants (ASVs) distribution across the groups
The lower fungal ASVs were expected, as studies show that heterofermentative and homofermentative mesophilic bacteria dominate the mashing process, as well as early stages of the fermentation in the mixed raw ingredients (i.e., MRI) (Tokpohozin 2018; Tyakht et al. 2021). The abundance of bacteria, particularly LAB, is attributed to the sorghum and maize used to prepare umqombothi (Liu et al. 2021). Differences in the lactic acid microbiota profiles in the brew can be attributed to the souring temperature (Van Der Walt 1956). Sequentially, lactic acid fermentation is carried out by several LAB, followed by yeast-mediated alcoholic fermentation (Katongole 2008). Cason et al. (2020) found a total of 46 fungal ASVs using deep identical sequencing. A loss in the number of reads and ASVs is often observed post-data analysis due to a lack of adequate fungal ASV classification in environmental studies. In the absence of reference sequences in the database, new species, genera, or families remain unidentified, even at a kingdom classification level (Heeger et al. 2019). As a result, fungal diversity is significantly underestimated and underrepresented (Cason et al. 2020). According to Needham et al. (2017), abundant taxa have a greater chance of displaying rare variants. While microbial variants can have a lower relative abundance, they usually show higher genetic and functional diversity (Cao et al. 2022). Conversely, the two bacterial ASVs shared between the CB and MRI samples (Fig. 7) suggest common genetic traits shared by a group of individuals.
Predictive functional profiles of bacterial and fungal communities
The metabolic activity observed in the biochemical pathways of bacterial communities is often attributed to LAB cereal-grain fermentation (Kayitesi et al. 2023). Cason et al. (2020), suggested that LAB such as Lactobacillus, Pediococcus, and Weisella in Sesotho beer was responsible for the high level of predicted fermentative metabolism. The LAB introduced in the mashing and souring stages of umqombothi preparation are highly dependent on the BCAA biosynthesis super pathway for optimal growth, adaptation to the alcoholic conditions, and maintenance of the internal pH (Smid and Hugenholtz 2010). In yeasts such as S. cerevisiae, this pathway is responsible for the metabolism of ketobutyrate to isoleucine, and pyruvate to valine, both essential precursors for growth, glucose metabolism, and producing vicinal diketones which impart a butter-tasting flavour (Krogerus and Gibson 2013). This was confirmed by the high enrichment in predicted superclasses related to amino acid biosynthesis and/ or degradation across all three samples (Table 2). The pentose phosphate pathway (PPP) is the catabolic pathway for xylose in naturally xylose-fermenting yeasts, as well as the catabolic pathway for xylulose in Candida shehatae and S. cerevisiae (Johansson and Hahn-Hägerdal 2002). In the cell, the PPP regulates the concentration of simple sugars and provides precursors for the biosynthesis of aromatic amino acids, nucleic acids, and fatty acids (Kloska et al. 2022). In yeasts such as S. cerevisiae, the PPP has a similar contribution to glucose degradation during fermentation (Bertels et al. 2021).
The breakdown of complex sugars such as hexitol (sorbitol and mannitol), l-rhamnose, glycogen, and l-fucose to lactate, formate, ethanol, and acetate (Table 2) were consistent with the findings of a preceding study, which found that there was a reduction in the concentration of the sugars arabitol, fructose, sorbitol, myo-inositol, and sucrose during the fermentation of umqombothi (Hlangwani et al. 2021b). In the same study, the MRI, CB, and OPB were shown to be rich in amino acids, and B-group vitamins (Hlangwani et al. 2021b). The fermented samples CB and OPB were particularly rich in valine, and folate (vitamin B9) (Hlangwani et al. 2021b). The availability of these compounds drives the metabolic activity of the associated microorganisms (Ledesma-Amaro et al. 2013). Specifically, carbohydrates are substrates used by both bacteria and yeast for biomass formation and the conversion of a spectrum of sugars into alcohol and organic acids (Maicas 2020). In umqombothi, amino acids (the main nitrogen source) are an essential component of protein synthesis, production of aromatic compounds, ATP generation, and beer flavour, especially by yeasts such as S. cerevisiae (Takagi 2019). Finally, vitamins such as thiamine (B1), biotin (B7), and pantothenic acid (B5) are necessary for yeast growth, enzyme function, and a successful alcoholic fermentation process in umqombothi production (Katongole 2008). For example, the absence of thiamine (B1) may lead to poor-quality fermentations due to sluggish yeast metabolism as observed in a preceding study (Hlangwani et al. 2021b).
