
Abstract
Zirconia-supported vanadium–copper catalysts (VCux:yZr) were used for the oxidative depolymerization of softwood LignoBoost Kraft lignin (LB). Various VCux:yZr catalysts were prepared (x:y = 0:1, 1:4, 1:2, 3:4, 1:1, and 1:0) by incipient wetness impregnation, and reactions were performed in alkaline water at 150 °C under an O2 pressure of 5 bar for 10 min. 1H–13C HSQC NMR spectroscopy was used for product identification and quantification. The most promising catalyst was VCu1:2Zr, giving a total monomer yield of 9 wt% and the highest selectivity for vanillin (59%). This catalyst was characterized before and after use by N2 physisorption, XRD, TGA, SEM-EDS, and XPS. Cleavage of the main interunit linkages in LB, including the β-O-4 bonds and recalcitrant C–C bonds, was also observed. The findings of this study demonstrate the potential of the V–Cu/ZrO2 catalyst system in the production of value-added aromatics from technical lignin under relatively mild conditions. This would contribute to the more sustainable use of an underutilized side-stream in forest-based industries, provided catalyst reuse can be successfully demonstrated.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Lignin is a complex aromatic biopolymer comprising up to 30 wt% of lignocellulosic biomass, and has significant potential as a renewable feedstock for the production of chemicals, fuels, and functional materials [1, 2]. Lignin is also a major byproduct in the pulp and paper and cellulosic ethanol industries, where the bulk raw material is referred to as technical lignin [3, 4]. The highly heterogeneous nature of lignin makes its valorization challenging and, as a result, most lignin side-streams in industry are burned for energy recuperation. However, the efficient utilization of lignin would improve the economic viability and the sustainability profiles of the pulp and paper and the biofuel industries [5, 6]. Developing effective catalysts for lignin conversion is therefore important for better utilization of this aromatic-rich resource [7,8,9].
Given the highly heterogeneous structure of lignin and the high recalcitrance of technical lignin, much attention has been devoted to studies using lignin model compounds, synthesized to mimic the linkages found in real lignin [10, 11]. While these investigations can provide some insight into the chemistry regarding bond cleavage, and are useful for mechanistic studies and screening of reaction conditions, the results are often not representative of the actual material, and similar performance is rarely achieved when extrapolating to real lignin [12, 13]. For this reason, more effort should be devoted to the valorization of real technical lignin streams.
Environmentally benign routes for lignin depolymerization are important for the sustainable production of chemicals from lignin. Oxidative depolymerization of lignin is an energy-efficient means of lignin conversion, providing targeted functional chemicals, including high-value chemicals such as aromatic aldehydes, aromatic acids, and alkyl carboxylic acids [14, 15]. Such functional chemicals can be used in numerous applications, for example, plastics, adhesives, coatings, textiles, and pharmaceutical precursors [16, 17]. Metallic catalysts can also be used to reduce energy consumption by lowering the temperature and pressure, and shortening the reaction time, while providing higher selectivity and monomer yields [18]. Lignin oxidation for vanillin production has been generally limited to lignosulfonates, but the implementation of new catalyst systems could provide novel opportunities for the oxidation of other lignin raw materials [19, 20].
The use of homogeneous metal-based catalysts including metal ions, oxovanadium complexes, and salen complexes for lignin oxidation has been reported in the literature [21]. An advantage of homogeneous catalytic systems is that a variety of ligands that increase the activity and stability of the catalysts and facilitate the selective cleavage of specific lignin linkages can be used. However, the separation and reuse of homogeneous catalysts is challenging. There is thus a need for new cost-efficient catalysts to overcome these problems [19, 22]. The solution may be to engineer inexpensive heterogeneous metallic catalysts. However, the challenges of metal leaching, loss of surface area, poisoning of active sites, and coking on the catalyst surface must be overcome, while ensuring high catalytic activity and selectivity [23, 24].
