
Abstract
The known strains of human cytomegalovirus (HCMV) represent genotypic variants of a single species, and HCMV genotypic variability has been studied in order to reveal correlations between different disease patterns and the presence of certain HCMV genotypes, either as single or as multiple infections. The methods used for the detection of HCMV genotypes have not always been sophisticated enough to achieve complete comprehensiveness, mainly because only one genotype is usually detected in a certain specimen, due to primer specificity and genome copy number. To improve detection of variant HCMV genotypes in mixed infections, we developed PCR assays with degenerate primers targeting two variable HCMV genes, glycoprotein B (gB, UL55) and the G-protein-coupled receptor gene UL33. Primers were designed to bind conserved sites in the genomes of HCMV variants and great ape CMVs. To analyse if samples contained one or more HCMV genotypic variants, PCR assays were supplemented with oligonucleotides containing locked nucleic acids. This broad-range PCR methodology and subsequent sequence analysis detected all gB/UL55 and UL33 genotypic variants known to date in primary clinical specimens, but also revealed that many samples contained genotype mixtures. Importantly, a novel UL33 genotypic variant could be discovered in several specimens, and one HCMV isolate was plaque-purified containing the novel UL33 genotype and a so far undescribed variant of gB.
Similar content being viewed by others


Introduction
The existence of genotypic variants of human cytomegalovirus (HCMV) was first reported by Chou & Dennison [1] by analysing glycoprotein B (gB) gene sequences spanning the gB cleavage site (CLS) of different HCMV isolates. Four distinct CLS genotypes (gBCLS-1 to -4) were reported. Later it was shown that gB exhibits variability also at the N-terminus (gBN-1 to -4) as well as at the C-terminus (gBC-1 to -2) [2]. Further studies provided evidence for homologous recombination between gBCLS, gBN and gBC genotypes [3]. A fifth gBCLS genotype was detected in several AIDS patients [4]. More recently, additional distinct gBN sequences were described in AIDS patients, but not assigned to a genotype numbering system [5, 6]. Several laboratories investigated HCMV genotype variability using PCR-based methods to unravel possible relationships between certain HCMV CLS genotypes and disease outcome, and although results have been conflicting at times, the occurrence of infections with more than one HCMV genotype and its impact on severity of HCMV disease has been considered crucial [reviewed in [7–14]].
For studies of these types, a comprehensive detection of all HCMV variants is highly desirable. In principle, this can be done with a set of independent and specific PCR assays, e.g., specific multiplex PCR, by restriction length polymorphism analysis, or by microarrays [1, 15–17]. However, sequences of unknown HCMV variants would remain undetected. This is a serious drawback, in particular because recent discoveries of non-human CMV in both chimpanzees and gorillas revealed a considerably higher diversity than in humans [18].
An unprejudiced way of analysing specimens for the presence of known and unknown herpesviruses is to perform PCR with degenerate primers targeting highly conserved sites within the herpesvirus genome. Such assays proved to be highly effective in the discovery of herpesviruses [reviewed in [19]]. However, in mixed herpesvirus infections, usually only one viral genome is preferentially amplified, due to genome copy number or preferential primer binding. Using oligonucleotides substituted with locked nucleic acids (LNA), we recently developed a variant of the universal herpesvirus PCR and applied it to the detection of unknown herpesviruses in Old World monkeys. Up to three different monkey herpesviruses could be amplified from single specimens, thereby revealing the presence of hitherto unknown herpesviruses [20].
The aim of the present study was to universally determine variable genotypes of two HCMV genes, gB (UL55) and UL33. The latter encodes a G-protein-coupled receptor (GPCR) and is present in human and animal CMVs. UL33 signals constitutively, interferes with cellular signaling networks and modulates cellular function after infection, thereby probably contributing to HCMV-associated pathology [[21, 22]; reviewed in [23]]. UL33 exhibits sequence variability but this information came from a limited number of strains [24]. Here we studied whether variable genotypes of HCMV gB and the UL33-encoded GPCR both can be universally and differentially identified in human clinical specimens by degenerate and LNA-substituted PCR.
Methods
Sample collection, cell culture, and DNA preparation
Twenty-eight blood samples pre-tested positive for HCMV with a pan-herpes PCR assay [25] were selected from a random collection of bone marrow transplanted, HCMV-seropositive children and from patients suffering from HIV infection or from different haematologic diseases. DNA was extracted by standard methods.
In addition, a collection of 89 HCMV patient isolates was analyzed after virus expansion on human MRC-5 fibroblasts (ATCC CCL-171, passages 3-15) upon HCMV isolation from various clinical specimens (throat washing, urine, EDTA blood, bronchoalveolar lavage fluid) which were simultaneously tested to be HCMV DNA-positive by routine PCR detecting HCMV polymerase. DNA was extracted from the pellets of infected cells after the second to sixth-passage. Plaque purification of HCMV isolates was performed using MRC-5 cells by limiting dilution of supernatants of infected cultures in log2 steps in a 96-well plate format. Wells containing a single plaque were harvested 7 days post infection followed by a second plaque purification using 48-well plates. HCMV plaques were then propagated in a 6-well plate the cells of which were used for DNA preparation by standard methods.
