
Abstract
The human cytomegalovirus (HCMV) genome was sequenced by hierarchical shotgun almost 30 years ago. Over these years, low and high passaged strains have been sequenced, improving, albeit still far from complete, the understanding of the coding potential, expression dynamics and diversity of wild-type HCMV strains. Next-generation sequencing (NGS) platforms have enabled a huge advancement, facilitating the comparison of differentially passaged strains, challenging diagnostics and research based on a single or reduced gene set genotyping. In addition, it allowed to link genetic features to different viral phenotypes as for example, correlating large genomic re-arrangements to viral attenuation or different mutations to antiviral resistance and cell tropism. NGS platforms provided the first high-resolution experiments to HCMV dynamics, allowing the study of intra-host viral population structures and the description of rare transcriptional events. Long-read sequencing has recently become available, helping to identify new genomic re-arrangements, partially accounting for the genetic variability displayed in clinical isolates, as well as, in changing the understanding of the HCMV transcriptome. Better knowledge of the transcriptome resulted in a vast number of new splicing events and alternative transcripts, although most of them still need additional validation. This review summarizes the sequencing efforts reached so far, discussing its approaches and providing a revision and new nuances on HCMV sequence variability in the sequencing field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 1881, Hugo Ribbert found the first evidence of cytomegalia and body inclusions in kidney and paratiroid gland cells [1]. Nevertheless, it was only in 1904, and in parallel with Jesionek and Kiolemenoglu, that the evidence was properly reported [1, 2]. Years later, between 1956 and 1957 Smith, Rowe and Weller collaborated in the isolation of the virus, known thereafter as “cytomegalovirus” [3,4,5]. In 1984, 28 years after its first isolation, the first sequence of human cytomegalovirus or HCMV (strain AD169) was published [6], and only 6 years after, in 1990, the first draft of an annotated HCMV genome was published [7], at that time the biggest contiguous genome sequenced (GenBank accession number BK000394.5, additional information in Table 1). Since 1990 and until the submission of this original work, 305 full-length distinct complete HCMV genomes have been published, including low and high passaged strains, lab-attenuated strains, or artificial genomes (NIAID Virus Pathogen Database and Analysis Resource, ViPR) [8].
Human herpesvirus 5 (HHV-5) or HCMV, a member of the family Herpesviridae subfamily Betaherpesvirinae, is a human-infecting ubiquitous host–restricted virus with a world-wide seroprevalence between 45 and 100% in adult population [29]. Primary infections of healthy children and adults are frequently asymptomatic but the virus can establish lifelong persistence as a latent infection, from which it can reactivate and spread new infectious particles [30]. Latency is characterized by an absence or low-level presence of virus replication and the appearance of viral genomes as circularized episomes inside the nuclei of bone-marrow cells CD33+ and CD34+ and peripheral blood mononuclear cells [31]. Reactivation of latent forms of the virus, as well as reinfections of the same are common [32], especially for susceptible groups, as immunocompromised patients, pregnant women, newborns, and elderly. Moreover, in some cases there can be sequelae after infection [33].
HCMV consists of a linear double-stranded DNA genome with an average longitude of 235 kbp ± 1.9 kbp (see genome size variation at Table 1), one of the largest of all human-infecting viruses. The GC content of HCMV genome (57.5%) is the highest among human Betaherpesvirinae, alike the GC content of Gammaherpesvirinae (53.8–59.5%) [34]. The genome is packaged in an icosahedral capsid (T = 16) surrounded by a matrix of proteins, the tegument, and enclosed by lipid bilayer, consisting of a mixture between host and virus proteins [35]. Although the genome is linear inside the nucleocapsid, it is circularized during replication; first through theta-like replication and subsequently by rolling circle amplification, generating multiple linked copies in tandem [35]. Thereafter, the genome is cleaved, linearized and introduced inside the nucleocapsid, following a headfull type packaging [35]. HCMV has a type E genome architecture [36], therefore composed by 2 big inverted domains: long (L) and short (S). In turn, each domain is composed of a central unique region (U, thus UL and US respectively) and by two repeated regions, one at the terminal end and the other at the intersection with the other unique domain (thus TRL/IRL and TRS/IRS, respectively), resulting in TRL–UL–IRL–IRS–US–TRS as a genome organization (Fig. 1). Recombination between repetitive regions is possible, changing the orientation of each unique domain, yielding four possible combinations, thereafter referred as genomic isomers [37, 38] (detailed in Fig. 1). All four genomic isomers can be found in any infective viral population in equimolar proportion [38].

Structure and isomerization of HCMV genome. Representation of the HCMV genome (not on scale) with its four possible isomers (panels a–d). In panel a, the orientation of UL and US is based on UL- and US- orientation of the HCMV wild-type reference Merlin (GenBank AY446894). Panels b–d correspond to the other three possible isomer orientations. Genome regions that are characteristic for HCMV: IRL, IRS, TRL, TRS, OriLyt repetition (OriLytRep) and a′ are colored in red, purple, gray, green, black and yellow, respectively. Small black arrows correspond to the direction of selected ORFs (UL1, UL145, US1 and US34) which help to illustrate the orientation of the unique regions (big black arrow) between the different isomers. Dashed gray lines connect the specific a′ sequences that contributed to the isomerization
In this review, an overview of HCMV next-generation sequencing (NGS) applications will be given, emphasizing the advances in genomic diversity, strain genotyping, full-length genome methodologies, and coding potential based on transcriptomic and translatomic analysis. In this review, we present the current state-of-the-art and promote future steps in the field.
HCMV variability
I am very concerned about the use of the same strains, such as Davis or AD169, in different countries and over long periods of time. I wonder how much these could have changed since their initial isolation.—T.H. Weller [39].
HCMV has been regarded as being highly variable between isolates. As early as 1960, T.H. Weller already stated that serological differences between cytomegalic inclusion disease (CID) isolates are sufficiently different to differentiate classes, thus being an antigenically heterogeneous group [5]. Later in 1976, Huang and colleagues quantified this variability using DNA–DNA hybridization of 12 different HCMV strains and herpes simplex 1 (HSV-1) and herpes simplex 2 (HSV-2) [40]. It was found that the similarity at nucleotide level was of at least 80%, when comparing different strains of HCMV, in comparison with the 50% when compared to either HSV-1 or HSV-2 [40]. Restriction endonuclease typing also supported moderate divergence between different clinical isolates, without any clear grouping or subtyping between isolates, diverging in concurrence of restriction sites, position and size of the digested fragments [37, 40]. In 1980, Pritchett and colleagues found similar results comparing HCMV AD169 and Towne strains by DNA–DNA hybridization and restriction profiling, implying a similarity of at least 90% at nucleotide level [41].
