
Abstract
Seasonal influenza viruses constitute a major global concern. Currently, H3N2 and H1N1pdm09 are the commonly circulating influenza A viruses. The haemagglutin and neuraminidase genes of influenza A(H3N2) and A(H1N1)pdm09 viruses from Egyptian paediatric patients with respiratory distress were sequenced. Mutational analysis of all published sequences from Egypt was evolutionary tracked for both HA and NA genes. Phylogenetic analysis of H3N2 HA showed that the Egyptian strains belong to 3C2 subclade while Egyptian A(H1N1)pdm09 strains belong to 6B1 subclade. Some Egyptian A(H1N1)pdm09, 2013–2014, strains form a new subclade; 6B3. High score of mutations were recorded in HA of H1N1pdm09 but higher was recorded in H3N2 strains. These findings confirmed a high mutation rate of influenza A subtypes specially H3N2 strains.
Similar content being viewed by others

Introduction
Seasonal influenza viruses continue to constitute a major global concern that is responsible for high morbidity (5–10% of the adults and 20–30% of children) and considerable mortality of approximately 290,000–650,000 deaths annually [27, 28]. Both influenza A virus (IAV) and influenza B virus (IBV) are the potential causes of seasonal influenza. Human influenza A viruses (IAVs) belong to the genus Alphainfluenzavirus within the family Orthomyxoviridae. Viruses of that genus possess eight segments of negative-sense RNA, PB2, PB1, P, HA, NP, NA, M and NS, which encode for ten essential proteins [1]. Both HA and NA proteins determine the antigenic properties and pathogenicity of the influenza viruses. HA is responsible for virus attachment, envelope fusion and neutralization while the virus uses NA for eluting the virus progeny from infected cells [1]. The HA protein is cleaved by cellular proteases into the HA1 and HA2. To date, there are eighteen different types of haemagglutinin (HA) and eleven types of the neuraminidase (NA) [1]. Although there are dozens of different subtypes of the IAV, only three IAV subtypes, H1N1, H2N2, and H3N2, have been reported to infect humans [31].
Currently, the circulating human influenza A viruses include H3N2 and H1N1pdm09. The latter replaced the seasonal influenza H1N1 virus which had circulated prior to 2009 [9, 27]. The current annual seasonal influenza vaccine is efficient in reducing the severity of the disease symptoms. It also guards against the considerable mortality especially in young children, elderly persons, pregnant women and immunocomprimized patients [15]. Accumulation of point mutations in the haemagglutinin (HA) leads to genetic drift and virus evolution that facilitates the escape from the host immune response with subsequent decrease in vaccine efficiency [22]. Neuraminidase (NA) is the second main surface protein that is also subjected to mutations that are recently linked to the mutation of HA [5]. Accordingly, continuous analysis of the sequence variations of the circulating seasonal influenza viruses is important for detecting variant strains. There is a shortage in the screening of influenza in Egypt a fact that diminish the understanding of influenza evolution and epidemiology [4, 16]. The current study aimed to molecularly characterize the influenza A viruses present in clinical samples from paediatric patients. It also intended to track the mutational changes of HA and NA genes in such strains and other Egyptian strains present in the EpiFlu database.
Materials and methods
Ethical approval
The study protocol was ethically approved by the Faculty of Medicine, Cairo University Ethical Committee. Written informed consents were collected from the guardian of the children.
Specimen collection
Sixty nasopharyngeal swabs were obtained from diseased children under the age of five years at the Paediatric Hospital, Aboelreesh, Giza, Egypt. The children suffered from influenza-like symptoms including fever, cough, sore throat and runny nose from Jan 2015 to Dec 2016. Each swab was kept in a universal virus transport medium (1 mL) and routinely processed. Processed samples were transferred to sterile RNase free tubes and stored at − 80 °C.
Nucleic acid extraction and real time RT-PCR
Viral RNA was extracted with 250 µL of the clarified supernatant of the nasopharyngeal fluid using the QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA). Viral RNA was used as a template for cDNA synthesis according to the manufacturer’s instructions (cDNA Synthesis Premix (Seegene, Korea). Samples were screened for influenza A virus, influenza B virus, adenovirus, parainfluenza viruses (1–4), rhinovirus, human respiratory syncytial virus (A and B), human bocavirus (HBoV), metapneumovirus, human coronaviruses (OC43, NL63, and 229E) and enterovirus (HEV) using respiratory virus detection kits-A and B (AnyplexTM II RV16 detection; Seegene, South Korea) according to the manufacturer’s instructions.