Conclusion
The culture-independent (i.e., PacBio single-molecule, real-time (SMRT) technology) techniques are highly effective for identifying microbial populations in fermented foods. This advantage was demonstrated through the abundance of data detailing the bacterial and fungal profiles in umqombothi. A mixture of harmful and beneficial microorganisms was observed in the MRI, CB, and OPB samples. The microbial diversity differed significantly between the MRI, CB, and OPB. The highest bacterial species diversity was observed in the MRI, while the highest fungal species diversity was observed in the OPB. The dominant bacterial species in the MRI, CB, and OPB were Kosakonia cowanii, Apilactobacillus pseudoficulneus, and Vibrio alginolyticus, respectively. The dominant fungal species across the MRI, CB, and OPB samples was Apiotrichum laibachii. The predicted functional annotations revealed fundamental differences in the pathways of the microbes in the fermented and unfermented samples. The most abundant pathways in the MRI were the branched-chain amino acid biosynthesis super pathway (p = 1 × 10–15), and the pentose phosphate pathway (p = 1 × 10–15). The CB sample was characterised by folate (vitamin B9) transformations III (p = 1 × 10–5), and mixed acid fermentation (p = 1 × 10–5). Biotin (vitamin B7) biosynthesis I (p = 1 × 10–15), and L-valine biosynthesis (p = 1 × 10–15) were characteristic of the OPB sample. Thus, this study contributes towards the advancement of our understanding of microbial communities in umqombothi through culture-independent techniques, offering insights into the diversity, dominant species, and functional pathways of the fermentation microbes. Furthermore, these findings can be used in assessing potential starter cultures for commercial production, upon which when adopted can facilitate the production of a quality and safe product. Further studies are however still required to isolate these potential starter cultures, investigate their genomic profile, utilize them for umqombothi processing and assess the overall quality of the resultant beverage.
Data availability
The nucleotide sequence data reported are available in the NCBI GenBank databases under the BioSample accession numbers SAMN26992645–SAMN26992650. BioProject ID: PRJNA845913.
References
Abe F, Minegishi H, Miura T, Nagahama T, Usami R, Horikoshi K (2006) Characterization of cold-and high-pressure-active polygalacturonases from a deep-sea yeast, Cryptococcus liquefaciens strain N6. Biosci Biotechnol Biochem 70:296–299
Adekoya I, Obadina A, Adaku CC, De Boevre M, Okoth S, De Saeger S, Njobeh P (2018a) Mycobiota and co-occurrence of mycotoxins in South African maize-based opaque beer. Int J Food Microbiol 270:22–30
Adekoya I, Obadina A, Phoku J, De Boevre M, De Saeger S, Njobeh P (2018b) Fungal and mycotoxin contamination of fermented foods from selected South African markets. Food Control 90:295–303
Akwa TE, Maingi JM, Birgen JK (2020) Characterisation of fungi of stored common bean cultivars grown in Menoua division, Cameroon. bioRxiv 10:2793
Bertels LK, Fernández Murillo L, Heinisch JJ (2021) The pentose phosphate pathway in yeasts–more than a poor cousin of glycolysis. Biomolecules 11:725
Bradshaw MJ, Aime MC, Rokas A, Maust A, Moparthi S, Jellings K, Pane AM, Hendricks D, Pandey B, Li Y, Pfister DH (2023) Extensive intragenomic variation in the internal transcribed spacer region of fungi. iScience 26:107317
Brenner D, Krieg N, Staley J (2005) Bergey’s manual® of systematic bacteriology. Springer, Berlin