Consensus-PCR assays for the amplification of genotypic variants of glycoprotein B (UL55)
For the comprehensive amplification of the CLS of HCMV gB genes, degenerate and deoxyinosine-substituted (deg/dI) primers were deduced from the gB/UL55 gene of HCMV strain AD169 in a nested format (primer set CMV-gBp1; Table 1). Binding sites were selected on the basis of a multiple alignment of all human and primate CMV gB genes available in Genbank. Primers were degenerated and substituted with deoxyinosine at their 3'-end. PCR was performed with up to 12.3 μl of DNA, containing a maximum of 250 ng DNA, in first round amplification. PCR mix (12,7 μl; containing 1× AmpliTaq™ buffer, 2,0 mM MgCl2, 2-5% DMSO, the four dNTPs at 200 mM each, 2.0 units of AmpliTaq Gold Polymerase and 1 μM of each sense- and antisense primer) and - if necessary - water was added to achieve a total volume of 25 μl. For second round amplification, 1 μl of the first-round amplification product was added. Thermal cycling was performed in thermocyclers of type TgradientS (Biometra, Germany), under the following conditions: Initial denaturation 95°C for 2 min, 40 cycles with denaturation at 95°C for 30 sec, annealing at 46°C for 30 sec and elongation at 72°C for 1 min; final elongation at 72°C for 10 min. The annealing-to-elongation phase (ramp-time) was prolonged to 0,09°C/min. The PCR reactions were performed in duplicate.
A second nested deg/dI primer set was selected (primer set CMV-gBp2; Table 1) which amplifies two thirds of the CMV gB/UL55 gene comprising the N-terminal variable region and the CLS of gB. PCR was performed with the TaKaRa-Ex PCR system (Takara Bio Inc., Japan) using the following protocol: The reaction mixture was set up on ice and contained 50 μL of ExTaq buffer with MgCl2, 400 μM of each dNTP, 400 nM of each primer, 2.5 units of TaKaRa ExTaq polymerase (TaKaRa Bio Inc, Japan). Thirty cycles of amplification were performed, each cycle consisting of a denaturing step at 98°C for 20 sec, an annealing step at 45°C for 30 sec and an elongation step at 72°C. The duration of the elongation step in the first 15 cycles was 7 minutes and in the last 15 cycles 10 min +5 sec ramp time for each cycle. The final elongation step was performed for 30 minutes at 72°C. The PCR reactions were performed in duplicate.
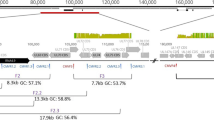
The primers of the second round of CMV-gBp1 and CMV-gBp2 amplified fragments of approximately 550 bp and 1590 bp, respectively. Binding regions of all gB primers are depicted in Figure 1.

Amplification of HCMV gB/ UL55 and UL33 using LNA-supplemented consensus PCR. Schematic diagram of the analysis strategy. Nested PCR is performed with deg/dI primers (black triangles). The primers CMV-gBp1 target the CLS of the gB gene, and the primers CMV-gBp2 target the gB gene from the N-terminus to the CLS (A). The complete UL33 gene is amplified by the CMV-UL33 primers (B). The binding regions of the LNA-oligonucleotides are present in the amplified sequences of both the first and the second PCR round, and represented by short thick lines (the position varies slightly for each LNA). The targeted HCMV genotypes are indicated.
Consensus-PCR assay for the amplification of genotypic variants of UL33
For the broad amplification of full-length HCMV UL33 genes (plus short, flanking regions of UL32 and UL34 sequences), nested deg/dI sense primers were deduced from the UL32 gene and antisense-primers from the UL34 gene of HCMV strain AD169 (primer set CMV-UL33; Table 1). Primer binding sites were selected on the basis of multiple alignments which comprised all UL32 and UL34 genes, respectively, of human and primate CMV available in Genbank. Primers were degenerated and substituted with deoxyinosine at their 3'-end. Primers used in the second round amplified fragments of approximately 1700 bp, and primer binding regions are depicted in Figure 1. PCR was performed with the TaKaRa-Ex PCR system as described above. The PCR reactions were performed in duplicate.
PCR for specific amplification of the UL33-5 gene
UL33-5 sequences were amplified with the primer set 4347-s (5'-CGTTCAACTTACAT AGTTTTGACG-3') and 4347-as (5'-GCGTCTGTAGCGCCACAAAAG-3') at 61°C annealing temperature. Amplified products had a size of 346 bp.
