
Abstract
Molecular surveillance of influenza viruses is essential for early detection of novel variants. The aim of the present study was to analyze the hemagglutinin gene of influenza A(H1N1)pdm09 and A(H3N2) viruses circulating during the 2017 season. To investigate the genetic diversity of hemagglutinin gene of influenza A(H1N1)pdm09 and A(H3N2) viruses from 2017 season, ten samples from each subtype were sequenced and analyzed. The season was predominated by influenza A(H1N1)pdm09 viruses. Ten samples were sequenced from each subtype and all sequenced influenza A(H1N1)pdm09 and A(H3N2) viruses belonged to clades 6B.1 and 3C.2a, respectively. Sequence analysis of H1 gene in comparison to 2010–2016 vaccine strain showed mutations K166Q and S188T (K180Q and S202T here) that most likely resulted in antigenic drift and emergence of variant viruses. H3 gene substitutions N137K, N187K, I422V, and G500E that define clade 3C.2a1 were detected during analysis of sequences in comparison to 2017–2018 vaccine strain of northern hemisphere. These substitutions contributed to the change of WHO’s recommendation of the 2018–2019 vaccine strain for northern hemisphere. The results of this study provide insights about the continuous genetic variability of the HA gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza viruses cause acute respiratory illness that can affect people of all age groups. Seasonal influenza viruses are a major threat to public health due to annual epidemics and the potential of pandemics [1, 2]. Influenza viruses belong to the Orthomyxoviridae family of viruses. The genome of the virus consists of negative-sense RNA segments encoding viral proteins. The family Orthomyxoviridae consists of seven different genera, Alphainfluenzavirus, Betainfluenzavirus, Deltainfluenzavirus, Gammainfluenzavirus, Isavirus, Thogotovirus, and Quaranjavirus [3]. Influenza A, B, and C viruses that belong to the genus Alphainfluenzavirus, Betainfluenzavirus, and Gammainfluenzavirus, respectively, cause human infections. Influenza A viruses are subtyped based on the two surface glycoproteins, namely hemagglutinin (HA) and neuraminidase (NA). The HA gene is under selective pressure and undergoes changes in the antigenic residues that result in the appearance of new strains that evade the host’s immune system by escaping pre-existing immunity [4, 5].
Annual outbreaks of influenza A(H1N1)pdm09 virus have been reported in the Indian subcontinent since the 2009 pandemic. A sudden increase in the virus activity was observed in the year 2015 with 42592 cases and 2990 deaths [6, 7]. A study from the Massachusetts Institute of Technology, USA, proposed that amino acid changes in the HA sequences of 2014–2015 influenza virus were responsible for the enhanced virulence and the disease severity of 2015 outbreak [8]. The integrated disease surveillance program reported a high activity of influenza A(H1N1)pdm09 virus in the year 2017 also, with 38811 cases and 2266 deaths [6]. In the Indian subcontinent, influenza A(H1N1)pdm09 virus data are well documented, a practice that was adopted in response to the 2009 pandemic, but data about the other influenza virus that is capable of causing outbreaks such as influenza A(H3N2) are sparse [7]. Thus, the objective of the study was to analyze HA gene of influenza A(H1N1)pdm09 and influenza A(H3N2) virus circulating in Southwest India during the year 2017.
Methodology
Amplification of HA gene and DNA sequencing
Manipal Centre for virus research (MCVR) is a regional reference laboratory for influenza virus for three Southwestern states, namely Karnataka, Kerala, and Goa (Fig. 1). From January 2017 to December 2017, a total of 17371 influenza suspected specimens (throat swab and/or nasopharyngeal swab) were received at MCVR. Nucleic acid was extracted from specimens and subtypes (H1 and H3) were identified using world health organization (WHO) recommended RT-PCR protocol (www.who.int/csr/resources/publications/swineflu/diagnostic_recommendations/en/). Amplification of full-length H1 gene was performed using RNA from positive culture supernatants according to previously published protocol [9]. Influenza A(H3N2) virus-positive specimens were directly amplified without culturing using WHO protocol (www.who.int/csr/resources/publications/swineflu/diagnosticrecommendations/en/).