Cao Q, Zhou Y, Zhao H, Bai Y (2022) Inconsistent response of abundant and rare bacterial communities to the developmental Chronosequence of Pinus massoniana. Forests 13:1904
Cason ED, Mahlomaholo BJ, Taole MM, Abong GO, Vermeulen JG, de Smidt O, Vermeulen M, Steyn L, Valverde A, Viljoen B (2020) Bacterial and fungal dynamics during the fermentation process of Sesotho, a traditional beer of Southern Africa. Front Microbiol 11:1451
Chen Y, Shen TJ (2017) Rarefaction and extrapolation of species richness using an area-based Fisher’s logseries. Ecol Evol 7:10066–10078
Cissé H, Kagambèga B, Sawadogo A, Tankoano A, Sangaré G, Traoré Y, Ouoba IIL, Savadogo A (2019) Molecular characterization of Bacillus, lactic acid bacteria and yeast as potential probiotic isolated from fermented food. Sci Afr 6:e00175
Cooper MB, Smith AG (2015) Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr Opin Plant Biol 26:147–153
Cui J, Lu Z, Xu G, Wang Y, Jin B (2020) Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 16:1–13
Cureau N, Threlfall R, Carbonero F, Howard L, Lavefve L (2021) Fungal diversity and dynamics during grape wine fermentations with different sulfite levels and yeast inoculations. Am J Enol Vitic 72:240–256
Deng Y, Huang D, Han B, Ning X, Yu D, Guo H, Zou Y, Jing W, Luo H (2021) Correlation: between autochthonous microbial diversity and volatile metabolites during the fermentation of Nongxiang Daqu. Front Microbiol 12:2117
Eltayeb MM, Eltigani SA, Taniguchi T (2020) Pyrosequencing scrutiny of bacterial and fungal communities in two Sudanese sorghum-based fermented foods. Ann Microbiol 70:1–10
Endo A, Maeno S, Tanizawa Y, Kneifel W, Arita M, Dicks L, Salminen S (2018) Fructophilic lactic acid bacteria, a unique group of fructose-fermenting microbes. Appl Environ Microbiol 84:e01290-e1318
Escalante A, Giles-Gómez M, Hernández G, Córdova-Aguilar MS, López-Munguía A, Gosset G, Bolívar F (2008) Analysis of bacterial community during the fermentation of pulque, a traditional Mexican alcoholic beverage, using a polyphasic approach. Int J Food Microbiol 124:126–134
Fonseca Á, Boekhout T, Fell JW (2011) Cryptococcus vuillemin (1901). In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts. Elsevier, Amsterdam, pp 1661–1737
Gambhir N, Harris SD, Everhart SE (2022) Evolutionary significance of fungal hypermutators: lessons learned from clinical strains and implications for fungal plant pathogens. mSphere 3:1–10
Geiger O, González-Silva N, López-Lara IM, Sohlenkamp C (2010) Amino acid-containing membrane lipids in bacteria. Prog Lipid Res 49:46–60
Gomes RJ, de Fatima BM, de Freitas RM, Castro-Gómez RJH, Spinosa WA (2018) Acetic acid bacteria in the food industry: systematics, characteristics and applications. Food Technol Biotechnol 56:139
Gomez-Gil B, Thompson CC, Matsumura Y, Sawabe T, Iida T, Christen R, Thompson F, Sawabe T (2014) The family Vibrionaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (eds) The prokaryotes: gammaproteobacteria. Springer, Berlin, pp 659–747
Gorte O, Aliyu H, Neumann A, Ochsenreither K (2019) Draft genome sequence of the oleaginous yeast Apiotrichum porosum (syn. Trichosporon porosum) DSM 27194. J Genomics 7:11
Heeger F, Wurzbacher C, Bourne EC, Mazzoni CJ, Monaghan MT (2019) Combining the 5.8 S and ITS2 to improve classification of fungi. Methods Ecol Evol 10:1702–1711
Hlangwani E, Adebiyi JA, Doorsamy W, Adebo OA (2020) Processing, characteristics and composition of umqombothi (a South African traditional beer). Processes 8:1451