Design of LNA-oligonucleotides specific for gB/UL55 and UL33 genes
Four LNA-oligonucleotides (TIB MOLBIOL GmbH, Berlin, Germany) were designed to specifically inhibit the amplification of one of the HCMV gB genotypes 1 to 4. Their binding sites were located in the region of the CLS. The four HCMV UL33 genotypes were targeted by only three LNA-oligonucleotides (LNA-HCMV-UL33-1/2, -3 and -4) because for the closely related genotypes UL33-1 and -2, selection of discriminating LNA-oligonucleotides was not possible. Therefore, the common LNA-oligonucleotide UL33-1/2 was used (Figures 1 and 2; Table 2).

Binding sites of LNA-oligonucleotides in the gB/ UL55 and UL33 genes of HCMV. The binding sites of the LNA used in this study for specific inhibition of HCMV gB/ UL55 (A-D) and UL33 (E-G) genotype amplification are boxed and aligned with the corresponding regions of the other three gB genotypes. The LNA- bases and sequences identical to the LNA sequence are highlighted in yellow/green. On top of alignments A-D, the positions in the gB genes of gB genotype CLS-1 [M60929] (A), gB genotype CLS-2 (strain AD169)(B), gB genotype CLS-3 [M85228](C) and gB genotype CLS-4 [M60926] (D) are shown. LNA-gB-1 and LNA-gB-4 bind to the sense strand, LNA-gB-2 and LNA-gB-3 bind to the antisense strand. On top of the alignments E-G, the positions in the genes of UL33 genotype 1 (strain Merlin) (E), UL33 genotype 3 (strain Toledo)(F) and UL33 genotype 4 (strain AD169)(G) are shown. LNA-UL33-1/2 binds to the antisense strand, LNA-UL33-3 and LNA-UL33-4 to the sense strand.
Sequence analysis and phylogenetic tree construction
PCR product purification, direct sequencing with dye terminator chemistry, as well as nucleotide and amino acid sequence analysis were performed as described [26]. Sequencing reactions were performed on both strands with a total redundancy of at least four-fold. Sequence chromatograms were assembled with the Seqman module of the Lasergene software (GATC, Konstanz, Germany). BLAST searches were performed using the NCBI database. ORF prediction, calculation of identity values and multiple sequence alignments (ClustalW) were performed using MacVector software (Version 10.0). For phylogenetic tree construction, a nucleotide sequence alignment was analysed with the neighbor-joining method (MacVector). Alternatively, the Proml module of the PHYLIP program package on the Trex server http://www.trex.uqam.ca was used for Maximum-Likelihood analysis.
Numerical designation of genotypes and nucleotide sequence accession numbers
The gB sequences of published HCMV strains were assigned to gBCLS genotype numbers 1-4 according to the numbering in earlier studies [1, 4]: Merlin [GenBank acc.-no. NC_006273], gBCLS genotype 1; Towne [AY315197], gBCLS genotype 1; AD169 [NC_001347], gBCLS genotype 2; Toledo [AC146905], gBCLS genotype 3; and isolate C194A [M60926] gBCLS genotype 4. The complete gB UL55 sequence of the HCMV S3 isolate was determined here for comparative purposes [GU180092]. The acc.-no. for chimpanzee cytomegalovirus (CCMV) UL-33 shown in figure 3 is [NC_003521].

Phylogenetic analysis of UL33 genotypes. UL33 proteins (Figure 4) were aligned to construct a phylogenetic tree using the Neighbor-joining method. A rooted phylogram is shown, with the UL33 of the chimpanzee cytomegalovirus CCMV as outlier. The branch length is proportional to evolutionary distance (scale bar). Results of bootstrap analysis (1000 replicates) are indicated at the nodes of the tree.
We did not find a numbering system for UL33 genotypes in the literature, but we observed by phylogenetic tree construction that the published HCMV UL33 genes clearly branch in four clades (not shown). Therefore, we assigned the UL33 genotypes to numbers 1-4 for the purpose of this study (Figure 3). The novel UL33 genotype, presented here, was tentatively named UL33-5. The gB UL55 and UL33 sequences of the novel HCMV variant (HCMV-C5) were deposited in GenBank under the acc.-nos. [GU180093] and [GU180094], respectively.
Results
Universal screening of human blood for the presence of variant HCMV gB/UL55 genotypes
In order to test the ability of the CMV-gBp1 PCR to universally detect all known CLS genotypes of HCMV gB, a random collection of 28 HCMV-positive clinical blood specimens (S1-S28) was tested with the CMV-gBp1 PCR (without LNA addition). Amplimers were sequenced and the genotype determined. All known gBCLS genotypes (types 1 to 5) were detected. The genotype gBCLS-1 was detected in 13/28, gBCLS-2 in 2/28, gBCLS-3 in 10/28, gBCLS-4 in 2/28 and gBCLS-5 in 1/28 specimens (Table 3). The sequences differed only slightly from the published sequences or were identical (data not shown).