RT-PCR for amplification of HA and NA genes
Viral RNA from samples showed strong positive for influenza A virus (IAV) (n: 10) was used as a template for the amplification of HA and NA from the H3N2 and H1N1pdm09 strains. One-step RT-PCR (Verso 1-Step RT-PCR kit ReddyMix) was used according to the manufacturer’s instructions and specific reference oligonucelotides primers [26]. PCR amplicons were purified using QIAquick gel extraction Kit (Qiagen). BigDye Terminator Cycle Sequencing was used to determine the nucleotide sequence of the purified PCR amplicon using the same primers.
Phylogenetic analysis
MEGA 5.2 freeware was used to process the raw sequences obtained in the current study prior to GenBank submission (accession numbers MG745919–MG745928). Maximum likelihood method and 1000 bootstrap replications and Tamura–Nei substitution model was used to construct the phylogenetic tree of different HA and NA genes. Representative influenza sequences representative to different virus subtypes were obtained from the influenza databases (GISAID Epiflu and GenBank) and used in building the phylogenetic tree.
Tracking of mutations of HA and NA proteins
Comparison of amino acid sequences of HA and NA proteins of both Egyptian H1N1pdm09 and H3N2 available in GISAID Epiflu database in addition to the sequence obtained in the current study were conducted. All Egyptian H3N2 strains from 2006 to 2017 and H1N1pdm09 from 2009 to 2017 were included in the analysis. Deduced amino acids for different genes were created using MEGA 5.2. The amino acid substitution was also analysed in the Influenza Research Database (https://www.fludb.org/brc/home.spg?decorator=influenza) and the scores of variations were recorded. Scores ranged from 0 (no polymorphism) to 200 (highest polymorphism) as previously described [7].
Results and discussion
Real time qPCR on clinical samples and RT-PCR for IAV
In the current study, 26 out of 60 (43.3%) samples of clinical paediatric patients’ samples were found positive for influenza A: H1N1pdm09 (six samples; 10%) and H3N2 seasonal influenza A subtype (twenty samples; 33.3%). Mixed infections were recorded with H3 subtypes including human respiratory syncytial virus A (hRSVA) in four samples (6.7%). Other mixed infections with either bocavirus, human coronavirus 229E or adenovirus were also recorded in four cases (6.7%). HA and NA gene sequencing were successful from five out of ten samples that showed strong positive real time PCR: three H3N2 and two H1N1pdm09 strains.
Genotyping and mutational analysis of H1N1pdm09 strains
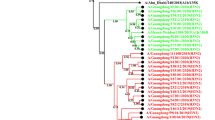
The genotype of the detected influenza A H1N1pdm09 strains, A/Egypt/BSU-13/2016 and A/Egypt/BSU-15/2016, detected from clinical cases of severe respiratory distress in Egypt, were found related to 6B1 subtype (Fig. 1a). Phylogenetic analysis of the neuraminidase of the same strains (Fig. 1b) revealed similar clustering profile to that of the HA (Fig. 1a). There are at least nince major genetic groups of H1N1pdm09 [14]. Since 2014, the genetic group 6 has prevailed. All the 2014–2016 Egyptian H1N1pdm09 strains found in the GISAID Epiflu database are related to genotype 6B and 6A but none related to 6C. Genetic group 6 harbours the amino acid residues characteristic to such genotype including: D97N, S185T, S203T, E374K and S451N. This group is subdivided into diverged into subgroups 6A (H138R, V249L), 6B (K163Q, A256T, K283E, E499K) and 6C (V2341, K283E, E499K). Interestingly, strains belong to 6C subgroup was not recorded in Egypt. Genotype 6B is further subdivided into 6B1(S84N, S162N) and 6B2(T13A, in the signal peptide and, N162S, N84S). In the current study, a newly detected 6B3 cluster was detected that contain signal peptide (L4T, T13A), N84S, N162S). Screening the score of variability of different amino acid residues of the current strains and those published in the influenza database, only 13 amino acid residues showed high score of variability, one in the signal peptide, and one in the S185T in Cb site and the rest in non-antigenic sites (Table 1).