Hlangwani E, Doorsamy W, Adebiyi JA, Fajimi LI, Adebo OA (2021a) A modeling method for the development of a bioprocess to optimally produce umqombothi (a South African traditional beer). Sci Rep 11:1–15
Hlangwani E, Adebiyi JA, Adebo OA (2021b) Nutritional compositions of optimally processed umqombothi (a South African indigenous beer). Fermentation 7:225
Hodzic J, Gurbeta L, Omanovic-Miklicanin E, Badnjevic A (2017) Overview of next-generation sequencing platforms used in published draft plant genomes in light of genotypization of immortelle plant (Helichrysium Arenarium). Med Arch 71:288
Houbraken JAMP, Samson R (2011) Phylogeny of Penicillium and the segregation of Trichocomaceae into three families. Stud Mycol 70:1–51
Hu Y, Dun Y, Li S, Fu B, Xiong X, Peng N, Liang Y, Zhao S (2017) Changes in microbial community during fermentation of high-temperature Daqu used in the production of Chinese ‘Baiyunbian’ liquor. J Inst Brew 123:594–599
Huang W, Xu H, Pan J, Dai C, Mintah BK, Dabbour M, Zhou R, He R, Ma H (2022) Mixed-strain fermentation conditions screening of polypeptides from rapeseed meal and the microbial diversity analysis by high-throughput sequencing. Foods 11:3285
Ikalafeng BK (2008) Microbiota and mycotoxins in traditional beer of the greater Kimberley area and associated brewing and consumption practices. Dissertation, Central University of Technology
Johansson B, Hahn-Hägerdal B (2002) The non-oxidative pentose phosphate pathway controls the fermentation rate of xylulose but not of xylose in Saccharomyces cerevisiae TMB3001. FEMS Yeast Res 2:277–282.
Johnson DB (2009) Extremophiles: acidic environments. In: Schaechter M (ed) Encyclopedia of microbiology. Academic Press, Massachusetts, pp 107–126
Johnson EA, Echavarri-Erasun C (2011) Yeast biotechnology. In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts. Elsevier, Amsterdam, pp 21–44
Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E (2019) Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun 10:1–11
Jung MJ, Nam YD, Roh SW, Bae JW (2012) Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters. Food Microbiol 30:112–123
Kaboré OD, Godreuil S, Drancourt M (2020) Planctomycetes as host-associated bacteria: a perspective that holds promise for their future isolations, by mimicking their native environmental niches in clinical microbiology laboratories. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2020.519301
Katongole JN (2008) The microbial succession in indigenous fermented maize products. Dissertation, University of the Free State
Kayitesi E, Onojakpor O, Moyo SM (2023) Highlighting the impact of lactic-acid-bacteria-derived flavours or aromas on sensory perception of African fermented cereals. Fermentation 9:111
Kersters K, De Vos P, Gillis M, Swings J, Vandamme P, Stackebrandt ERKO (2006a) Introduction to the proteobacteria. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds) The prokaryotes: a handbook on the biology of bacteria. Springer, New York, pp 3–37
Kersters K, Lisdiyanti P, Komagata K, Swings J (2006b) The family Acetobacteraceae: the genera Acetobacter, Acidomonas, Asaia, Gluconacetobacter, Gluconobacter, and Kozakia. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds) The prokaryotes: a handbook on the biology of bacteria. Springer, New York, pp 163–200
Kim M, Morrison M, Yu Z (2011) Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J Microbiol Methods 84:81–87
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Rese 4:1–11
Kloska SM, Pałczyński K, Marciniak T, Talaśka T, Miller M, Wysocki BJ, Davis P, Wysocki TA (2022) Queueing theory model of pentose phosphate pathway. Sci Rep 12:1–9