The existence of mixed infections with different HCMV variants was analysed by repeating the PCR in the presence of an LNA-oligonucleotide perfectly matching the gB genotype detected in the first experiment. Prior to use of these LNA-oligonucleotides, we confirmed that they specifically inhibited the amplification of a perfectly matching gB genotype in the CMV-gBp1 PCR (not shown). In additional pre-tests, we achieved selective amplification of a single CLS genotype from mixtures of strains, representing four CLS genotypes, using CMV-gBp1 PCR and up to three LNA oligonucleotides. These pre-tests were carried out in the same manner as reported by Prepens et al. [20], and are not described in detail.
In the presence of the genotype-specific LNA-oligonucleotides, an amplimer was obtained by CMV-gBp1 PCR from 9/28 HCMV-positive blood specimens. Sequence analysis revealed the presence of a second CLS genotype in each of the nine specimens. The PCR assay was repeated again, this time in the presence of two LNA-oligonucleotides in each reaction mixture. In one sample, a third CLS genotype was found. Thus, upon addition of the respective LNA, gBCLS-2 and -3 were detected in initially gBCLS -1-positive specimens, gBCLS-3 and/or gBCLS-5 in gBCLS-2-positive specimens, gBCLS-1 or gBCLS-4 in gBCLS-3-positive specimens and gBCLS-1 or gBCLS-3 in gBCLS-4-positive specimens (Table 3). In summary, LNA supplementation of the PCR enabled the differentiation of a total of 38 CLS sequences, ten more than with degenerate PCR alone.
Universal screening of human blood samples for the presence of HCMV UL33 genotypes
The same set of 28 primary specimens was tested for the presence of different UL33 genotypes with the CMV-UL33 primer set and - in subsequent experiments - in the presence of UL33-type-specific LNA-oligonucleotides. Single UL33 genotypes were detected in 23 samples, and 2 genotypes in five samples. All known UL33 genotypes known to date (types 1 to 4; Table 3) were detected. Without LNA addition, UL33-1, UL33-2, UL33-3 and UL33-4 were detected in 9, 11, 3 and 5 samples, respectively. The sequences differed only slightly from the published sequences or were identical (data not shown). Upon addition of the respective LNA, UL33-4 was detected in initially UL33-1-positive samples and UL33-1 in UL33-3-positive samples. Most importantly, a hitherto unknown HCMV UL33 genotype was discovered: from a primary blood specimen (S12), genotype 1 was initially amplified, but after addition of LNA-UL33-1 to the PCR reaction the novel UL33 sequence was detected (Table 3). It was tentatively named HCMV UL33 genotype 5 (UL33-5) (Table 3).
In pairwise sequence comparisons, UL33-5 was the most distant genotype, compared to the other four genotypes. It was most closely related to UL33-2 (88% at the nucleic acid level and 93% at the aa level). In phylogenetic analysis using the Neighbor-joining method, the UL33-5 genotype formed a branch with UL33-1 and -2 (Figure 3). In Maximum-Likelihood analysis, the same tree toplogy was obtained (not shown).
Since UL33 has been reported to be a spliced gene with two exons [21], we inspected the UL33-5 ORF for the presence of splice sites. In sample S12, a contiguous consensus sequence of 1715 bp spanning the 5'-end of the inversely oriented UL32, the complete UL33-5 ORF and the 5'-end of UL34 identified strong and conserved splice donor and acceptor signals (not shown). The spliced UL33-5 gene was determined to be 1242 bp in length (Exon 1: 27 bp; exon 2: 1212 bp) with a coding capacity of 413 amino acids. Based on a multiple amino acid alignment of UL33 proteins, putative structural receptor domains are shown in Figure 4.

Multiple alignment of HCMV UL33 genotypes. Amino acid sequences were deduced from the complete, spliced HCMV UL33-1 to UL33-5 ORFs. An alignment of the complete HCMV UL33 proteins was performed using the ClustalW module of MacVector version 10.6.0. Identical and similar amino acids are boxed and the latter additionally inversed. Mismatches are shown in lower case. The structural domains of the UL33 GPCR were determined as described in the Results section, and depicted in white (transmembrane domains), green (extracellular domains and loops) and red (intracellular domains and loops).