In 1990, when first feasible applications of sequencing came available, Chee and colleagues published the first version of HCMV genome (AD169 strain, GenBank X17403, Table 1 for related information) [7], which lead to sequence and genome-wide comparison of different isolates and its coding potential [7, 9, 42]. Based on comparative genomics and open-reading frame (ORF) analyses, Cha and colleagues in 1996 discovered 19 genes that were missing from high-passage isolates (strains AD169 and Towne) compared to the low-passage Toledo strain and five clinical isolates. As depicted in Fig. 2, large genomic re-arrangements between AD169 (GenBank AC146999), Towne (GenBank FJ616285) and Toledo (GenBank AC146905) bacterial artificial chromosomes (BACs) can be observed. These re-arrangements, excluding the different possible genome isomers fixed into BACs, are inversions and deletions at the internal end of the UL region, known as the UL/b′ sequence, and correspond to missing genes ranging from UL133 to UL154, where several HCMV specific glycoproteins are found in clinical isolates [42, 43]. Likely, UL/b′ is lost by recombination and excision with the terminal a′ sequence during long-term passage of clinical isolates, thus changing the levels of virulence and cell tropism of the viral population [42]. AD169 and Towne attenuation is thought to have appeared, partially, as consequence of UL/b′ deletion [43]. Later works by Hahn et al. and Bradley et al. described heterogeneous populations of both Towne and AD169 in regards to UL/b′ deletion, as well as other mutations [13, 44]. Hahn et al. provided a method for cloning both the Towne varS (GenBank AC146851) and varL (GenBank FJ616285), short and long Towne variants, into BACs as a mean to produce genetically stable viral stocks. Towne varS, as AD169, lacks the UL/b′ region, meanwhile Towne varL contains UL/b′, resembling an uninverted UL/b′ sequence from Toledo and clinical isolates obtained in that period [42, 44]. A similar phenomenon is also observed in AD169, one of the most extensively passaged HCMV isolates [13]. In Bradley et al. three AD169 stocks were sequenced: AD169 varUK (GenBank BK000394), AD169 varATCC VR-997 (GenBank AC146999), both derived from NIH 76559 original stock, and AD169 varUC (GenBank FJ527563), using for the first time in HCMV genomics an Illumina sequencing platform. AD169 varATCC proved to be a mixture population of two variants: varS and varL, the later containing UL/b′ region, as AD169 varUC. In 2004, Dolan and colleagues sequenced using Sanger method what would become the reference genome for the wild-type HCMV, the highly productive Merlin strain (GenBank AY446894), isolated from urine of a congenital infected infant and passaged three times on human foreskin fibroblasts (HFFs) [14]. In addition, Dolan et al. expanded the comparison between isolates with different passage histories, complementing Cha et al. results [42], by defining the genomic features of a wild-type HCMV, as opposed to high-passage attenuated HCMV strains. The Merlin strain has been extensively used as wild-type HCMV reference genome, especially as a backbone for genome annotation and annotation transfer. Since the publication of the first HCMV genome and its coding potential, heterogeneity has been studied either through the genotyping of a selected list of genes, viral markers, or through whole-genome comparisons.

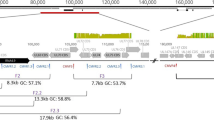
Genome structure of classic HCMV strains. Whole-genome alignments of classical HCMV strains (AD169, Merlin, TB40/E, Toledo and Towne) are presented. Linear maps were build using AliTV visualization software [45], based on whole-genome alignments with Lastz aligner [46]. Both panels a, b depict pair-wise comparisons, expressed as percentage of similarity (green to red), that connect different homologous genomic regions. Genomes are pictured in blue. As shown in the legend, the different type E genome repetitive regions (IRL, IRS, TRL, TRS, OriLyt and a′ sequence) are colored. Color pattern is shared with Fig. 1 for comparison purposes. Genome length and repetitive regions are on scale. Length units are expressed in base pairs, as shown in the superior part of both panels. Genomes are ordered by descending genome length. Panel a represents the pair-wise genome comparison of AD169, Merlin, TB40/E, Toledo and Towne genomes sequenced from BACs (excluding wild-type reference Merlin). GenBank accession numbers, ordered as represented in panel a, are AY446894, FJ616285, AC146999, EF999921, and AC146905. Panel b illustrates the pair-wise comparison of the same strains from panel a, but sequenced using NGS short-read technology (with the exception of Towne). GenBank accession numbers, ordered as represented in panel b are F297339, AY446894, FJ616285, GU937742, and FJ527563
Genotyping of viral markers
HCMV co-evolved with its human host since diverging from other Betaherpesvirinae, circa 120 million years ago [47], and displays a wide array of molecular strategies that allow for survival and perpetuity. All members of the family Herpesviridae, but especially HCMV, have acquired functions that favor persistency, immune evasion and molecular mimicry. Some of those functions have been co-opted from host pre-existing machinery [43], as well as other viruses [11, 48] which may account for their considerable genome size. Genes that are linked to persistency, evasion, resistance, or mimicry have been recurrently genotyped in different populations, to assess HCMV variability and its potential thread. These genes of interest, also known as viral markers, can be classified between (i) drug-resistance genes, (ii) virulence, immune evasion, molecular mimicry, and (iii) surface glycoprotein receptors.
Genotyping of HCMV can be distinguished in two approaches: (i) non-PCR and (ii) PCR-based methods. Non-PCR-based methods group direct restriction enzyme digestion [37, 40] and southern blot [41, 42]. Both methods were mostly used in the early days to analyze HCMV variability and to generate the first genetic maps [49]. PCR-based methods group (i) amplicon sequencing and (ii) molecular amplification. Amplicon sequencing has preferentially been conducted with Sanger/dye terminator chemistry sequencing [50,51,52,53,54,55,56], whereas variability assessments have been performed using NGS, concretely with second-generation 454 pyrosequencing [55, 57, 58]. Molecular amplification groups PCR techniques that (i) qualitatively and (ii) quantitatively characterize mutations. Qualitative genotyping was predominantly conducted by RFLPs [59, 60]. Quantitative or semi-quantitative genotyping has been exclusively conducted by qPCR [61, 62].
Methods based on restriction enzymes (enzyme digestion, Southern blot, or RFLPs) can fail to detect sequence variability, as only sites sensible to restriction enzymes are analyzed. Conversely, PCR-based methods (including amplicon sequencing) are less prone to miss sequence variability, although only variability found in the amplified region can be studied and poor primer design may reduce the sensibility to detect new variants. Amplicon sequencing has preferentially been conducted with Sanger sequencing, as sequencing base accuracy can reach a maximum of 99.999% with this technique [63]. NGS, specifically second-generation pyrosequencing, has also been used for genotype exploration [55, 57]. Despite having a lower base accuracy and read length, it can provide more reads, hence more sequencing depth of the sample to call for multiple variants. Under this scenario, NGS platforms are more informative, due to the higher read yield and their increased sensibility to multiple variants. Genotyping of multiple loci from clinical isolates can be scalable by using amplicon NGS. These sequencing platforms can analyze and later reconstruct different sequences, while keeping traceability of sequence origin by using molecular identifiers, or barcodes. Complete gene genotyping should be considered, as genotyping only specific regions of the gene increases the likelihood to lose unknown polymorphic sites [64] or to overlook new recombining genotypes between different polymorphic sites, as already been described for UL55 (gB) [65]. Other existing sequencing platforms have yet to be tested on HCMV amplicon genotyping, as sequencing technologies improved fast and full-length genomes where soon available.
Currently, there is no consensus on the classification of HCMV strains based on genotype, evolutionary relationship, or clinical relevance. Loci genotyping should proceed with caution, as the costs of sequencing a full-length HCMV genome have decreased in the last years. Not aiming to sequence a full clinical isolate genome might be an unrepairable opportunity to understand this complex pathogen. Whole-genome sequencing can simultaneously capture all variants and remove the need to design and optimize PCR assays for multiple variant detection, allowing e.g., for a parallel antiviral-resistance testing in a single experiment [66] or for predicting changes to epitopes for vaccine development [67].
Next-generation sequencing in HCMV research
…[A] knowledge of sequences could contribute much to our understanding of living matter—F. Sanger [68].
Since the apparition of the first massively parallel sequencing technologies in the 2000s, new possibilities for HCMV research emerged after each technological breakthrough. 454 Life Sciences, later known as Roche 454, and Illumina Inc independently created the first massively parallel sequencing platforms, used in the first deep sequencing on HCMV [13]. These technologies, not only created a new way to recompose full-length HCMV genomes without sequence cloning, but allowed a better understanding of its population variation and coding capacity during infection [69]. Recently, a new opportunity to differently understand the HCMV genome has appeared with the application of third-generation sequencing, based on long-read real-time sequencing [27, 70, 71].
Whole-genome sequencing
Up to the submission of this review, 305 full-length distinct HCMV genomes have been published (NIAID Virus Pathogen Database and Analysis Resource, ViPR) [8], 251 of them derived from clinical isolates (GenBank accession numbers and sequence relevant information can be consulted at Table 1), and of these sequences only 205 correspond to unpassaged or low-passage isolates (< 4 passages). Since Chee et al. published the first HCMV genome, Sanger sequencing has been regarded as the standard for HCMV drug-resistance detection [72].