Study population of the study. MCVR, the regional reference laboratory for influenza virus, is indicated by
 and the three Indian states that are part of the influenza surveillance network are mapped
and the three Indian states that are part of the influenza surveillance network are mapped
Sequence analysis
For genetic characterization of HA gene, a total of 10 H1 and H3 gene-amplified PCR products selected by purposive sampling from different months were sequenced using 3500xL genetic analyzer (Applied Biosystems, CA, USA). For analysis, WHO recommended vaccine strain sequences and HA clade reference sequences were downloaded from the influenza research database (www.fludb.org/). H1 (MG572208.1-MG572216.1 and MH236898.1) and H3 (MH327515.1-MH327524.1) gene sequences have been submitted to the GenBank. Phylogenetic analysis was performed using MEGA 7 software (www.megasoftware.net/) using the maximum-likelihood method. Mutation analysis was performed using flusurver online tool (www.flusurver.bii.a-star.edu.sg/). A 3D model of representative H1 (template PDB ID: 4M5Z) and H3 (template PDB ID: 4W28.1) protein was built using Swiss-Model protein homology modeling online server (https://www.swissmodel.expasy.org/). Mutations and antigenic sites of H1 and H3 proteins [4, 10,11,12] were mapped and visualized using PyMOL (https://www.pymol.org/) software.
Results
Influenza virus detection
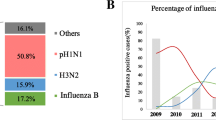
During the 2017 influenza season, influenza A virus activity was high in the three Indian states (Kerala, Karnataka, and Goa) that form a part of MCVR’s influenza surveillance network. Influenza A virus positivity was 28.3% (4924 cases) among the 17371 clinically suspected cases. Among influenza A cases, 72.9% (3590) were influenza A(H1N1)pdm09 virus and 27.1% (1334) were influenza A(H3N2) virus. The maximum number of positive cases was detected in the month of July.
Genetic characterization of HA gene of influenza A(H1N1)pdm09 virus
H1 gene sequences of the 10 MCVR isolates were compared with the sequence of 2017–2018 vaccine strain of northern hemisphere, A/Michigan/45/2015(H1N1) virus, and percentage amino acid identity was found to be 98.74–99.28%. Phylogenetic tree built using amino acid sequences showed clustering of all the 10 HA genes within the genetic clade 6B.1 (Fig. 2a). Amino acid mutations were detected in all the 10 sequences (Table 1). The highest number of mutations (seven) was detected in isolate A/Kolar/MCVRAF3154/2017(H1N1). The residue numbers used for analysis are continuous amino acid residue numbers. Mutations R240Q, S91R, S181T, and I312V were detected in most of the isolates, but only mutation R240Q (first detected in September 2015) has been detected worldwide with a global prevalence of 99.83% (www.flusurver.bii.a-star.edu.sg/). Mutations A26S, H198N, T408S, and S551R are newer mutation detected with no documented reports to date (www.flusurver.bii.a-star.edu.sg/). In comparison to the 2010–2016 vaccine strain A/California/7/2009(H1N1), substitutions D114N, S202T, S220T, E391K, and S468N that define viruses in clade six [13] were detected in all the ten isolates. Mutations K180Q, A273T, K300E, and E516K that are characteristic of clade 6B viruses were also detected in all the isolates. Mutations S101N, S179N, and I233T that define clade 6B.1 viruses were detected in 100, 80, and 90% of isolates, respectively. Antigenic sites Sa, Sb, Ca 1/2, Cb of H1 protein were mapped on the 3D homology model of representative H1 (template PDB ID: 4M5Z) protein (Fig. 3a). Among the amino acid substitutions detected in comparison to the 2017–2018 vaccine strain, two were located in antigenic sites: S91R in site Cb, and S181T in site Sa (Fig. 3b). Analysis of mutations detected in comparison to 2010–2016 vaccine strain showed that four (33.3%) out of 12 amino acid substitutions, which resulted in the evolution of viruses and generation of new clades 6, 6B, and 6B.1, were located in the antigenic sites Sa, Sb, and Ca1 (Fig. 3c).