Krogerus K, Gibson BR (2013) Influence of valine and other amino acids on total diacetyl and 2, 3-pentanedione levels during fermentation of brewer’s wort. Appl Microbiol Biotechnol 97:6919–6930
Kulakovskaya TV, Golubev WI, Tomashevskaya MA, Kulakovskaya EV, Shashkov AS, Grachev AA, Chizhov AS, Nifantiev NE (2010) Production of antifungal cellobiose lipids by Trichosporon porosum. Mycopathologia 169:117–123
Kumar P, Dobriyal M, Kale A, Pandey AK, Tomar RS, Thounaojam E (2022) Calculating forest species diversity with information-theory based indices using sentinel-2A sensor’s of Mahavir Swami Wildlife Sanctuary. PLoS ONE 17:e0268018
Ledesma-Amaro R, Santos MA, Jiménez A, Revuelta JL (2013) Microbial production of vitamins. In: McNeil B, Archer D, Giavasis I, Harvey L (eds) Microbial production of food ingredients, enzymes and nutraceuticals. Woodhead Publishing, Sawston, pp 571–594
Lim SL, Tay ST (2011) Diversity and killer activity of yeasts in Malaysian fermented food samples. Trop Biomed 28:438–443
Liu C, Gong X, Zhao G, Htet MNS, Jia Z, Yan Z, Liu L, Zhai Q, Huang T, Deng X, Feng B (2021) Liquor flavour is associated with the physicochemical property and microbial diversity of fermented grains in waxy and non-waxy sorghum (Sorghum bicolor) during fermentation. Front Microbiol. https://doi.org/10.3389/fmicb.2021.618458
Loman NJ, Quick J, Simpson JT (2015) A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12:733–735
Lues JFR, Ikalafeng BK, Maharasoa M, Shale K, Pool E (2009) Brewing and consumptions practices of indigenous traditional beer in a typical South African semi-urban area: indigenous knowledge systems, health, illness and healing. IAJIKS 8:163–174
Lues JFR, Ikalafeng BK, Maharasoa M, Shale K, Malebo NJ, Pool E (2011) Staphylococci and other selected microbiota associated with indigenous traditional beer. Afr J Microbiol Res 5:1691–1696
Maeno S, Nishimura H, Tanizawa Y, Dicks L, Arita M, Endo A (2021) Unique niche-specific adaptation of fructophilic lactic acid bacteria and proposal of three Apilactobacillus species as novel members of the group. BMC Microbiol 21:1–14
Maicas S (2020) The role of yeasts in fermentation processes. Microorganisms 8:1142
Mareque C, da Silva TF, Vollú RE, Beracochea M, Seldin L, Battistoni F (2018) The endophytic bacterial microbiota associated with sweet sorghum (Sorghum bicolor) is modulated by the application of chemical N fertilizer to the field. Int J Genomics. https://doi.org/10.1155/2018/7403670
Martínez-Herrera E, Duarte-Escalante E, del Rocío R-M, Arenas R, Acosta-Altamirano G, Moreno-Coutiño G, Vite-Garín TM, Meza-Robles A, Frías-De-León MG (2021) Molecular identification of yeasts from the order Trichosporonales causing superficial infections. Rev Iberoam Micol 38:119–124
Meera Krishna B, Khan MA, Khan ST (2019) Next-generation sequencing (NGS) platforms: an exciting era of genome sequence analysis. In: Tripathi V, Kumar P, Tripathi P, Kishore A, Kamle M (eds) Microbial genomics in sustainable agroecosystems. Springer, Singapore, pp 89–109
Moguel IS, Yamakawa CK, Pessoa A Jr, Mussatto SI (2020) L-asparaginase production by Leucosporidium scottii in a bench-scale bioreactor with co-production of lipids. Front Bioeng Biotechnol 8:576511
Molina GES, Shetty R, Bang-Berthelsen CH (2023) Genotypical and phenotypical characterization of Apilactobacillus kunkeei NFICC 2128 for high-mannitol scale-up production using apple pulp and sugar beet molasses as fermentable biological side streams. Bioresour Technol Rep 21:101350