Phylogenetic trees of haemagglutinin and neuraminidase of Egyptian H3N2 and H1N1pdm09 strains in comparison to reference strains. Maximum likelihood method with 1000 bootstrap replications were used to construct the phylogenetic trees. Red colour refers to strains sequenced in the current study. a Haemagglutin of the H1N1pdm09 (grey shaded area is the new subclade 6B3), b neuraminidase of the H1N1pdm09, c haemagglutin of the H3N2 subtype and d neuraminidase of the H3N2 subtype (colour figure online)
The HA1 subunit of the HA possesses the receptor binding site which is surrounded by five antigenic sites named: Sa, Sb, Ca (Ca1 and Ca2) and Cb [30]. Antigenic drift at such amino acids leads to evolution of immune escape mutants or antigenic variants [23, 24]. These antigenic sites were found conserved in H1N1pdm09 Egyptian strains; however, considerable scores of mutations were detected in Ca site (D222E/G/N) Sb site (N162S, Q163K) and in Cb site (T185S) (Fig. 2). Ser 162 to Asn in the Sb site results in increasing the number of N-glycosylation site which is assumed to increase the virus virulence [21].

Different antigenic sites of the H1N1pdm09 haemagglutinin of all Egyptian isolates
Increasing the affinity to α-2,6-linked sialic acids is expected by increasing the mutations in the H1N1pdm09 haemagglutinin [8]. Egyptian strains possess Asp 187 in all published isolates and Asp 222 in the majority of isolates (Fig. 2), such amino acid residue provide the affinity to the upper respiratory tract receptor, α-2,6-sialic acid [3].
Genotyping and mutational analysis of H3N2 strains
Both phylogenetic trees of the HA and NA of the H3N2 showed similar pattern of strains distributions (Fig. 1c, d). The characterized H3N2 in the current study and the Egyptian strains found in the different influenza databases during 2016–2017 were found to be related to subgroup 3C2. Such strains contain S45N and T48I (3C) as well as Q33R, N145S, N278K, D489N(3C2) (Table 2). They possess L3I, N144S, F159Y, K160T, N225D, Q311H (Table 2) which were found to be linked to 3C.2b subclade as previously described [14]. Two unique amino acid substitutions were detected in A/Egypt/BSU-8/2015 (H3N2): Y178D and N230T (data not shown) with yet unknown influence of pathogenicity or antigenicity.
The cell-based influenza seasonal vaccine (2016–2017) used a seed virus that had undergone egg passage that possessed T160K HA amino acid mutation assumed to be related to the egg passage [32]. This speculation could be not true since the T160K was found in most of the circulating Egyptian H3N2 strains. The majority of 2006–2014 strains (62/64) harbour T160K. While 2015–2017 H3N2 strains, possess T160 (Table 2).
Five antigenic sites (A–E) are present in the H3N2 haemagglutinin: A (122, 124, 126, 131, 133, 135, 137, 142–146), B (155–160, 163, 186, 188–190, 192, 193, 196, 197), C (50, 53, 54, 275, 276, 278, 299), D (121, 172, 174, 201, 207, 213, 217, 222, 225–227, 242, 244, 248), E (57, 62, 63, 75, 78, 82, 83, 94) [12, 19, 20]. High score of mutations were recorded in the amino acid residues of epitopes: A (T135K, R142G/K/A, N144K/S, N145S), B (L157S, F159Y, K160T), C (D53N/G, N278K) and D (N121D/K/S, N225D) while low score of mutations were recorded with amino acid residues of epitope E (Table 2).
It was presumed that the co-mutation of NA and HA of the H3N2 strains is utilized for the selection of the virus [5]. Amino acid combinational co-mutation in both HA and NA genes was detected mainly in H3N2 of the season 2013 and NA93G–HA278K was found to be predominant in Japan that was then in the following seasons, NA93D–HA278N was then become more evident. The latter combination was presumed to be more stable in structure than NA93G–HA278K [5]. It is known that amino acid residue HA 278 is associated with the epitope C [29] whereas; NA 93 is not related to any functional sites of the NA including the catalytic, antigenic epitope, or even the N-linked glycosylation sites [2, 6]. N278K is present in the H3 of 2015–2014 Egyptian isolates while the N278 was found in the H3 of 2006–2013 strains (Table 2). The NA 93 amino acid residue was changed from D in most of the 2006–2013 isolates (39/42) to be G in 22/22 of the 2014–2017 (data not shown). Amino acid mutation in the residue 43 of the NA was found to change the virus antigenicity and hence contribute to antigenic drift of the virus [17, 25].