Currently, the most precise full-genome cloning system consists of an embedded complete genome in a BAC with Cre/LoxP self-excising system, amplifying the genome in a bacterial system with very low mutation rates, as the BAC is amplified by the bacterial DNA polymerase. Cre/LoxP self-excising system does not modify the original virus sequence with the exception of a 34 bp insertion downstream of the US28 gene [73]. Although, BAC cloning can produce long-lasting stable strain amplification systems [20, 73], cloning and sequencing by primer walking can be time-consuming, inefficient and might not be an optimal method for exploring virus diversity within a clinical sample. Interestingly, HCMV genome BAC cloning captures genomes individually, as they are contained inside the viral particles, creating fixed genetically stable viral genomes. These stable genomes faithfully represent the individual variants of that viral particle, including its genome isomerization (represented in Fig. 1), as well as multiple genomic variants, which may not necessarily represent the most abundant form in the viral population or its infective capacity. In Fig. 2, many genomic re-organizations become apparent when comparing different HCMV strain genomes. In Fig. 2a, different BAC isolated HCMVs are represented, characterizing inversions spanning the entirety of unique regions when comparing two genomes. These apparent inversions are in fact a result of comparing different HCMV isomered genome sequences, fixed and stabilized in BACs (i.e. TB40/E—BAC vs. Toledo—BAC, in which UL has different directions). In addition, in Fig. 2a, inversions or translocations that reorganize the classical structure TRL–UL–IRL–IRS–US–TRS should be taken with caution, as they may arise from the introduction of the genome into the BAC vector. AD169-BAC (GenBank AC146999) offers a clear example, as its US region appears to be fragmented and translocated to the terminus of the genome. Once these previous re-organizations are considered, other re-arrangements can be recognized in Fig. 2. These re-organizations arise from imperfect homologous recombination during HCMV genome replication, being focus of HCMV infective variation studies. Interestingly, these re-organizations can be found in both Fig. 2a, b, as the same strains are illustrated in both panels but with their genome sequence is derived from either BAC cloning and posterior sequencing or by second-generation sequencing from a pool of viral particles. This comparison between both sequencing methods exemplifies the differences between re-organizations derived from (i) technical procedures (BAC cloning), (ii) viral replication (genome isomers), or by (iii) imperfect homologous recombination and mutation. In this regard, the deletion (and/or inversion) at the UL/b′ region, characteristic for high-passage attenuated strains, can be observed when comparing AD169, Towne and Toledo strains in both panels of Fig. 2, as previously discussed in this review.
Despite the benefit of capturing, fixing and genetically stabilize single viral genomes that BAC cloning can offer, most of the partial or full-length genomes have been derived from second-generation sequencing platforms, mainly due to BACs poor scalability for viral population research. These platforms enabled the discovery of different variants in HCMV viral populations (as previously discussed for Towne varS and varL) [13], and a substantial decrease in time and resources needed for genome sequencing. High-throughput NGS allowed to increase the number of clinical HCMV genomes to more than 170. Sequencing of full-length HCMV genomes was initially performed with Roche 454 pyrosequencing [17, 19, 25], coupled to either or both Sanger and Illumina sequencing to polish low quality regions, producing 57 HCMV genomes. Illumina sequencing platform rapidly outperformed its competitors with its improved chemistry, yield and base quality, generating most of the available genomes (158 out of 251) [15, 20, 22,23,24, 26]. Albeit NGS boosted the sequencing of HCMV genomes, direct sequencing of clinical HCMV remained an issue, due to its low viral particle yield of during infection. Common sources of clinical material for HCMV sequencing include: blood, urine, bronchoalveolar liquid, tissue (mostly kidney or liver), and amniotic fluid (a relationship between sequenced genomes and tissue of isolation can be found in Table 1).
Short-read second-generation sequencing provides a solid working approach to the study of single-nucleotide variants (SNVs) due to its high read yield, increased read coverage along the genome and improved sequence error (correction, improving variant detection). Unfortunately, the characterization of genome re-arrangements or structural variants (SVs) with second-generation sequencing can be challenging due to (i) its association with (low-complexity) repetitive regions, (ii) the difficulty of short-reads to span large genomic events, and (iii) to precisely localize breakpoint coordinates [74].
The reconstruction or assembly of a HCMV genome can be a complex task as (i) clinical material has low viral genome copy numbers, directly affecting sequencing coverage and the overall genome quality. Additionally, (ii) HCMV genome contains three regions with low-complexity repetitions at the unique terminal and unique internal end, increasing the difficulty to correctly align and recruit reads during genome assembly. Finally, (iii) mixed HCMV populations are expected, either as a result of co-infection of different strains or activation of latent HCMV infections, generating a genetically heterogeneous (or heteroclonal) population [55]. Discerning which variants co-concur (co-linearize) and belong to the same viral genome may benefit the examination of clonal heterogeneity of the viral population.
Different techniques have been coupled to second-generation sequencing platforms, to increase the yield of viral reads. Most strategies use (i) multiple sequence displacement amplification (MDA) [19, 22] to increase the input viral DNA in the sample, or (ii) target enrichment to enrich the sample by capturing viral DNA using DNA or RNA probes (also known as bait libraries) [26, 66, 70, 75]. MDA kits use high-fidelity polymerases (generally a ф29 polymerase) in conjunction with a set of random hexamers to amplify DNA at isothermal conditions [19, 76, 77]. Although, this technique amplifies viral genomic fragments between one to three orders of magnitude [19], biases have been reported specifically linked to a high allelic drop-out effect (ADO, preferential amplification of a subset of alleles in a heteroallelic sample) and non-uniform amplification of linear double-stranded DNA (related to the GC content of the amplified sequence) [19, 76, 78]. Both Marine et al. and Roux et al. conducted genome coverage analysis on MDA dsDNA amplified viruses [76, 78], providing clear evidence that MDA amplification is one of the disturbing factors in completing a full-length genome. A recent study by Borgström and colleagues compared four different MDA available kits during single-cell human DNA amplification: AMPLI1, MALBAC, Repli-G and PicoPlex, taking coverage, SNP calling and ADO to test the reliability of the kits [77]. Borgström et al. showed that Repli-G produced the most uneven low coverage genome amplification, followed by PicoPlex. AMPLI1 and MALBAC had comparable even coverages [77]. SNP calling performed poorly by Repli-G kit, only 3% of the variants were detected, in comparison with the 25% detected by MALBAC [77]. The Repli-G ADO effect is probably linked to the poor performance during SNP calling. Only one allele in all studied loci and replicates was detected [77]. Target enrichment, conducted mainly with SureSelectXT library enrichment, has been used to obtain over 50 unpassaged HCMV genomes [26, 66, 70]. By designing custom bait libraries that cover the entire HCMV genome, it is possible to capture (by hybridization and streptavidin bead separation) the fraction of a given NGS library that corresponds to the virus, and further amplify it [79]. This technique allows to sequence viral genomes directly from clinical samples, avoiding virus culturing (used to increase the yield of the virus at cost of virus adaptation to the growing cell line [16]). SureSelectXT enrichment has been extensively used in the last years [23, 26, 66, 70, 80]. There are at least two different custom bait libraries being currently used, one developed at the Center for Virus Research, University of Glasgow [26] and a second designed by the PATHSEEK consortium, jointly with Oxford Gene Technology™ [23, 66, 70], albeit none of them is publicly available. Both MDA and target enrichment rely on additional PCR amplifications, hence susceptible to introduce a new sequence bias to the sequencing library. Regardless of the increased HCMV sequencing performance that both techniques offer, the omission of infrequent viral variants should be a cause of concern. MDA methods, especially Repli-G, have a known preference to amplify certain regions and variants, leading to uneven low coverage regions and narrowed variant diversity, hence likely over-looking the intrinsic variation in a viral population. Theoretically, a narrowed variant diversity could also be found if target enrichment was used, as its efficiency relies on a library design for known but also unknown variants.
Assembly of herpesviruses, such as HCMV, can be inaccurate due to its low-complexity repetitive regions as well as its local deviant GC content, producing discontinuities, or gaps, in the assembly [15]. This inaccuracy is linked to the read length [74]. A longer read size is more likely to produce reads that can span or bridge regions where the library or sequencing platform might be infra-represented, and correctly characterize repetitious regions, both its boundaries and number of repeats [74].