Isolation and plaque-purification of a virus containing the novel genotype UL33-5
Next, we wanted to isolate HCMV carrying the novel UL33-5 genotype. Since the clinical specimen S12 was not suitable for cell culture experiments, we collected eighty-nine additional specimens which had been tested positive for HCMV in routine diagnostics, and cultured them on MRC-5 cells. Early-passage cultures were analysed with the primer set 4347 which specifically detects UL33-5 but not UL33-1 to UL33-4. Fifteen samples (17%) were positive. Since these cultures were also mixtures of strains (data not shown), we re-tested five of them (C1-C5) with the degenerate primer set CMV-UL33 in order to identify a sample which predominantly contains a virus with the UL33-5 genotype. This was the case with culture C5. It was UL33-5-positive in both PCR assays and therefore contained a virus with the UL33-5 genotype as a majority in the strain mixture (Table 3). HCMV-C5 was then plaque-purified by limiting dilution. After two rounds of plaque purification, seven plaques were isolated and passaged once. PCR followed by sequence analysis revealed that all contained UL33-5. To study the gB gene of this virus in more detail, we used two degenerate primer sets, CMV-gBp1 and CMV-gBp2. The latter amplified two thirds (roughly 1.6 kb) of the gB gene including both the N-terminus and the CLS. Sequence analysis revealed a CLS sequence similar to that of AD169 (gBCLS-2). However, it differed from AD169 at the N-terminus, and was identical to a divergent gB sequence detected by Steininger et al. in an AIDS patient [5]. Thus, we concluded that HCMV-C5 contains a so far undescribed gBCLS/gBN combination. Re-inspection of the UL33-1/UL33-5-positive specimen S12 with the CMV-gBp2 PCR revealed the same gBCLS/gBN combination (although in the previous experiment with CMV-gBp1 PCR gBCLS-3 was detected; Table 3). This indicated for S12 that this gBCLS/gBN combination might be part of HCMV genomes which contain either the UL33-1 or the UL33-5 genotype. A comparison of the HCMV-C5 gB gene with those of published strains is shown in Figure 5.

Multiple alignment of HCMV gB/ UL55 variants. An alignment of HCMV gB amino acid sequences was performed as described in the legend to Figure 4. The alignment starts with Gly20 of the gB protein (the first 19 residues were not determined because their coding sequence is not part of the amplified gB fragment). The core region of the antigenic domain 2 [29] and the CLS are marked in green and red, respectively.
Discussion
Here we present the application of a broad-spectrum technique for the universal detection and differentiation of variable genotypes of the gB and UL33 genes of HCMV. All known HCMV gB and UL33 genotypes could be amplified with two sets (CMV-gBp1, CMV-UL33) of universal primate CMV primers (Table 1). Most importantly, a novel UL33 genotype (UL33-5) was discovered that differs significantly from the known genotypes (Figure 3, 4). UL33-5 could be identified in a total of 16 clinical HCMV isolates, one of which was plaque-purified (HCMV-C5).
Both the isolate HCMV-C5 and the UL33-5-positive specimen S12 contained the same gBCLS/gBN combination. While gBCLS was similar to that found in AD169, gBN was not (Figure 5). An identical gBN sequence was detected previously in an HCMV isolate of an AIDS patient [5], however, the CLS sequence of that isolate was not analysed. The gBCLS/gBN found in HCMV-C5 (and the UL33-5-positive specimen S12) are reported here for the first time. During the preparation of this manuscript, two HCMV isolates were deposited in GenBank with almost identical genotype sequences for UL33 and UL55 (GQ396662 and GQ221975, respectively). It is inferred that the novel UL33 and gB sequences may be not rare.
The discovery of this novel HCMV variant indicates that the primer sets are of a sufficiently broad detection range. This is underscored by our recent discoveries of novel CMV gB and UL33 sequences in chimpanzees and gorillas where these primer sets were employed (CMV-gBp1: [18]; CMV-gBp2 and CMV-UL33: B. Ehlers, unpublished data). We infer from these findings that the universal primers would also detect unknown, even more aberrant HCMV genotypes and variants.
The differentiation of HCMV genotypes from mixed infections was achieved by performing PCR in the presence of LNA-oligonucleotides. Previously, LNA-supplemented PCR was used for the discovery and differentiation of non-human primate herpesviruses in multi-infected samples [20]. It was adapted here to differentiate HCMV genotypes, and proved to be equally effective. By this approach, single samples revealed up to three genotypes. The most important finding was the discovery of the novel UL33-5 genotype with LNA-supplemented consensus PCR in a clinical sample (S12) in which only the UL33-1 genotype was detected with consensus PCR alone (Tab. 3).
In principle, known HCMV genotypes can be amplified with specific primers. However, with more than ten N-terminal and CLS genotypes already known, this could become very laborious, making LNA-supplemented PCR approaches with degenerate primers as those presented here feasible alternatives. With this method, every genotype should be detectable, including unknown ones. In addition, for genotypes preferentially present in mixed infections or for the comprehensive analysis of sample material from rare patients, the LNA-based differentiation of genotypes from single samples is the method of choice.