Screening of possible drug resistant mutations in NA proteins
The clinical resistance to the neuraminidase inhibitors were found to be associated with Arg 292 to Lys and Glu 119 to Val amino acid substitution in H3N2 while His 275 to Tyr and Asp 294 to Ser amino acid substitutions in H1N1 virus subtypes [10, 13]. All the Egyptian H3N2 strains harbour Arg at 292, and Glu at 119 while Egyptian H1N1pdm09 possess His 275 and Asp 294 except for a single strain found in the database (EPI824334|A/Egypt/1424/2016) that possesses 275 Tyr)Data not shown). Meanwhile, neuraminidase D79G, and S247G/N amino acid substitutions of the H1N1 neuraminidase were associated with oseltamivir resistance [18] while H126N or Q136K were found to confer resistance to zanamivir [11, 18]. None of these mutations were found in the Egyptian H1N1pdm09 strains found in the database.
It was concluded that detected H3N2 and A(H1N1)pdm09 Egyptian strains belong to 3C2 and 6B1 subclades, respectively. Some A(H1N1)pdm09, from 2013 to 2014, constitutes a new subclade; 6B3. High scores of amino acid substitutions were detected in both HA of H1N1pdm09 and H3N2 however, higher scores were found among H3N2 strains.
References
Abdelwhab EM, Abdel-Moneim AS. Orthomyxoviruses. In: Malik YS, Singh RK, Yadav MP, editors. Recent advances in animal virology. Springer: Berlin; 2019. p. 351–78.
Air GM, Els MC, Brown LE, Laver WG, Webster RG. Location of antigenic sites on the three-dimensional structure of the influenza N2 virus neuraminidase. Virology. 1985;145(2):237–48.
Baldo V, Bertoncello C, Cocchio S, Fonzo M, Pillon P, Buja A, et al. The new pandemic influenza A/(H1N1)pdm09 virus: is it really “new”? J Prev Med Hyg. 2016;57(1):E19–22.
Barakat A, Benjouad A, Manuguerra JC, El Aouad R, Van der Werf S. Virological surveillance in Africa can contribute to early detection of new genetic and antigenic lineages of influenza viruses. J Infect Dev Ctries. 2011;5(4):270–7.
Chong Y, Ikematsu H. Spread of predominant neuraminidase and hemagglutinin co-mutations in the influenza A/H3N2 virus genome. J Infect Chemother. 2018;24(3):193–8. https://doi.org/10.1016/j.jiac.2017.10.010.
Colman PM, Varghese JN, Laver WG. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature. 1983;303(5912):41–4.
Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90. https://doi.org/10.1101/gr.849004.
Elderfield RA, Watson SJ, Godlee A, Adamson WE, Thompson CI, Dunning J, et al. Accumulation of human-adapting mutations during circulation of A(H1N1)pdm09 influenza virus in humans in the United Kingdom. J Virol. 2014;88(22):13269–83. https://doi.org/10.1128/jvi.01636-14.
Grohskopf LA, Sokolow LZ, Broder KR, Olsen SJ, Karron RA, Jernigan DB, et al. Prevention and control of seasonal influenza with vaccines. MMWR Recomm Rep. 2016;65(5):1–54. https://doi.org/10.15585/mmwr.rr6505a1.
Gubareva LV. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res. 2004;103(1–2):199–203. https://doi.org/10.1016/j.virusres.2004.02.034.
Hurt AC, Holien JK, Parker M, Kelso A, Barr IG. Zanamivir-resistant influenza viruses with a novel neuraminidase mutation. J Virol. 2009;83(20):10366–73. https://doi.org/10.1128/jvi.01200-09.
Lees WD, Moss DS, Shepherd AJ. Analysis of antigenically important residues in human influenza A virus in terms of B-cell epitopes. J Virol. 2011;85(17):8548–55. https://doi.org/10.1128/jvi.00579-11.