Poor connectivity between distinct assembled regions, or contigs, is a major challenge for assembling full-length HCMV genomes. As previously stated, assembly inaccuracy can be linked to low-complexity repetitive regions as well as a local deviant GC content [74, 81], such is the case for type E genomes like HCMV. GC deviant regions may produce read miss-representation in those regions, potentially failing to assemble a full-length genome. Different parts of the sequencing scheme can be responsible for this phenomenon, PCR amplification of the library, cluster amplification, or the reading during sequencing [81]. However, library amplification by PCR plays the major role in generating GC bias, especially affecting short-read synthesis-based sequencing (i.e., Illumina sequencing platform) [81]. Likewise, low-complexity repetitive regions can impair an assembly by generating multiple possible positions where reads could align and in that way, generating new sub-alignments, increasing the complexity of the assembly and reducing the accurateness of certain regions [74]. Poor region connectivity (i.e., an assembly with too many short contigs) can challenge full-length HCMV genome assembly when different viral variants are present in the same sample. Improving this connectivity would increase the recovery of complete and distinct genomes at sub-strain level, as well as sequencing through repetitive regions [74]. Consequently, longer reads are desirable, as the longer a given read is, the longer the contigs in the assembly would be, hence increasing the assembly connectivity. Furthermore, improved read-length would likely increase the chance to contain multiple (distant) variants in the same read, providing direct evidence of their co-linearity in a given single virus genome from a clonal heterogeneous HCMV population.
Third-generation long-read sequencing platforms, such as SMRT™ from Pacific Biosciences™ and Nanopore sequencing from Oxford Nanopore Technologies™ open the possibility to improve assembly connectivity, providing a promising platform for single virus (partial) genome sequencing, due to its extended read-length. Until the submission of this review, only 3 different long-read HCMV sequencing projects have been published, 2 in 2017: (i) Balázs and colleagues with a hybrid approach using the Pacific Biosciences™, PacBio RS II system and Oxford Nanopore Technologies™, MinION™ platform [27, 28, 82], and (ii) Eckert et al. with the Oxford Nanopore Technologies™, MinION™ platform [70]. The last project, published in 2018, by Karamitros and colleagues, also used Oxford Nanopore Technologies™, MinION™ platform [71]. In Balázs et al., HCMV Towne varS strain (passaged more than 125 times with the 180,887–191,406 region of the original genome substituted with the 1–11,996 region, GenBank LT907985) full-length genome was assembled using cDNA transcripts and the reference Towne (GenBank FJ616285) [27, 28]. In Eckert et al., HCMV genomes were sequenced, directly from clinical material, with and without target enrichment (SureSelectXT, without downstream amplification). In contraposition with Balázs et al., only 1.2% of the reads (from non-enriched samples) could be assigned to HCMV; meanwhile for viral enriched samples, 98.7% of the reads were assigned to the virus, reconstructing the HCMV genome up to 99.4% with a mean coverage of 89.9× [70]. In Karamitros et al., HCMV TB40/E strain (GenBank EF999921) clone Nano, was sequenced using cell-associated replication, without in vitro amplification, and a posterior viral concentration with ultracentrifugation. A 48-h run produced close to 47,000 reads with a uniform average coverage of 100×. The full-length genome was obtained by a co-assembly (or “hybrid de novo assembly”, as the authors refer to) of the same filtered reads by a fast long-read assembler, SMARTdenovo [83], and a short-read assembler, SPAdes [84]. With this approach Karamitros et al. (raw MinION™ data are available at NCBI—SRA project number PRJEB25285) provide for the first time, using NGS, evidence of viral genome isomers from a type E genome [71]. Long-read sequencing enabled Karamitros and colleagues to find several SVs in a BAC-derived TB40/E polyclonal sample: (i) a deletion spanning UL144–UL145, (ii) one inversion, (iii) two relocations, and (iii) several short insertions or deletions (deriving in local misalignments) [71].
Hitherto, no method combines all characteristics to analyze variation in a HCMV polyclonal infection as, or close to, single virus genomes. The ideal method for studying HCMV would have to (i) sequence directly from clinical material (no cell or in vitro amplification), (ii) to be unbiased (either by enrichment or uneven amplification), and (iii) to provide direct evidence of variant co-linearity to an individual viral genome.
Transcription, translation and regulation analysis through RNA-sequencing
Since the publication of the first studies of HCMV transcriptomics by Gatherer and colleagues, the advancements on HCMV RNA-sequencing have highlighted new aspects on its behavior: regulatory small RNAs [85, 86], new RNA splice variants [27, 69] and newly detected ORFs [69, 87]. Early estimates ranged from 164 ORFs [14, 69, 88] to 220 ORFs [89], although ribosome profiling identified up to 751 individual ORFs [87]. Those 751 translationally active ORFs may be a more precise estimate of coding capacity, as it is likely to account for over the (i) 100 splice junctions that HCMV genome contains [69, 87], (ii) transcript polycistrony (i.e., UL138) [90] and (iii) short ORFs [87]. Despite the obvious codifying complexity of HCMV, the wild-type reference Merlin (GenBank NC_006273.2) currently has 173 annotated genes, of which 168 are protein-coding genes and 5 non-protein-coding genes.
According to Gatherer et al. (BioProject PRJEB2543, see Table 2 for additional project information) 3 different types of transcripts can be expected when analyzing HCMV infections: (i) protein-coding transcripts, (ii) non-coding non-overlapping transcripts (RNA2.7, RNA5.0, and RNA1.2 long non-coding RNA or lncRNA), and (iii) antisense transcripts (transcribed antisense with respect to protein-coding regions) [69, 85]. Studying the infectious behavior of HCMV Merlin strain in HFFs at 72 h post infection showed that the presence of antisense transcripts throughout the HCMV genome, by strand-specific RNA-seq (in [69] referred as “directional sequencing or DDS”) and strand-unspecific RNA-seq (in [69] referred as “non-directional transcript sequencing NDS”), represented a 8.7% of the overall transcription [69]. In addition, RNA2.7, RNA5.0, and RNA1.2 transcripts account for 65.1% of the overall transcription, especially RNA2.7 that represents the 46.8% of the overall transcription. Strikingly, protein-coding transcripts only account for a third of the overall transcript production [69]. New splicing sites were discovered, leading to the description of new alternative splicing events and confirmation of four novel gene transcripts (RL8A, RL9A, UL150, and US33A) [69]. A year later, Stern-Ginossar and colleagues (BioProject PRJNA177721) used ribosome profiling (sequencing of mRNA protected within the ribosomes) to study Merlin transcription in HFFs at 5, 24, and 72 h post infection [87]. 751 translated ORFs were found with only 147 being previously described. These novel putative ORFs where derived from (i) nested ORFs (in and out of frame), (ii) upstream short ORFs, (iii) antisense ORFs, and (iv) short unpredicted ORFs (ORFs coding for protein between 100 and 200 aminoacids [91]). Multiple ORFs were translated from RNA1.2, RNA2.7 and RNA4.9 lncRNA, acting as a precursor polycistronic mRNA [87].