Several heterogeneous metallic catalysts have been reported in the literature for oxidative lignin conversion, including supported noble metals such as Pd and Au, supported transition metals such as Cu and Mn, as well as non-supported mixed oxides such as perovskites containing Mn and Co [25,26,27,28,29]. Although promising results have been obtained, loss of catalytic activity upon the reuse of the catalyst, due to leaching of active metals, has been reported in the majority of these studies. Therefore, a major challenge in the field of lignin oxidation lies in finding an effective heterogeneous catalyst that is stable under the reaction conditions required.
In the present study, zirconia-supported vanadium–copper catalysts (VCux:yZr) were prepared and used for the oxidative conversion of Kraft lignin into vanillin and other high-value aromatic monomers. The active metals used in this study were chosen based on our previous study [30], in which a combination of Cu and V salts was used as a homogeneous catalyst in the oxidative conversion of Kraft lignin into aromatics. ZrO2 was used as the catalyst support due to its relatively high surface area and redox properties, as well as its mechanical, thermal, and alkaline stability. ZrO2 facilitates the dispersion of CuO, increases reducibility, and improves the adsorption of reactants [31]. ZrO2 has also been used as a support material in oxidative lignin depolymerization with Co as the active phase [32].
2 Experimental Methods
2.1 Chemicals and Materials
The following chemicals were used in the experiments; they were of reagent grade and were used without further purification, unless otherwise stated: vanadyl acetylacetonate (VO(acac)2, 95%, Sigma–Aldrich), copper(II) acetate monohydrate (Cu(OAc)2·H2O, ≥ 99%, Sigma–Aldrich), zirconium oxide (99.5%, Daiichi Kigenso Kagaku Kogyo Co., Ltd.), vanillin (99%, Sigma–Aldrich), vanillic acid (97%, Sigma–Aldrich), 4-hydroxybenzaldehyde (98%, Sigma–Aldrich), 4-hydroxybenzoic acid (99%, Fluka Chemicals), acetovanillone (≥ 98%, Sigma–Aldrich), hydrochloric acid (37% aqueous HCl solution, Fisher Scientific), sodium hydroxide (NaOH, 98.6%, VWR chemicals), dimethyl sulfoxide (DMSO-d6, 99.8%), ethyl acetate (EtOAc, HPLC grade, VWR chemicals), acetone (HPLC grade, VWR chemicals), sodium sulfate (Na2SO4, anhydrous for analysis, Merck KGaA), dioxygen (99.5%, Air Liquide Denmark). Softwood LignoBoost Kraft lignin (LB) was obtained as dry powder from Innventia’s LignoBoost demonstration plant in Bäckhammar, Sweden.
2.2 Catalyst Preparation
Catalysts were prepared by incipient wetness impregnation with a metal salt loading of 5 wt% on the support material ZrO2 at different molar ratios. The theoretical metal loadings of the catalysts were in the range of 1–1.6 wt%. A mixture of appropriate amounts of Cu(OAc)2·H2O and VO(acac)2 was first dissolved in a small volume of deionized water. ZrO2 powder was then added, and the mixture was stirred. It was important to maintain a suitable water-to-support ratio to obtain a paste with a consistency that would ensure the even distribution of the metals on the surface of the support. The paste was then dried at 110 °C overnight, and was subsequently calcined at 450 °C for 6 h. After calcination, the catalysts were fractionated to a size range of 24–18 mesh (0.71–1 mm). This range was chosen to facilitate the recovery of the catalyst from the reaction mixture.