McKimm-Breschkin JL. Resistance of influenza viruses to neuraminidase inhibitors—a review. Antivir Res. 2000;47(1):1–17.
Nkwembe E, Cintron R, Sessions W, Kavunga H, Babakazo P, Manya L, et al. Molecular analysis of influenza A(H3N2) and A(H1N1)pdm09 viruses circulating in the Democratic Republic of Congo, 2014. J Harmon Res Med Health Sci. 2016;3(4):247–64.
Osterholm MT, Kelley NS, Sommer A, Belongia EA. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12(1):36–44. https://doi.org/10.1016/s1473-3099(11)70295-x.
Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, Gregory V, et al. The global circulation of seasonal influenza A(H3N2) viruses. Science. 2008;320(5874):340–6. https://doi.org/10.1126/science.1154137.
Sandbulte MR, Westgeest KB, Gao J, Xu X, Klimov AI, Russell CA, et al. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc Natl Acad Sci U S A. 2011;108(51):20748–53. https://doi.org/10.1073/pnas.1113801108.
Sheu TG, Deyde VM, Okomo-Adhiambo M, Garten RJ, Xu X, Bright RA, et al. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob Agents Chemother. 2008;52(9):3284–92. https://doi.org/10.1128/aac.00555-08.
Shih AC, Hsiao TC, Ho MS, Li WH. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc Natl Acad Sci U S A. 2007;104(15):6283–8. https://doi.org/10.1073/pnas.0701396104.
Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, et al. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305(5682):371–6. https://doi.org/10.1126/science.1097211.
Tate MD, Job ER, Deng YM, Gunalan V, Maurer-Stroh S, Reading PC. Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses. 2014;6(3):1294–316. https://doi.org/10.3390/v6031294.
Tewawong N, Prachayangprecha S, Vichiwattana P, Korkong S, Klinfueng S, Vongpunsawad S, et al. Assessing antigenic drift of seasonal influenza A(H3N2) and A(H1N1)pdm09 viruses. PLoS ONE. 2015;10(10):e0139958. https://doi.org/10.1371/journal.pone.0139958.
Treanor J. Influenza vaccine—outmaneuvering antigenic shift and drift. N Engl J Med. 2004;350(3):218–20.
Webster RG, Govorkova EA. Continuing challenges in influenza. Ann N Y Acad Sci. 2014;1323(1):115.
Westgeest KB, de Graaf M, Fourment M, Bestebroer TM, van Beek R, Spronken MI, et al. Genetic evolution of the neuraminidase of influenza A(H3N2) viruses from 1968 to 2009 and its correspondence to haemagglutinin evolution. J Gen Virol. 2012;93(Pt 9):1996–2007. https://doi.org/10.1099/vir.0.043059-0.
WHO. Cartographer WHO information for laboratory diagnosis of pandemic (H1N1) 2009 virus in humans. Washington, DC: World Health Organization; 2009.
WHO. Influenza (Seasonal). World Health Orgamization, WHO. 2018. https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal). Accessed 6 Nov 2018.
WHO. Sesonal influenza. World Health Organization. 2020. https://www.who.int/ith/diseases/influenza_seasonal/en/.
Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289(5796):373–8.
Xu H, Yang Y, Wang S, Zhu R, Qiu T, Qiu J, et al. Predicting the mutating distribution at antigenic sites of the influenza virus. Sci Rep. 2016;6:20239. https://doi.org/10.1038/srep20239.
Yoon S-W, Webby RJ, Webster RG. Evolution and ecology of influenza A viruses. In: Compans RW, Oldstone MBA, editors. Influenza Pathogenesis and Control—Volume I. Springer: Berlin; 2014. p. 359–75.
Zost SJ, Parkhouse K, Gumina ME, Kim K, Diaz Perez S, Wilson PC, et al. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc Natl Acad Sci. 2017;114(47):12578–83. https://doi.org/10.1073/pnas.1712377114.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Soliman, M.S., Kamel, M.M., Alorabi, J.A. et al. Characterization and mutational analysis of haemagglutinin and neuraminidase of H3N2 and H1N1pdm09 human influenza A viruses in Egypt. VirusDis. 31, 262–269 (2020). https://doi.org/10.1007/s13337-020-00609-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-020-00609-8