Of note, the gBCLS-5 genotype has been detected in five HIV-positive patients [4]. In accordance with this, we detected gBCLS-5 in two HIV-positive patients (S1, S3; Table 3). At the N-terminus, the gB sequence of S3 was identical to gBN sequences which were retrieved from two HIV-infected patients [5] (since these authors did not analyse the CLS, further comparisons could not be done). These findings warrant further investigation on a possible association of HCMV carrying the gBCLS-5 genotype with HIV.
For a long time, gB has been in the focus as a suitable target antigen that might be utilized for vaccination and thus prevention of maternal cytomegalovirus infection. In a phase 2 placebo controlled, randomized and double-blind trial, a gB-based vaccine was found to have a vaccination efficacy of 50% [27]. Although the key epitopes for neutralizing antibodies are supposed to be conserved between the presently known gB genotypes [28], the core region of the antigenic determinant 2 [29] is not entirely conserved (Figure 5). HCMV variants may exist that are even more diverse and only insufficiently neutralized by the antibodies elicited by the gB vaccine. The broad-spectrum PCR methodology presented here may prove useful in detecting these variants.
Whether the GPCR protein encoded by UL33 has a natural ligand or functions merely through constitutive, i.e., ligand-independent intracellular signalling is not known. All published functional tests on HCMV UL33 were undertaken with the UL33 GPCR encoded by the laboratory strain AD169 [30, 31]. When the five UL33 amino acid sequences were compared, intra-genotypic variation was most pronounced in certain sections. These correspond to the putative extracellular parts of the GPCR, whereas the intracellular parts show very low variability (Figure 4). Ligand interaction was attributed to the extracellular domain and extracellular loops (primarily loops 2 and 3) [32]. Since these domains revealed the highest variability in UL33, it may be useful to additionally examine UL33 genotypes other than AD169. This might help to identify a ligand for UL33.
Mixed infections with several herpesvirus genotypes are frequent, and especially immunocompromised patients are affected [8, 13–19, 33–35]]. In the latter situation, infections with multiple HCMV gBCLS genotypes correlate with an increased rate of graft rejection, higher viral loads, and frequency of infection with other herpesviruses [33, 34, 36, 37]. For therapeutic purposes, it is therefore important to early identify genotypes that bear a higher virulence potential. Relationships between certain HCMV glycoprotein genotypes and organ manifestations or outcome of HCMV infections have been looked into. However, establishing correlations has frequently been difficult because of limited testing sensitivity with the majority of methods using a defined, narrow detection spectrum. Only two groups of investigators used partially degenerate primers [36, 37]. Methods enabling the detection of multiple genotypes in a single sample were applied in two studies where approximately 25% to 29% of the samples were found to be multi-infected with HCMV strains of different gB genotypes [8, 38]. In accordance with this, more than one HCMV gB genotype was detected in 32% of the samples S1-S28 in the present study. Thus, an improved technique for universally detecting and differentiating known and unknown genotypic HCMV variants in clinical specimens would enhance understanding multiple HCMV infections, the reasons for their occurrence, and the pathogenicity they cause. In contrast to detection systems used in earlier studies [1, 8, 36–39], the methodology presented here is the first that meets this requirement. It allows to comprehensively detect and differentiate cytomegaloviruses and might help to identify a correlation between a given genotype or a certain mixture of genotypes and disease.
References
Chou SW, Dennison KM: Analysis of interstrain variation in cytomegalovirus glycoprotein B sequences encoding neutralization-related epitopes. J Infect Dis 1991, 163: 1229-1234.
Meyer-Konig U, Haberland M, von Laer D, Haller O, Hufert FT: Intragenic variability of human cytomegalovirus glycoprotein B in clinical strains. J Infect Dis 1998, 177: 1162-1169. 10.1086/515262
Haberland M, Meyer-Konig U, Hufert FT: Variation within the glycoprotein B gene of human cytomegalovirus is due to homologous recombination. J Gen Virol 1999, 80: 1495-1500.
Shepp DH, Match ME, Lipson SM, Pergolizzi RG: A fifth human cytomegalovirus glycoprotein B genotype. Res Virol 1998, 149: 109-114. 10.1016/S0923-2516(98)80086-1
Steininger C, Schmied B, Sarcletti M, Geit M, Puchhammer-Stockl E: Cytomegalovirus genotypes present in cerebrospinal fluid of HIV-infected patients. AIDS 2005, 19: 273-278.