MicroRNAs (miRNAs) are small RNAs of 22 nucleotides long, transcribed by RNA polymerase II [92], related to RNA silencing and post-transcriptional regulation of gene expression. Both functions have been studied for their possible regulatory role during an HCMV infection [10, 85, 103]. While miRNAs are known to be non-immunogenic, some are known to have a regulatory function in viruses [86]. HCMV is known to produced mature miRNAs during infection [10, 103]. Stark and colleagues (BioProject PRJNA148583) studied host (HFFs) and HCMV Towne miRNAs profiles at 24 and 72 h post infection. Up to 20% of the miRNAs were from viral origin, providing evidence of 22 miRNAs being incorporated into the endogenous host silencing machinery [85]. In contrast, Meshesha and colleagues described the fraction of miRNA dropped to only 5% [86]. Even if Stark et al. and Meshesha et al. identified the same seven top most abundant transcripts (miR US5-2-3p, US25-1-5p, US25-2-3p, US25-2-5p, UL22A-3p, UL22A-5p, and UL36-5p), their abundances substantially differed. Those changes in abundance could be attributed to 3 different causes: firstly, (i) two different HCMV strains were used (Towne in Stark et al. vs. AD169 and 3 clinical isolates in Meshesha et al.). Secondly, (ii) RNA was collected at different time points (72 h vs. 96 h post infection), and finally (iii) different methods of miRNA assignment were used (mapping reads with Bowtie v0.12 to miRBase v16.0 vs. mapping reads with BWA v0.5 miRBase v17.0) [85, 86]. Using previously published ribosome profiling data by Stern-Ginossar et al., Ingolia and colleagues (BioProject PRJNA257463) found evidence of novel polypeptide production in RNA2.7 transcript, capable to induce immune responses from the host [101]. Kim et al. (BioProject PRJNA269099) found that a large fraction of human miRNAs targets was shared with viral miRNAs in HFFs infected with Towne varL after 24, 48 and 72 h post infection [99]. In 2016, Buzdin and colleagues (BioProject PRJNA304028) could link a complete suppression of host miRNAs regulation during early stages (3 h) of an HCMV infection, by infecting embryonic lung fibroblasts (HELF-977) and skin fibroblasts (HAF-1608) with AD169 [98]. Lastly, Stark et al. found evidence of miRNAs being derived from the lncRNA RNA2.7, contributing to profile HCMV long RNAs as precursors to other functional RNAs. Interestingly, Stern-Ginossar et al. found similar results applying ribosome profiling techniques, describing lncRNAs as precursors for putative short proteins [87].
Transcriptomics can also be used to understand the processes of cell tropism and infection in different cell types. Van Damme et al. (BioProject PRJEB15199) compared differences in expression between TB40/E infected macrophages and dendritic cells (DCs) derived from whole blood donations [96]. Interestingly, in primary cell types, differentially expressed genes often belong to clusters, suggesting a functional coordination between those transcripts coming from genes of the same family. Concretely, the decrease in expression of RL11–RL13–RL14–UL1 and UL4–UL11; and the increase of UL120–UL121, UL148D–UL149, and US33–US34A in DCs were strikingly pronounced [96]. In macrophages type 1, the cluster UL81–UL86 appeared to have its expression generally decreased (although UL81–UL82 and UL85 did not reach clear significance). Contrary, in macrophages type 2, the same cluster, UL82–UL86 had its expression increased (UL84 was not significant), as well as RL11–RL12, UL2-3, and UL148A–UL149 loci. Similarly, the unique short region US7–US9 had their expression increased [96]. Possibly, US1–US6 region would have its expression increased, as the whole region is generally related to immunomodulation, but was deleted in the production of TB40/E BAC on which Van Damme and colleagues based their study. As expected, most of the differentially expressed genes were related to immunomodulation, cell tropism (prominently UL74, US9, and US27) and adaptability to different cell types (as UL4 and UL5) [96].
Batra et al. (BioProject PRJNA342503) proposed some advances on alternative therapeutic targets in 2016 [97]. Cytoplasmic polyadenylation element binding protein 1 (CPEB1), responsible for cytoplasmic polyadenylation, was found to have a major role in infection-related cytopathology and post-transcriptional changes in different strains of HCMV (TB40/E and Towne) and in Herpes Simplex virus 2 in different tissue types [97]. Decreased transcription levels of CPEB1 reduced viral RNA polyadenylation (shortening poly-A tails), alternative splicing and other RNA processing events, which leaded to a decrease of HCMV titers and shift in the transcription profile in comparison with a mock infection [97].
Although pending for experimental validation, Zhang and colleagues (BioProject PRJNA340198) described the latent HCMV Towne infection cell transcriptome in THP-1 cells [100], defining more than 2000 host differentially expressed genes, with approximately half of them with an upregulated expression profile. As expected, those differentially expressed genes were involved in pathways of apoptosis, inflammatory response and cell cycle progression [100]. Interestingly, lncRNAs were differentially expressed with an ongoing HCMV latent infection [100]. A year later, Cheng et al. (BioProject PRJNA389726) compared the expression of natural infection (healthy peripheral blood mononuclear cells latently infected with clinically uncharacterized HCMV) and experimental latency system in a transcriptome-wide study using positive strand SureSelectXT target enrichment [80]. The experimentally latent system used mutated TB40/E strains: ΔUL135–TB40/E (latent-like) and ΔUL138–TB40/E (strict-lytic). The SureSelectXT enrichment represented a viral RNA increase between 74.35 and 81.2%, increasing viral read yield more than 6000 fold, without biasing the read distribution of the transcriptome [80]. Strikingly, wild-type TB40/E and recombinant ΔUL135 were very similar in transcript composition and abundances [80]. Alternatively, recombinant ΔUL138 infected cells harbored transcripts being antagonistically expressed in wild-type or ΔUL135 infected cells. Finally, the authors proposed a list of 30 core differentially “low to moderate levels” expressed genes in HCMV latent samples (ΔUL135 or clinical latent samples). Unfortunately, no lncRNA were analyzed [80]. Shnyder and colleagues (BioProject PRJNA394123) used publicly available datasets and single-cell transcriptomics to define HCMV latency dynamics in infected cell populations. Interestingly, Shnyder et al. did not find any “clear restricted latency-associated expression program” [102] or set of genes, that could clearly explain the transitions from lytic-to-latent or latent-to-lytic during infection. Furthermore, transcription levels in latent cells resembled more those of very late infection, with low to medium transcription rates. This overall change in transcriptional rate, as cause of transition between the two states, apparently conflicts with Cheng et al. 30 latency-associated candidate genes list [80]. Further research is needed to understand the dynamics of latency in HCMV.
In 2017, Balázs and colleagues (BioProject PRJEB22072) reported the first HCMV transcriptome sequenced with long-read technology, the SMRT Bell™ Pacific Biosciences™ single molecule consensus platform. In this study, more than 291 novel transcript isoforms, 13 transcriptional starting sites (TSS), 22 transcriptional ending sites (TES) and 11 novel splicing events were characterized [27]. Most isoforms displayed unique combinations of ORFs, modifying the length of the transcript. Most of the length differences between isoforms were caused by an N-terminal truncation, losing an additional ORF upstream of the main ORF. Moreover, 8 novel antisense transcripts to canonical ORFs (UL20, UL36, UL38, UL54, UL115, US1, US17, and US30), and a new partially antisense transcript (RS2) in the short repeat region [27]. Balázs and colleagues also described transcript diversity in UL38 locus (i.e., hypothetical UL38A, longer form of UL38 with a putative non-canonical start codon), which has already been hypothesized to have a role in latency-to-replication transition in Cheng et al. [80]. Oxford Nanopore Technologies™ direct RNA-sequencing could provide extra evidence, as it can allow for (i) sequencing RNA directly (no retrotranscription or amplification) and (ii) keeping strand specificity. Additionally, (iii) it permits sequence transcripts with very different fragment sizes (as opposed to SMRT Bell™ Pacific Biosciences™ approach in Balázs et al. where analyzed transcripts range between 1 and 2 kbp).
Conclusions and future challenges
Almost 30 years have passed since the first full-length genome of HCMV was published [7], and the amount of knowledge gathered with different NGS experiments has been invaluable to detangle the nature of this ubiquitous virus. Even with all information that has been collected and technologies developed, some challenges are yet to be addressed: (i) the centralization and integration of information, and (ii) the production of improved assemblies, notably in complex clonally heterogeneous samples.
HCMV genome-, expression- and translation information is scattered in literature, but by (i) improving protein orthology, (ii) collecting and unifying clinical data, and (iii) creating a dynamic and collaborative annotation environment, the scattered available information may be reconstructed and contextualized, providing a valuable broad picture of HCMV. Different annotation nomenclatures have existed for the past years [7, 104] and recently, a new protein orthology has been published [8]. This new annotation, promoted by ViPR, is based on Domain-architecture Aware Inference of Orthologs (DAIO, Forester library) [105], and already available phylogenetic classifications, offering a manually high-quality curated database of Strict Orthology Groups (SOG). Orthology groups may help to identify and classify new Herpesviridae genes and to understand the functional differences between the different orthologs.