Moretti A, Sarrocco S (2016) Fungi. In: Caballero B, Finglas PM, Toldrá F (eds) Encyclopedia of food and health. Academic Press, Massachusetts, pp 162–168
Mukisa IM, Porcellato D, Byaruhanga YB, Muyanja CM, Rudi K, Langsrud T, Narvhus JA (2012) The dominant microbial community associated with fermentation of Obushera (sorghum and millet beverages) determined by culture-dependent and culture-independent methods. Int J Food Microbiol 160:1–10
N’Guessan FK, N’Dri DY, Camara F, Djè MK (2010) Saccharomyces cerevisiae and Candida tropicalis as starter cultures for the alcoholic fermentation of tchapalo, a traditional sorghum beer. World J Microbiol Biotechnol 26:693–699
Needham DM, Sachdeva R, Fuhrman JA (2017) Ecological dynamics and co-occurrence among marine phytoplankton, bacteria and myoviruses shows microdiversity matters. ISME J 11:1614–1629
Oberholster T, Vikram S, Cowan D, Valverde A (2018) Key microbial taxa in the rhizosphere of sorghum and sunflower grown in crop rotation. Sci Total Environ 624:530–539
Obi CN (2017) Brewery contaminants challenges and remedies-a review. Nigerian J Microbiol 31:3926–3940
Odhav B, Naicker V (2002) Mycotoxins in South African traditionally brewed beers. Food Addit Contam 19:55–61
Quilodrán-Vega S, Albarracin L, Mansilla F, Arce L, Zhou B, Islam MA, Tomokiyo M, Al Kassaa I, Suda Y, Kitazawa H, Villena J (2020) Functional and genomic characterization of Ligilactobacillus salivarius TUCO-L2 isolated from Lama glama milk: a promising immunobiotic strain to combat infections. Front Microbiol 11:3123
Sansone G, Rezza I, Calvente V, Benuzzi D, de Tosetti MIS (2005) Control of Botrytis cinerea strains resistant to iprodione in apple with rhodotorulic acid and yeasts. Postharvest Biol Technol 35:245–251
Schloss PD, Jenior ML, Koumpouras CC, Westcott SL, Highlander SK (2016) Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 4:e1869
Sha SP, Jani K, Sharma A, Anupma A, Pradhan P, Shouche Y, Tamang JP (2017) Analysis of bacterial and fungal communities in Marcha and Thiat, traditionally prepared amylolytic starters of India. Sci Rep 7:1–7
Shivaprakash MR, Singh G, Gupta P, Dhaliwal M, Kanwar AJ, Chakrabarti A (2011) Extensive white piedra of the scalp caused by Trichosporon inkin: a case report and review of literature. Mycopathologia 172:481–486
Shoff CJ, Perfect JR (2021) Uncommon yeasts and molds causing human disease. In: Zaragoza Ó, Casadevall A (eds) Encyclopedia of mycology. Elsevier, Amsterdam, pp 813–834
Smid EJ, Hugenholtz J (2010) Functional genomics for food fermentation processes. Annu Rev Food Sci Technol 1:497–519
Sohlberg E, Sarlin T, Juvonen R (2022) Fungal diversity on brewery filling hall surfaces and quality control samples. Yeast. https://doi.org/10.1002/yea.3687
Sun XD, Shan H, Li L, Su P, Lan J, Zhao L, Yang HC (2019) Field fungal diversity in freshly harvested maize. Int J Biochem Res Rev 26:1–13
Takagi H (2019) Metabolic regulatory mechanisms and physiological roles of functional amino acids and their applications in yeast. Biosci Biotechnol Biochem 83:1449–1462
Takashima M, Manabe RI, Nishimura Y, Endoh R, Ohkuma M, Sriswasdi S, Sugita T, Iwasaki W (2019) Recognition and delineation of yeast genera based on genomic data: lessons from Trichosporonales. Fungal Genet Biol 130:31–42
Tamari F, Hinkley CS, Ramprashad N (2013) A comparison of DNA extraction methods using Petunia hybrida tissues. J Biomol Tech 24:113–118
Tchakounté GVT, Berger B, Patz S, Fankem H, Ruppel S (2018) Data on molecular identification, phylogeny and in vitro characterization of bacteria isolated from maize rhizosphere in Cameroon. Data Br 19:1410–1417