2.3 Catalytic Oxidation
The catalytic oxidation reactions were performed in a 300 mL mechanically stirred autoclave equipped with a Parr 4848 reactor controller (Parr Instrument Company, Moline, IL, USA). In a typical experiment, 0.5 g of LB was dissolved in 50 mL of an aqueous 2 M NaOH solution. The solution was then loaded into the autoclave, together with 0.25 g of the solid catalyst (only catalyzed reactions), resulting in a catalyst-to-lignin ratio of 1:2 w/w. The reactor was purged (three times) and pressurized with molecular O2 to 5 bar, and heated to 150 °C (± 2 °C) under stirring at 500 rpm. The heating time was approximately 25–30 min. After the desired temperature had been reached, it was maintained for a reaction time of 10 min. The reaction time, temperature, and O2 pressure were chosen based on the optimized conditions reported in our previous study using a homogeneous V–Cu catalyst system [30]. After reaction, the autoclave was cooled in an ice bath for approximately 75 min until it reached room temperature, and was then depressurized after the final pressure had been noted. The product mixture was then transferred to a Büchner funnel using 4 mL H2O, where the spent catalyst was filtered off using a filter paper. The reaction mixture was stored in a freezer at -18 °C before further workup.
2.4 Workup Procedure
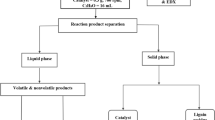
The product mixture (pH > 13) was thawed and acidified with aqueous HCl to pH ~ 2, to precipitate the residual lignin. The mixture was centrifuged at 3900 rpm for 20 min to separate the liquid phase from the solid fraction. The liquid phase was then transferred to a separation funnel and the solid fraction was stored in the freezer until further treatment. The liquid phase was extracted three times with EtOAc (2 × 10 mL and 1 × 30 mL). The volume of the aqueous fraction was measured before storing it at 5 °C until further analysis. The organic phase was dried over anhydrous Na2SO4, and after 15 min filtered with filter paper that was washed with 2 × 2.5 mL EtOAc. The EtOAc was removed by evaporation for 40 min in a rotary evaporator with a bump trap (IKA RV 10) at 40 °C and 210 mbar. The bump trap was then emptied, and evaporation continued at 40 °C and 200 mbar for approximately 4 h, until a dark-brown, viscous bio-oil was obtained. Figure 1 shows a block flow diagram of the workup procedure used to extract the bio-oil fraction from the reaction mixture.

Block flow diagram of the workup procedure used to extract the bio-oil fraction from the reaction mixture, following the catalytic oxidative depolymerization of LB. (EtOAc = ethyl acetate)
2.5 Product Analysis
Samples were prepared for NMR analysis by redissolving 50 mg of the bio-oil in 550 µL of DMSO-d6 and transferring the solution to 5 mm NMR tubes. The samples were quantitatively analyzed to determine the contents of the aromatic monomers vanillin, vanillic acid, acetovanillone, 4-hydroxybenzoic acid, and 4-hydroxybenzaldehyde. The monomer yield (wt%) relative to the initial lignin mass, and the monomer selectivity (%) relative to the total yield of these five monomers were determined. Three standard solutions were prepared, each containing gravimetrically determined amounts of monomers in DMSO-d6. All spectra were acquired at 25 °C using an 800 MHz Bruker Avance III instrument equipped with a TCI CryoProbe and a SampleJet sample changer. 1H–13C heteronuclear single-quantum coherence (HSQC) NMR spectra with 140 ppm spectral width in the 13C dimension were acquired as data matrices of 1024 (1H) × 256 (13C) complex data points to probe the linkage region and the chemical composition of the substrate. Reaction products were identified and quantified by the acquisition of 1H–13C HSQC NMR spectra with 60 ppm spectral width centered in the aromatic spectral region, sampling 2048 (1H) × 512 (13C) complex data points with 2 accumulations per increment, using an interscan recycle delay of 1.2 s and non-uniform sampling of 50% of the data points in the indirect dimension. All NMR spectra were processed with ample zero filling and baseline corrections in all dimensions using Bruker Topspin 3.5 pl 7 and were integrated in the same software.