Over the years, the number of sequenced clinical isolates has greatly increased, albeit clinical metadata linked to the viral isolates (i.e., gender, age, patient cohort, ethology of the disease or isolation year) has not. Most of this information remains unavailable or heavily scattered in the bibliography. Some resources, as ViPR [8], provide centralized access to part of this metadata by automatically accessing GenBank records, although it remains incomplete as relies on non-standardized GenBank entries. An environment to deposit relevant clinical data with the corresponding viral information (i.e., isolate characteristics and genome) would provide high-quality information, helping to identify pathogenic determinants, as already has occurred for other viruses [106]. Currently, most of HCMV genome and transcriptome are annotated by automatic or semi-automatic tools, based on pre-existing references (custom databases or annotation transfer tools, such as RATT [107]). Unfortunately, not all annotations are updated with the current discoveries in HCMV expression and translation. A centralized and integrative RNA-seq platform would benefit the current state of annotation, as it would offer a constantly updated HCMV annotation contextualizing the available evidence from different experiments.
Finally, assemblies can be improved using different strategies, although connectivity, as previously discussed in this review, is one of its key aspects. Long-read technologies cannot only, connect scattered or unfinished regions of HCMV assemblies and characterize complete transcription events; but it can also provide a better understanding of structural and point variation in HCMV infections. Recently the term “ultra-long reads” (ULR), reads longer than 100 kbp, has been introduced [108]. Theoretically, ULR could (partially) cover any of the unique regions (UL or US) of HCMV, or in exceptionally cases, bridging both unique regions, as reads longer than 1 Mbp have already been reported [108]. Reads longer than 100 kbp may help to unambiguously connect distant variants from a clonally heterogeneous HCMV population.
References
Ribbert H (1904) Ueber protozoenartige Zellen in der Niere eines syphilitischen Neugeborenen und in der Parotis von Kindern. Zbl All Pathol 15:945–948
Jesionek AKB (1904) Ueber einen Befund von protozoenartigen Gebilden in den Organen eines hereditar-luetis- chen Foetus. Muenchner Med Wochenschr 51:1905–1907
Smith MG (1956) Propagation in tissue cultures of a cytopathogenic virus from human salivary gland virus (SGV) disease. Proc Soc Exp Biol Med 92:424–430. https://doi.org/10.3181/00379727-92-22498
Rowe WP, Hartley JW, Waterman S, Turner HC, Huebner RJ (1956) Cytopathogenic agents resembling human salivary gland virus recovered from tissue cultures of human adenoids. Proc Soc Exp Biol 92:418–424
Weller TH, MacAuley JC, Craig JM, Wirth P (1957) Isolation of intranuclear inclusion producing agents from infants with illnesses resembling cytomegalic inclusion disease. Exp Biol Med 94:4–12. https://doi.org/10.3181/00379727-94-22841
Nelson JA, Fleckenstein B, Jahn G et al (1984) Structure of the transforming region of human cytomegalovirus AD169. J Virol 49:109–115
Chee MS, Bankier AT, Beck S et al (1990) Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol 154:125–169. https://doi.org/10.1007/978-3-642-74980-3_6
Pickett BE, Sadat EL, Zhang Y et al (2012) ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 40:593–598. https://doi.org/10.1093/nar/gkr859
Murphy E, Yu D, Grimwood J et al (2003) Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci 100:14976–14981. https://doi.org/10.1073/pnas.2136652100
Dunn W, Chou C, Li H et al (2003) Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci 100:14223–14228. https://doi.org/10.1073/pnas.2334032100
Davison AJ, Akter P, Cunningham C et al (2003) Homology between the human cytomegalovirus RL11 gene family and human adenovirus E3 genes. J Gen Virol 84:657–663. https://doi.org/10.1099/vir.0.18856-0
Sinzger C, Hahn G, Digel M et al (2008) Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J Gen Virol 89:359–368. https://doi.org/10.1099/vir.0.83286-0
Bradley AJ, Lurain NS, Ghazal P et al (2009) High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J Gen Virol 90:2375–2380. https://doi.org/10.1099/vir.0.013250-0
Dolan A, Cunningham C, Hector RD et al (2004) Genetic content of wild-type human cytomegalovirus. J Gen Virol 85:1301–1312. https://doi.org/10.1099/vir.0.79888-0
Cunningham C, Gatherer D, Hilfrich B et al (2010) Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J Gen Virol 91:605–615. https://doi.org/10.1099/vir.0.015891-0
Dargan DJ, Douglas E, Cunningham C et al (2010) Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J Gen Virol 91:1535–1546. https://doi.org/10.1099/vir.0.018994-0
Jung GS, Kim YY, Kim JI et al (2011) Full genome sequencing and analysis of human cytomegalovirus strain JHC isolated from a Korean patient. Virus Res 156:113–120. https://doi.org/10.1016/j.virusres.2011.01.005
Renzette N, Bhattacharjee B, Jensen JD et al (2011) Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1001344
Sijmons S, Thys K, Corthout M et al (2014) A method enabling high-throughput sequencing of human cytomegalovirus complete genomes from clinical isolates. PLoS ONE 9:e95501. https://doi.org/10.1371/journal.pone.0095501
Murrell I, Wilkie GS, Davison AJ et al (2016) Genetic stability of bacterial artificial chromosome-derived human cytomegalovirus during culture in vitro. J Virol 90:3929–3943. https://doi.org/10.1128/JVI.02858-15
Tomasec P, Wang ECY, Davison AJ et al (2005) Downregulation of natural killer cell-activating ligang CD155 by human cytomegalovirus UL141. Nat Immunol 6:181–188. https://doi.org/10.1038/ni1156
Sijmons S, Thys K, Mbong Ngwese M et al (2015) High-throughput analysis of human cytomegalovirus genome diversity highlights the widespread occurrence of gene-disrupting mutations and pervasive recombination. J Virol 89:7673–7695. https://doi.org/10.1128/JVI.00578-15
Lassalle F, Depledge DP, Reeves MB et al (2016) Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol 2:vew017. https://doi.org/10.1093/ve/vew017
Khan KA, Coaquette A, Davrinche C, Herbein G (2009) Bcl-3-regulated transcription from major immediate-early promoter of human cytomegalovirus in monocyte-derived macrophages. J Immunol 182:7784–7794. https://doi.org/10.4049/jimmunol.0803800
Corcoran K, Sherrod CJ, Perkowski EF et al (2017) Genome sequences of diverse human cytomegalovirus strains with utility in drug screening and vaccine evaluation. Genome Announc 5:e01433–e01416. https://doi.org/10.1128/genomeA.01433-16
Hage E, Wilkie GS, Linnenweber-Held S et al (2017) Characterization of human cytomegalovirus genome diversity in immunocompromised hosts by whole-genome sequencing directly from clinical specimens. J Infect Dis 215:1673–1683. https://doi.org/10.1093/infdis/jix157
Balázs Z, Tombácz D, Szucs A et al (2017) Long-read sequencing of human cytomegalovirus transcriptome reveals RNA isoforms carrying distinct coding potentials. Sci Rep 7:1–9. https://doi.org/10.1038/s41598-017-16262-z
Balázs Z, Tombácz D, Sz A et al (2018) Data descriptor: long-read sequencing of the human cytomegalovirus transcriptome with the Paci fi c Biosciences RSII platform. Sci Data. https://doi.org/10.1038/s41598-017-16262-z
Cannon MJ, Schmid DS, Hyde TB (2010) Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol 20:202–213. https://doi.org/10.1002/rmv.655