Phylogenetic analysis of HA gene sequences. Protein coding amino acid sequence phylogenetic tree was built using maximum likelihood method with bootstrap values of 1000. Vaccine strains and clade reference strains are indicated by blue and brown boxes, respectively. a Phylogenetic tree of influenza A(H1N1)pdm09 virus sequences. b Phylogenetic tree of influenza A(H3N2) virus sequences

Homology model of HA protein mapped with antigenic sites and mutations in antigenic sites. H1 protein model based on PDB ID: 4M5Z and H3 protein model based on PDB ID: 4WE8.1 are shown in cartoon. Chains A, B, and C are shown in gray, cyan, and magenta, respectively. a Antigenic sites Sa (yellow), Sb (green), Ca1/2 (blue), and Cb (red) are shown in spheres [10]. b Globular head of H1 protein in chain A is shown in surface and mutations S181T and S91R detected in comparison to the 2017–2018 vaccine strain are colored according to the antigenic sites they constitute. c Mutations detected in comparison to the 2010–2016 vaccine strain are colored according to the antigenic site they constitute. d Antigenic sites A (red), B (blue), C (green), D (yellow), and E (pink) are shown in spheres [11]. e Globular head of H3 protein in chain A is shown in surface and mutations D69N, E78G, K108R, N137K, N138D, T151K, R158G, S160K/R, I208T (hidden in site B), and Q213H detected in comparison to the 2017–2018 vaccine strain are colored according to the antigenic sites they constitute. f Mutations detected in comparison to the 2014–2015 vaccine strain are colored according to the antigenic site they constitute
Genetic characterization of HA gene of influenza A(H3N2) virus
H3 gene sequences of the ten samples were compared with the 2017–2018 vaccine strain of northern hemisphere A/Hong Kong/4801/2014(H3N2) cell-derived virus. Percentage amino acid identity was found to be 98.37–99.45%. Phylogenetic tree built using amino acid sequences showed clustering in clade 3C.2a (Fig. 2b). Amino acid residues were numbered with continuous numbers and used for analysis. Mutations were detected in all the 10 H3 protein sequences in comparison to the 2017–2018 vaccine strain (Table 1). The highest number of mutations (nine) was detected in two strains A/Trivandrum/MCVRAG1073/2017(H3N2) and A/Udupi/MCVRAG1600/2017(H3N2). Mutation N137K, N187K, I422V, and G500E were detected in most of the samples. Among the mutations detected, R158G (four samples) was the most widely detected substitution with a global prevalence of 26.76% (detected in February 2014) (www.flusurver.bii.a-star.edu.sg/). Mutation N22H is a newer mutation detected in this study with no documented reports to date (www.flusurver.bii.a-star.edu.sg/). In comparison to the 2014–2015 vaccine strain of northern hemisphere A/Texas/50/2012(H3N2) cell-derived virus, substitutions N161S and D505N that define clade 3C.2 [13] were detected in all samples. Mutations L19I, N160S/K/R, F175Y, K176T, N241D that define clade 3C.2a were detected in all samples, but mutation Q327H that also defines clade 3C.2a viruses was detected only in six samples. Antigenic sites A–E of H3 protein were mapped on the 3D homology model of representative H3 (template PDB ID: 4WE8.1) protein (Fig. 3d). Among the amino acid substitutions that were detected in comparison to the 2017–2018 vaccine strain of northern hemisphere, 10 (41.6%) were located in antigenic sites: mutations N137K, N138D, S160K/R, T151K, and R158G in antigenic site A; mutations I208T and Q213H in site B; D69N in site C; E78G and K108R in site E (Fig. 3e). Analysis of mutations detected in comparison to the 2014–2015 vaccine strain of northern hemisphere showed that five (62.5%) out of eight amino acid substitutions, which resulted in the evolution of viruses and generation of new clades 3C.2 and 3C.2a, were located in the antigenic sites (Fig. 3f).
Discussion
Influenza A(H1N1)pdm09 virus was the predominant subtype that circulated in the states of Kerala, Karnataka, and Goa during the 2017 season accounting for 72.9% of the influenza A viruses. According to the CDC’s influenza activity updates, influenza A(H1N1)pdm09 viruses predominated Southern Asia with elevated activity reported in India, Nepal, and the Maldives. In Southern Asia, increased virus activity was observed in the months of August and September [14]. In our study, elevated activity of the influenza A(H1N1)pdm09 virus was observed in the month of July. The maximum number of influenza A virus-positive cases was also detected in July, which corresponds with the peak rainy season in Southwestern India.