Nye MB, Leman AR, Meyer ME, Menegus MA, Rothberg PG: Sequence diversity in the glycoprotein B gene complicates real-time PCR assays for detection and quantification of cytomegalovirus. J Clin Microbiol 2005, 43: 4968-4971. 10.1128/JCM.43.10.4968-4971.2005
Pignatelli S, Dal Monte P, Rossini G, Landini MP: Genetic polymorphisms among human cytomegalovirus (HCMV) wild-type strains. Rev Med Virol 2004, 14: 383-410. 10.1002/rmv.438
Coaquette A, Bourgeois A, Dirand C, Varin A, Chen W, Herbein G: Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin Infect Dis 2004, 39: 155-161. 10.1086/421496
Dal Monte P, Pignatelli S, Rossini G, Landini MP: Genomic variants among human cytomegalovirus (HCMV) clinical isolates: the glycoprotein n (gN) paradigm. Hum Immunol 2004, 65: 387-394. 10.1016/j.humimm.2004.02.023
Pignatelli S, Dal Monte P, Landini MP: gpUL73 (gN) genomic variants of human cytomegalovirus isolates are clustered into four distinct genotypes. J Gen Virol 2001, 82: 2777-2784.
Rasmussen L, Geissler A, Winters M: Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J Infect Dis 2003, 187: 809-819. 10.1086/367900
Bradley AJ, Kovács IJ, Gatherer D, Dargan DJ, Alkharsah KR, Chan PK, Carman WF, Dedicoat M, Emery VC, Geddes CC, Gerna G, Ben-Ismaeil B, Kaye S, McGregor A, Moss PA, Pusztai R, Rawlinson WD, Scott GM, Wilkinson GW, Schulz TF, Davison AJ: Genotypic analysis of two hypervariable human cytomegalovirus genes. J Med Virol 2008, 80: 1615-1623. 10.1002/jmv.21241
Arav-Boger R, Willoughby RE, Pass RF, Zong JC, Jang WJ, Alcendor D, Hayward GS: Polymorphisms of the cytomegalovirus (CMV)-encoded tumor necrosis factor-alpha and beta-chemokine receptors in congenital CMV disease. J Infect Dis 2002, 186: 1057-1064. 10.1086/344238
Pignatelli S, Dal Monte P: Epidemiology of human cytomegalovirus strains through comparison of methodological approaches to explore gN variants. New Microbiol 2009, 32: 1-10.
Tafreshi NK, Sadeghizadeh M, Amini-Bavil-Olyaee S, Ahadi AM, Jahanzad I, Roostaee MH: Development of a multiplex nested consensus PCR for detection and identification of major human herpesviruses in CNS infections. J Clin Virol 2005, 32: 318-324. 10.1016/j.jcv.2004.05.018
Chantaraarphonkun S, Bhattarakosol P: Intra- and intergenotypic variations among human cytomegalovirus gB genotypes. Intervirology 2007, 50: 78-84. 10.1159/000097393
Jääskeläinen AJ, Piiparinen H, Lappalainen M, Koskiniemi M, Vaheri A: Multiplex-PCR and oligonucleotide microarray for detection of eight different herpesviruses from clinical specimens. J Clin Virol 2006, 37: 83-90. 10.1016/j.jcv.2006.05.010
Leendertz FH, Deckers M, Schempp W, Lankester F, Boesch C, Mugisha L, Dolan A, Gatherer D, McGeoch DJ, Ehlers B: Novel cytomegaloviruses in free-ranging and captive great apes: phylogenetic evidence for bidirectional horizontal transmission. J Gen Virol 2009, 90: 2386-2394. 10.1099/vir.0.011866-0
Ehlers B: Discovery of herpesviruses. In Encyclopedia of Virology. 3rd edition. Edited by: Mahy B, Van Regenmortel M. Amsterdam: Elsevier; 2008:421-429.
Prepens S, Kreuzer KA, Leendertz F, Nitsche A, Ehlers B: Discovery of herpesviruses in multi-infected primates using locked nucleic acids (LNA) and a bigenic PCR approach. Virol J 2007, 4: 84. 10.1186/1743-422X-4-84
Davis-Poynter NJ, Lynch DM, Vally H, Shellam GR, Rawlinson WD, Barrell BG, Farrell HE: Identification and characterization of a G protein-coupled receptor homolog encoded by murine cytomegalovirus. J Virol 1997, 71: 1521-1529.
Beisser PS, Vink C, Van Dam JG, Grauls G, Vanherle SJ, Bruggeman CA: The R33 G protein-coupled receptor gene of rat cytomegalovirus plays an essential role in the pathogenesis of viral infection. J Virol 1998, 72: 2352-2363.