Reeves M, Sinclair J (2008) Aspects of human cytomegalovirus latency and reactivation. In: Shenk TE, Stinski MF (eds) Human cytomegalovirus. Current topics in microbiology and immunology, vol 325. Springer, Berlin, pp 297–313
Schottstedt V, Blümel J, Burger R et al (2010) Human cytomegalovirus (HCMV)—revised. Transfus Med Hemother 37:365–375. https://doi.org/10.1159/000322141
Bale JF, Petheram SJ, Souza IE, Murph JR (1996) Cytomegalovirus reinfection in young children. J Pediatr 128:347–352. https://doi.org/10.1016/S0022-3476(96)70279-2
Boeckh M, Geballe AP (2011) Cytomegalovirus: pathogen, paradigm, and puzzle. J Clin Investig 121:1673–1680. https://doi.org/10.1172/JCI45449
Sharma V, Mobeen F, Prakash T (2016) Comparative genomics of herpesviridae family to look for potential signatures of human infecting strains. Int J Genom 2016:9543274. https://doi.org/10.1155/2016/9543274
Murphy E, Shenk TE (2008) Human cytomegalovirus genome. Curr Top Microbiol Immunol 325:1–19. https://doi.org/10.1007/978-3-540-77349-8_1
Roizmann B, Carmichael LE, Deinhardt F et al (1981) Herpesviridae: definition, provisional nomenclature, and taxonomy. Intervirology 16:201–217. https://doi.org/10.1159/000149269
Kilpatrick BA, Huang ES, Pagano JS (1976) Analysis of cytomegalovirus genomes with restriction endonucleases Hin D III and EcoR-1. J Virol 18:1095–1105
Weststrate MW, Geelen JLMC, van der Noordaa J (1980) Human cytomegalovirus DNA: Physical maps for the restriction endonucleases BglII, HindIII and XbaI. J Gen Virol 49:1–21. https://doi.org/10.1099/0022-1317-49-1-1
Weller TH (1991) Pathogenesis of human cytomegalovirus-associated diseases. Historical perspective. Transplant Proc 23:5–6 (discussion 6–7)
Huang ES, Kilpatrick BA, Huang YT, Pagano JS (1976) Detection of human cytomegalovirus and analysis of strain variation. Yale J Biol Med 49:29–43
Pritchett RF (1980) DNA nucleotide sequence heterogeneity between the Towne and AD169 strains of cytomegalovirus. J Virol 36:152–161
Cha T, Tom E, Kemble GW et al (1996) Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol 70:78–83
Prichard MN, Penfold MET, Duke GM et al (2001) A review of genetic differences between limited and extensively passaged human cytomegalovirus strains. Rev Med Virol 11:191–200. https://doi.org/10.1002/rmv.315
Hahn G, Rose D, Wagner M et al (2003) Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Townelongas BACs and site-directed mutagenesis using a PCR-based technique. Virology 307:164–177. https://doi.org/10.1016/S0042-6822(02)00061-2
Ankenbrand MJ, Hohlfeld S, Hackl T, Förster F (2017) AliTV—interactive visualization of whole genome comparisons. PeerJ Comput Sci 3:e116. https://doi.org/10.7717/peerj-cs.116
Harris RS (2007) Improved pairwise alignment of genomic DNA. Pennsylvania State University
McGeoch DJ, Cook S, Dolan A et al (1995) Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol 247:443–458. https://doi.org/10.1006/jmbi.1995.0152
Davison AJ, Bhella D (2007) Comparative betaherpes viral genome and virion structure. In: Arvin A, Campadelli-Fiume G, Mocarski E et al (eds) Human herpesviruses: biology, therapy, and immunoprophylaxis, first. Cambridge University Press, Cambridge, pp 177–203
Kilpatrick BA, Huang ES (1977) Human cytomegalovirus genome: partial denaturation map and organization of genome sequences. J Virol 24:261–276
Bar M, Shannon-Lowe C, Geballe AP (2001) Differentiation of human cytomegalovirus genotypes in immunocompromised patients on the basis of UL4 gene polymorphisms. J Infect Dis 183:218–225. https://doi.org/10.1086/317939
Rasmussen L, Geissler A, Winters M (2003) Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J Infect Dis 187:809–819. https://doi.org/10.1086/367900
Lurain N, Fox A, Lichy H et al (2006) Analysis of the human cytomegalovirus genomic region from UL146 through UL147A reveals sequence hypervariability, genotypic stability, and overlapping transcripts. Virol J 3:4. https://doi.org/10.1186/1743-422X-3-4
Sun ZR, Ji YH, Ruan Q et al (2006) Genetic variability of human cytomegalovirus UL132 gene in strains from infected infants. Microbiol Immunol 50:773–779
Deckers M, Hofmann J, Kreuzer KA et al (2009) High genotypic diversity and a novel variant of human cytomegalovirus revealed by combined UL33/UL55 genotyping with broad-range PCR. Virol J 6:1–12. https://doi.org/10.1186/1743-422X-6-210
Görzer I, Guelly C, Trajanoski S, Puchhammer-Stockl E (2010) Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J Virol 84:7195–7203. https://doi.org/10.1128/JVI.00475-10
Faure-Della Corte M, Samot J, Garrigue I et al (2010) Variability and recombination of clinical human cytomegalovirus strains from transplantation recipients. J Clin Virol 47:161–169. https://doi.org/10.1016/j.jcv.2009.11.023
Sahoo MK, Lefterova MI, Yamamoto F et al (2013) Detection of cytomegalovirus drug resistance mutations by next-generation sequencing. J Clin Microbiol 51:3700–3710. https://doi.org/10.1128/JCM.01605-13
Görzer I, Guelly C, Trajanoski S, Puchhammer-Stöckl E (2010) The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. J Virol Methods 169:248–252. https://doi.org/10.1016/j.jviromet.2010.07.040
Zipeto D, Hong C, Gerna G et al (1998) Geographic and demographic differences in the frequency of human cytomegalovirus gB genotypes 1–4 in immunocompromised patients. AIDS Res Hum Retrovir 14:533–536. https://doi.org/10.1089/aid.1998.14.533
Madi N, Al-Nakib W, Pacsa A, Saeed T (2011) Cytomegalovirus genotypes gB1 and gH1 Are the most predominant genotypes among renal transplant recipients in Kuwait. Transplant Proc 43:1634–1637. https://doi.org/10.1016/j.transproceed.2011.02.053
Coaquette A, Bourgeois A, Dirand C et al (2004) Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin Infect Dis 39:155–161. https://doi.org/10.1086/421496
Madi N, Al-Nakib W, Mustafa AS et al (2007) Detection and monitoring of cytomegalovirus infection in renal transplant patients by quantitative real-time PCR. Med Princ Pract 16:268–273. https://doi.org/10.1159/000102148
Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26:1135–1145. https://doi.org/10.1038/nbt1486
Shepp DH, Match ME, Lipson SM, Pergolizzi RG (1998) A fifth human cytomegalovirus glycoprotein B genotype. Res Virol 149:109–114. https://doi.org/10.1016/S0923-2516(98)80086-1
Haberland M, Meyer-Konig U, Hufert FT (1999) Variation within the glycoprotein B gene of human cytomegalovirus is due to homologous recombination. J Gen Virol 80(Pt 6):1495–1500
Houldcroft CJ, Bryant JM, Depledge DP et al (2016) Detection of low frequency multi-drug resistance and novel putative maribavir resistance in immunocompromised pediatric patients with cytomegalovirus. Front Microbiol 7:1–11. https://doi.org/10.3389/fmicb.2016.01317
Backert L, Kohlbacher O (2015) Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med 7:1–12. https://doi.org/10.1186/s13073-015-0245-0
Sanger F (2005) Frederick Sanger—Biographical. Nobelprize.org 1–4
Gatherer D, Seirafian S, Cunningham C et al (2011) High-resolution human cytomegalovirus transcriptome. Proc Natl Acad Sci 108:19755–19760. https://doi.org/10.1073/pnas.1115861108
Eckert SE, Chan JZ-M, Houniet D et al (2016) Enrichment by hybridisation of long DNA fragments for Nanopore sequencing. Microb Genom. https://doi.org/10.1099/mgen.0.000087
Karamitros T, van Wilgenburg B, Wills M et al (2018) Nanopore sequencing and full genome de novo assembly of human cytomegalovirus TB40/E reveals clonal diversity and structural variations. BMC Genom 19:577. https://doi.org/10.1186/s12864-018-4949-6
Sijmons S, Van Ranst M, Maes P (2014) Genomic and functional characteristics of human cytomegalovirus revealed by next-generation sequencing. Viruses 6:1049–1072