Phylogenetic analysis of the H1 gene sequences showed that all the ten sequences belonged to subclade 6B.1, with high similarity to 2017–2018 vaccine strain of northern hemisphere. Blanton et al. showed that most of the H1 gene sequences from all over the world, collected since May 2017, were of subclade 6B.1 [14]. In comparison to the 2017–2018 vaccine strain, mutations R240Q, S91R, S181T, and I312V were detected in most of the isolates. Residue Q240 (226 in H5 numbering) in H5N1 viruses is a key residue that contributes to the binding specificity of HA [15], and thus the mutation may be increasing the receptor specificity of the virus. According to a report by Bedford and Neher, clade 6B.1 has been diversified further, and mutations S74R and I295V (S91R and I312V in our study) form a subclade that had started to increase since January 2017. Within the subclade is a more recent subclade with the additional mutation S164T (S181T here) that increased in frequency since April 2017. The frequency of these mutations started to rise globally in recent times (www.nextflu.org/reports/sep-2017/). Analysis of antigenic sites showed that substitution S91R was in site Cb, and S181T was in site Sa. A report suggests that there is no visible antigenic effect of these substitutions with ferret antisera (www.nextflu.org/reports/sep-2017/). Mutations A26S, H198N, T408S, and S551R are newer mutation detected in this study with no documented reports to date. These substitutions were not present in antigenic sites and thus might not affect antibody recognition. In comparison to the 2010–2016 vaccine strain A/California/7/2009(H1N1), substitutions that define clades 6 and 6B were detected in all the isolates. Mutations that define clade 6B.1 were present in 80% of the isolates. Isolate A/Mysore/MCVRAF7729/2017(H1N1) possessed only S101N substitution of clade 6B.1 viruses and formed a diversified subclade (Fig. 2a), but the amino acid identity to 2017–2018 vaccine strain was 98.9%. In isolate A/Kolar/MCVRAF3154/2017(H1N1), substitution S179N was absent resulting in a diversified subclade (Fig. 2a). Among the substitutions that define clades 6, 6B, and 6B.1 virus, four mutations (S179N, K180Q, S202T, and S220T) were present in antigenic sites. S179N substitution creates a potential glycosylation site at residues 179–181 which may affect antigenic and other properties of the strain (www.flusurver.bii.a-star.edu.sg/). Mutations K166Q and S188T (K180Q and S202T here) are the substitutions that likely resulted in antigenic drift and emergence of variant viruses. Free-energy analysis of mutation S206T (S220T here) showed that the mutation has a slightly destabilizing effect on the structure of HA [16].
Phylogenetic analysis of the H3 gene sequences showed that all the ten sequences belonged to subclade 3C.2a with close similarity to 2017–2018 vaccine strain of northern hemisphere. Phylogenetic analysis of the HA genes from viruses collected globally showed diversity and sequences belonged to clade 3C.2a or 3C.3a, but 3C.2a viruses predominated [14]. Within clade 3C.2a, diversification has occurred in recent years (www.nextflu.org/reports/sep-2017/). In comparison to the 2017–2018 vaccine strain, mutations N137K, N187K, I422V, and G500E were detected in most of the samples. Substitutions N121K and N171K in HA1 (N137K and N187K here), and I77V and G155E in HA2 (I422V and G500E) are substitutions that define clade 3C.2a.1 viruses represented by A/Singapore/INFIMH-16-0019/2016(H3N2) virus. A report suggests that these mutations had started to rise in early 2016 (www.nextflu.org/reports/sep-2017/). In February 2018, WHO recommended A/Singapore/INFIMH-16-0019/2016(H3N2) virus as the vaccine strain for the year 2018–2019 of northern hemisphere (www.who.int/influenza/vaccines/virus/recommendations/2018_19north/en/). Strains A/Kannur/MCVRAG0207/2017(H3N2) and A/Thrissur/MCVRAF2695/2017(H3N2) viruses possessed only N137K substitution of clade 3C.2a1 and formed a diversified subclade (Fig. 2b). Strain A/Mysore/MCVRAF8301/2017(H3N2) virus possessed only N187K substitution and formed a diversified subclade (Fig. 2b). Among the mutations detected substitutions N138D and T151K/N are reported to remove potential N-glycosylation sites which can affect antigenic and other properties of virus (www.flusurver.bii.a-star.edu.sg/). R158G substitution, most widely detected substitution worldwide, that was detected in four samples was present in antigenic site A and might contribute to antigenic drift/escape mutant (www.flusurver.bii.a-star.edu.sg/). N22H a newer mutation detected in this study was not present in antigenic site and thus might not affect antibody binding. Analysis of antigenic site residues, in comparison to the 2017–2018 vaccine strain of northern hemisphere, showed that 41.6% of substitutions were located in antigenic sites. In comparison to the 2014–2015 vaccine strain of northern hemisphere, substitutions that define clade 3C.2 were detected in all samples. Mutations that define clade 3C.2a except Q327H substitution were detected in all samples. The amino acid residue at 327 position is reported to be involved in binding host proteins and report suggests that in the year 2017 frequency of appearance of Q327 was more than H327 in South Asia (www.nextstrain.org/flu/h3n2/ha/3y?c=gt-HA1311&f_region=south_asia). Four strains which lacked the Q327H substitution diversified within clade 3C.2a (Fig. 2b). Among the mutations that define clades 3C.2 and 3C.2a, 62.5% were present in antigenic sites. These substitutions most likely resulted in antigenic drift and the emergence of variant viruses.