Vischer HF, Leurs R, Smit MJ: HCMV-encoded G-protein-coupled receptors as constitutively active modulators of cellular signaling networks. Trends Pharmacol Sci 2006, 27: 56-63. 10.1016/j.tips.2005.11.006
Dolan A, Cunningham C, Hector RD, Hassan-Walker AF, Lee L, Addison C, Dargan DJ, McGeoch DJ, Gatherer D, Emery VC, Griffiths PD, Sinzger C, McSharry BP, Wilkinson GW, Davison AJ: Genetic content of wild-type human cytomegalovirus. J Gen Virol 2004, 85: 1301-1312. 10.1099/vir.0.79888-0
Ehlers B, Borchers K, Grund C, Frölich K, Ludwig H, Buhk HJ: Detection of new DNA polymerase genes of known and potentially novel herpesviruses by PCR with degenerate and deoxyinosine-substituted primers. Virus Genes 1999, 18: 211-220. 10.1023/A:1008064118057
Goltz M, Ericsson T, Patience C, Huang CA, Noack S, Sachs DH, Ehlers B: Sequence analysis of the genome of porcine lymphotropic herpesvirus 1 and gene expression during post-transplant lymphoproliferative disease of pigs. Virology 2002, 294: 383-393. 10.1006/viro.2002.1390
Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang ML, Corey L, Hill J, Davis E, Flanigan C, Cloud G: Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 2009, 360: 1191-1199. 10.1056/NEJMoa0804749
Pass RF: Development and evidence for efficacy of CMV glycoprotein B vaccine with MF59 adjuvant. J Clin Virol 2009, 46: S73-S76. 10.1016/j.jcv.2009.07.002
Meyer H, Masuho Y, Mach M: The gpll6 of the gp58/116 complex of human cytomegalovirus represents the amino-terminal part of the precursor molecule and contains a neutralizing epitope. J Gen Virol 1992, 71: 2443-2450. 10.1099/0022-1317-71-10-2443
Casarosa P, Gruijthuijsen YK, Michel D: Constitutive signaling of the human cytomegalovirus-encoded receptor UL33 differs from that of its rat cytomegalovirus homolog R33 by promiscuous activation of G proteins of the Gq, Gi, and Gs classes. J Biol Chem 2003, 278: 50010-50023. 10.1074/jbc.M306530200
Waldhoer M, Kledal TN, Farrell H, Schwartz TW: Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J Virol 2002, 76: 8161-8168. 10.1128/JVI.76.16.8161-8168.2002
Mirzadegan T, Benko G, Filipek S, Palczewski K: Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry 2003, 42: 2759-2767. 10.1021/bi027224+
Hentrich M, Oruzio D, Jager G, Schlemmer M, Schleuning M, Schiel X, Hiddemann W, Kolb HJ: Impact of human herpesvirus-6 after haematopoietic stem cell transplantation. Brit J Haematol 2005, 128: 66-72. 10.1111/j.1365-2141.2004.05254.x
Sowmya P, Madhavan HN: Analysis of mixed infections by multiple genotypes of human cytomegalovirus in immunocompromised patients. J Med Virol 2009, 81: 861-869. 10.1002/jmv.21459
Puchhammer-Stöckl E, Görzer I, Zoufaly A, Jaksch P, Bauer CC, Klepetko W, Popow-Kraupp T: Emergence of multiple cytomegalovirus strains in blood and lung of lung transplant recipients. Transplantation 2006, 81: 187-194. 10.1097/01.tp.0000194858.50812.cb
Binder T, Siegert W, Kruse A, Oettle H, Wilborn F, Peng R, Timm H, Neuhaus P, Schmidt CA: Identification of human cytomegalovirus variants by analysis of single strand conformation polymorphism and DNA sequencing of the envelope glycoprotein B gene region-distribution frequency in liver transplant recipients. J Virol Methods 1997, 78: 153-162. 10.1016/S0166-0934(98)00173-6
Zheng SS, Zhou L, Qian J, Cai T, Fan J, Ma WH: Cytomegalovirus glycoprotein B sequence variation in Chinese liver transplant recipients. Hepatobiliary Pancreat Dis Int 2002, 1: 26-29.
Tarrago T, Quereda C, Tenorio A: Different Cytomegalovirus Glycoprotein B Genotype Distribution in Serum and Cerebrospinal Fluid Specimens Determined by a Novel Multiplex Nested PCR. J Clin Microbiol 2003, 41: 2872-2877. 10.1128/JCM.41.7.2872-2877.2003
VanDevanter DR, Warrener P, Bennett L, Schultz ER, Coulter S, Garber RL, Rose TM: Detection and Analysis of diverse herpesviral species by consensus primer PCR. J Clin Microbiol 1996, 34: 1666-1676.
Acknowledgements
The authors acknowledge the excellent technical assistance of Sonja Liebmann, Maria Thieme, Anja Voges and Nezlisah Yasmum.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SV and BE designed research, MD and HR performed experiments, JH, AE, KAK and HH collected, pretested, and provided samples, and SV and BE wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Deckers, M., Hofmann, J., Kreuzer, KA. et al. High genotypic diversity and a novel variant of human cytomegalovirus revealed by combined UL33/UL55 genotyping with broad-range PCR. Virol J 6, 210 (2009). https://doi.org/10.1186/1743-422X-6-210
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-6-210