Tokpohozin SE (2018) Microbial biodiversity of traditional Beninese sorghum beer starter and multi-stage fermentation for beer safety and flavor improvement. Dissertation, Technische Universität München
Touijer H, Benchemsi N, Ettayebi M, Janati Idrissi A, Chaouni B, Bekkari H (2019) Thermostable Cellulases from the yeast Trichosporon sp. Enzyme Res. https://doi.org/10.1155/2019/2790414
Tyakht A, Kopeliovich A, Klimenko N, Efimova D, Dovidchenko N, Odintsova V, Kleimenov M, Toshchakov S, Popova A, Khomyakova M, Merkel A (2021) Characteristics of bacterial and yeast microbiomes in spontaneous and mixed-fermentation beer and cider. Food Microbiol 94:103658
van der Klei I, Veenhuis M, Brul S, Klis FM, De Groot PW, Müller WH, van Driel KG, Boekhout T (2011) Cytology, cell walls and septa: a summary of yeast cell biology from a phylogenetic perspective. In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts. Elsevier, Amsterdam, pp 111–128
Van Der Walt JP (1956) Kaffircorn malting and brewing studies. II.—studies on the microbiology of Kaffir beer. J Sci Food Agric 7:105–113
Vujanovic V (2021) Tremellomycetes yeasts in kernel ecological niche: early indicators of enhanced competitiveness of endophytic and mycoparasitic symbionts against wheat pathobiota. Plants 10:905
Walsh AM, Crispie F, Daari K, O’Sullivan O, Martin JC, Arthur CT, Claesson MJ, Scott KP, Cotter PD (2017) Strain-level metagenomic analysis of the fermented dairy beverage nunu highlights potential food safety risks. Appl Environ Microbiol 83:e01144-e1217
Wang H, Tomasch J, Jarek M, Wagner-Döbler I (2014) A dual-species co-cultivation system to study the interactions between Roseobacters and dinoflagellates. Front Microbiol 5:311
Weiss M, Bauer R, Sampaio JP, Oberwinkler F (2014) 12 Tremellomycetes and related groups. In: McLaughlin D, Spatafora J (eds) Systematics and evolution. Springer, Berlin, pp 331–355
Yang B, Wang Y, Qian PY (2016) Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform 17:1–8
Yap M, Ercolini D, Álvarez-Ordóñez A, O’Toole PW, O’Sullivan O, Cotter PD (2022) Next-generation food research: use of meta-omic approaches for characterizing microbial communities along the food chain. Annu Rev Food Sci Technol 13:361–384
Acknowledgements
We would like to thank Ms. Cebile Ndwandwe for her recommendations and technical assistance.
Funding
Open access funding provided by University of Johannesburg. This study was supported by University of Johannesburg (UJ) Global Excellence and Stature (GES) Masters’ Fellowship, UJ GES 4.0 Catalytic Initiative Grant and, National Research Foundation (NRF) South Africa Thuthuka Grant (121826).
Author information
Authors and Affiliations
Contributions
EH: data curation, formal analysis, investigation, methodology, writing—original draft. AA: data curation, methodology, validation, writing—review editing. KM: data curation, methodology, software, validation, visualization, writing—review editing. OAA: conceptualisation, data curation, funding acquisition, methodology, project administration, resources, supervision, validation, writing—review editing. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hlangwani, E., Abrahams, A., Masenya, K. et al. Analysis of the bacterial and fungal populations in South African sorghum beer (umqombothi) using full-length 16S rRNA amplicon sequencing. World J Microbiol Biotechnol 39, 350 (2023). https://doi.org/10.1007/s11274-023-03764-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-023-03764-4