The study suggests that there are more frequent substitutions in the antigenic sites of H3 gene sequences in comparison to H1 gene sequences. Accumulation of mutations in the HA genes can result in the emergence of new strains that imposes a challenge towards the recommendation of strains for vaccine development. Therefore, continuous monitoring of the circulating strain is important for detecting variant viruses and recommending vaccine strains exhibiting high similarity with the circulating virus.
References
Oliveira TFDMS, Yokosawa J, Motta FC, Siqueira MM, da Silveira HL, Queiróz DAO (2015) Molecular characterization of influenza viruses collected from young children in Uberlandia, Brazil-from 2001 to 2010. BMC Infect Dis 15(1):71. https://doi.org/10.1186/s12879-015-0817-z
WHO | influenza (seasonal) [cited 2018 Feb 10]. http://www.who.int/mediacentre/factsheets/fs211/en/
Orthomyxoviridae—Negative Sense RNA Viruses—Negative Sense RNA Viruses (2011) Int Comm Taxon Viruses ICTV [cited 2019 May 17]. https://talk.ictvonline.org/ictv-reports/ictv_9th_report/negative-sense-rna-viruses-2011/w/negrna_viruses/209/orthomyxoviridae
Caton AJ, Brownlee GG, Yewdell JW, Gerhard W (1982) The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427
Korsun N, Angelova S, Gregory V, Daniels R, Georgieva I, McCauley J (2017) Antigenic and genetic characterization of influenza viruses circulating in Bulgaria during the 2015/2016 season. Infect Genet Evol 49:241–250
State/UT—wise, Year-wise number of cases and deaths from 2010-2017: Ministry of Health and Family Welfare [cited 2018 Mar 6]. http://idsp.nic.in/showfile.php?lid=3908
The cold facts: on tracking influenza outbreak. The Hindu 2017 [cited 2018 Mar 3]. http://www.thehindu.com/opinion/editorial/the-cold-facts/article19791906.ece
Tharakaraman K, Sasisekharan R (2015) Influenza surveillance: 2014–2015 H1N1 “Swine”-derived influenza viruses from India. Cell Host Microbe 17:279–282
Chan C-H, Lin K-L, Chan Y, Wang Y-L, Chi Y-T, Tu H-L et al (2006) Amplification of the entire genome of influenza A virus H1N1 and H3N2 subtypes by reverse-transcription polymerase chain reaction. J Virol Methods 136:38–43
Igarashi M, Ito K, Yoshida R, Tomabechi D, Kida H, Takada A (2010) Predicting the antigenic structure of the pandemic (H1N1) 2009 influenza virus hemagglutinin. PLoS ONE 5:e8553
Stray SJ, Pittman LB (2012) Subtype- and antigenic site-specific differences in biophysical influences on evolution of influenza virus hemagglutinin. Virol J 9:91
Wiley DC, Wilson IA, Skehel JJ (1981) Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–378
Influenza virus characterisation, Summary Europe, June 2017. Eur Cent Dis Prev Control 2017 [cited 2018 May 8]. http://ecdc.europa.eu/en/publications-data/influenza-virus-characterisation-summary-europe-june-2017
Blanton L (2017) Update: influenza activity—United States and Worldwide, May 21–September 23, 2017. MMWR Morb Mortal Wkly Rep. https://doi.org/10.15585/mmwr.mm6639a3
Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, Raguram S, Tumpey TM et al (2008) Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol 26:107–113
Ribas-Aparicio RM, Castelan-Vega JA, Jimenez-Alberto A, Magana-Hernandez A (2014) The hemagglutinin of the influenza A(H1N1)pdm09 is mutating towards stability. Adv Appl Bioinform Chem 7:37
Acknowledgement
This work was supported by the Indian Council of Medical Research (ICMR), New Delhi, India (File No: 5/8/7/15/2010/ECD-1).
Funding
Indian Council of Medical Research (ICMR), New Delhi, India (File No:5/8/7/15/2010/ECD-1).
Author information
Authors and Affiliations
Contributions
All authors have contributed equally to the research work of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the Institutional Ethics Committee (MAHE EC/010/2018).
Additional information
Edited by Zhen F. Fu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jagadesh, A., Krishnan, A., Nair, S. et al. Genetic characterization of hemagglutinin (HA) gene of influenza A viruses circulating in Southwest India during 2017 season. Virus Genes 55, 458–464 (2019). https://doi.org/10.1007/s11262-019-01675-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-019-01675-x