Wilkinson GWG, Davison AJ, Tomasec P et al (2015) Human cytomegalovirus: taking the strain. Med Microbiol Immunol 204:273–284. https://doi.org/10.1007/s00430-015-0411-4
Pollard MO, Gurdasani D, Mentzer AJ et al (2018) Long reads: their purpose and place. Hum Mol Genet 27:R234–R241. https://doi.org/10.1093/hmg/ddy177
Houldcroft CJ, Breuer J (2015) Tales from the crypt and coral reef: the successes and challenges of identifying new herpesviruses using metagenomics. Front Microbiol 6:1–6. https://doi.org/10.3389/fmicb.2015.00188
Marine R, McCarren C, Vorrasane V et al (2014) Caught in the middle with multiple displacement amplification: the myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2:1–8. https://doi.org/10.1186/2049-2618-2-3
Börgstrom E, Paterlini M, Mold JE et al (2017) Comparison of whole genome amplification techniques for human single cell exome sequencing. PLoS ONE 12:1–15. https://doi.org/10.1371/journal.pone.0171566
Roux S, Solonenko NE, Dang VT et al (2016) Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 4:e2777. https://doi.org/10.7717/peerj.2777
Houldcroft CJ, Beale MA, Breuer J (2017) Clinical and biological insights from viral genome sequencing. Nat Rev Microbiol 15:183–192. https://doi.org/10.1038/nrmicro.2016.182
Cheng S, Caviness K, Buehler J et al (2017) Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc Natl Acad Sci 114:E10586–E10595. https://doi.org/10.1073/pnas.1710522114
Aird D, Ross MG, Chen WS et al (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. https://doi.org/10.1186/gb-2011-12-2-r18
Balázs Z, Tombácz D, Szűcs A et al (2018) Dual platform long-read RNA-sequencing dataset of the human cytomegalovirus lytic transcriptome. Front Genet 9:432. https://doi.org/10.3389/FGENE.2018.00432
Ruan J (2015) SMARTdenovo: Ultra-fast de novo assembler using long noisy reads
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Stark TJ, Arnold JD, Spector DH, Yeo GW (2012) High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J Virol 86:226–235. https://doi.org/10.1128/JVI.05903-11
Meshesha MK et al. (2012) The microRNA transcriptome of human cytomegalovirus (HCMV). Open Virol J 6:38–48. https://doi.org/10.2174/1874357901206010038
Stern-Ginossar N, Weisburd B, Michalski A et al (2012) Decoding human cytomegalovirus. Science 338:1088–1093. https://doi.org/10.1126/science.1227919
Davison AJ, Dolan A, Akter P et al (2003) The human cytomegalovirus genome revisited: Comparison with the chimpanzee cytomegalovirus genome. J Gen Virol 84:17–28. https://doi.org/10.1099/vir.0.18606-0
Murphy E, Rigoutsos I, Shibuya T, Shenk TE (2003) Reevaluation of human cytomegalovirus coding potential. Proc Natl Acad Sci 100:13585–13590. https://doi.org/10.1073/pnas.1735466100
Vincent HA, Ziehr B, Moorman NJ (2016) Human cytomegalovirus strategies to maintain and promote mRNA translation. Viruses 8:1–16. https://doi.org/10.3390/v8040097
Finkel Y, Stern-Ginossar N, Schwartz M (2018) Viral short ORFs and their possible functions. Proteomics 18:1–8. https://doi.org/10.1002/pmic.201700255
Møller R, Schwarz TM, Noriega VM et al (2018) miRNA-mediated targeting of human cytomegalovirus reveals biological host and viral targets of IE2. Proc Natl Acad Sci USA 115:1069–1074. https://doi.org/10.1073/pnas.1719036115
Marques-Bonet T, Kidd JM, Ventura M et al (2009) A burst of segmental duplications in the genome of the African great ape ancestor. Nature 457:877–881. https://doi.org/10.1038/nature07744
Oberstein A, Shenk T (2017) Cellular responses to human cytomegalovirus infection: induction of a mesenchymal-to-epithelial transition (MET) phenotype. Proc Natl Acad Sci 114:E8244–E8253. https://doi.org/10.1073/pnas.1710799114
Weisblum Y, Oiknine-Djian E, Zakay-Rones Z et al (2017) APOBEC3A is upregulated by human cytomegalovirus (HCMV) in the maternal-fetal interface, acting as an innate anti-hcmv effector. J Virol 91:e01296–e01217. https://doi.org/10.1128/JVI.01296-17
Van Damme E, Thys K, Tuefferd M et al (2016) HCMV displays a unique transcriptome of immunomodulatory genes in primary monocyte-derived cell types. PLoS ONE 11:1–20. https://doi.org/10.1371/journal.pone.0164843
Batra R, Stark TJ, Clark E et al (2016) RNA-binding protein CPEB1 remodels host and viral RNA landscapes. Nat Struct Mol Biol 23:1101–1110. https://doi.org/10.1038/nsmb.3310
Buzdin AA, Artcibasova AV, Fedorova NF et al (2016) Early stage of cytomegalovirus infection suppresses host microRNA expression regulation in human fibroblasts. Cell Cycle 15:3378–3389. https://doi.org/10.1080/15384101.2016.1241928
Kim S, Seo D, Kim D et al (2015) Temporal landscape of MicroRNA-mediated host-virus crosstalk during productive human cytomegalovirus infection. Cell Host Microbe 17:838–851. https://doi.org/10.1016/j.chom.2015.05.014
Zhang Q, Lai MM, Lou YY et al (2016) Transcriptome altered by latent human cytomegalovirus infection on THP-1 cells using RNA-sEq. Gene 594:144–150. https://doi.org/10.1016/j.gene.2016.09.014
Ingolia NT, Brar GA, Stern-Ginossar N et al (2014) Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep 8:1365–1379. https://doi.org/10.1016/j.celrep.2014.07.045
Shnayder M, Nachshon A, Krishna B et al (2018) Defining the transcriptional landscape during cytomegalovirus latency with single-cell RNA sequencing. MBio 9:e00013–e18. https://doi.org/10.1128/mBio.00013-18
Grey F, Antoniewicz A, Allen E et al (2005) Identification and characterization of human cytomegalovirus-encoded microRNAs. J Virol 79:12095–12099. https://doi.org/10.1128/JVI.79.18.12095-12099.2005
Spaete RR, Gehrz RC, Landini MP (1994) Human cytomegalovirus structural proteins. J Gen Virol 75(Pt 12):3287–3308. https://doi.org/10.1099/0022-1317-75-12-3287
Zmasek C (2018) Forester: Software libraries for evolutionary biology and comparative genomics research. Unpublished. https://github.com/cmzmasek/forester/
Niebel M, Singer JB, Nickbakhsh S et al (2017) Hepatitis C and the absence of genomic data in low-income countries: a barrier on the road to elimination? Lancet Gastroenterol Hepatol 2:700–701. https://doi.org/10.1016/S2468-1253(17)30257-1
Otto TD, Dillon GP, Degrave WS, Berriman M (2011) RATT: rapid annotation transfer tool. Nucleic Acids Res 39:e57–e57. https://doi.org/10.1093/nar/gkq1268
Jain M, Koren S, Miga KH et al (2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36:338–345. https://doi.org/10.1038/nbt.4060
Acknowledgements
JMC is supported by a doctoral grant from HONOURs Marie-Sklodowska-Curie training network (721367). The authors would like to thank their colleagues from the Laboratory of Clinical Virology for their helpful comments and insightful discussions. In addition, the authors would like to thank all members of HONOURs Marie-Sklodowska-Curie training network program for training provided. The authors would like to acknowledge the three anonymous reviewers for their help in improving the quality of this review.
Funding
This study was funded by HONOURs Marie-Sklodowska-Curie training network (grant number 721367).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human and animal participants
This article does not contain any studies with human participants performed by any of the authors.
Additional information
Edited by Hartmut Hengel.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Martí-Carreras, J., Maes, P. Human cytomegalovirus genomics and transcriptomics through the lens of next-generation sequencing: revision and future challenges. Virus Genes 55, 138–164 (2019). https://doi.org/10.1007/s11262-018-1627-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-018-1627-3