2.6 Catalyst Characterization
N2 physisorption measurements were performed on an ASAP 2020 Micromeritics instrument using liquid nitrogen. Samples were degassed under vacuum at 90 °C for 1 h, and then at 200 °C for 4 h prior to measurements. The specific surface area, pore size, and pore volume of the various catalysts were determined using the Brunauer–Emmett–Teller and Barrett–Joyner–Halenda methods [33, 34].
Powder X-ray diffraction (XRD) patterns were obtained with a Huber G670 powder diffractometer in the 2θ range 3–100° in steps of 0.005°, using CuKα1 radiation (λ = 1.54056 Å) emitted from a focusing quartz monochromator, for 10 min. The data were collected in transmission mode from a rotating flat plate sample inclined 45° relative to the primary beam.
Thermogravimetric analysis (TGA) was performed in an atmospheric air environment using a Mettler Toledo TGA/DSC 1 Stare system. The heating rate was 10 °C/min from room temperature to 600 °C, which was then maintained for 1 h.
Scanning electron microscopy (SEM) was conducted on a high-resolution FEI Quanta FEG 250 ESEM microscope fitted with an Everhart–Thornley detector operated at 10–20 kV, with a spot size of 4, and 500–2000 times magnification. Energy-dispersive spectroscopy (EDS) was coupled with SEM for elemental mapping. SEM-EDS data were obtained when operating the equipment at 20 kV with a spot size of 3.5 and 4000 times magnification with the same microscope, coupled to an Oxford Instruments X-Mas 50 mm2 EDS analyzer.
X-ray photoelectron spectroscopy (XPS) was carried out with a Thermo Scientific system at room temperature using monochromatic AlKα radiation (1484.6 eV). The base pressure in the analysis chamber was maintained at 2 × 10− 7 mbar. The number of scans used was 5, 10, 15, and 15 for O, Zr, V, and Cu, respectively, for the fresh catalyst samples, and 5, 10, 20, 20, and 20 for O, Zr, V, Cu, and S, respectively, for the spent catalysts.
3 Results and Discussion
3.1 Catalyst Performance
All product mixtures (bio-oils) from the oxidative depolymerization reactions were subjected to 1H–13C HSQC NMR analysis to identify and quantify the aromatic monomers produced (Fig. 2). The five main aromatics identified in the oxidatively depolymerized samples were vanillin, vanillic acid, acetovanillone, 4-hydroxybenzoic acid, and 4-hydroxybenzaldehyde.

Monomer yield and vanillin selectivity in oxidative depolymerization of LB without an added catalyst (control), with the ZrO2 support, and catalyzed with fresh VCux:yZr catalysts, and with the reused VCu1:2Zr catalyst (VCu1:2Zr-RU) and the same catalyst reused twice (VCu1:2Zr-RU II).
The optimum molar ratio of V and Cu supported on ZrO2 was determined from screening loadings with pure Cu, V:Cu molar ratios of 1:4, 1:2, 3:4, 1:1, and pure V. The total monomer yields obtained from all the catalyzed reactions were 7.8–9.3 wt%, and were generally higher than the control reaction without an added catalyst. The lowest monomer yield from the catalyzed reactions was obtained with the VCu1:0Zr catalyst; this could be due to the formation of other product species that could not be detected.
The VCu1:2Zr catalyst afforded the highest selectivity for vanillin (59.1%), while the other catalysts had selectivities in the range of 50.6–53.9%. The vanillin selectivity was only 1.7 wt% higher than in the control reaction. However, in comparison with the control reaction, the VCu1:2Zr catalyst gave a considerably higher yield of 5.3 wt% vanillin, and a total monomer yield of 9.0 wt%, which was about 20% higher than in the control reaction. These results indicate that the VCu1:2Zr catalyst was favorable in terms of high vanillin selectivity and high monomer yield from LB. Table 1 presents the total monomer yield in this study, together with the results of other similar studies presented in the literature for comparison.
The maximum vanillin yield obtained from the oxidative depolymerization of LB reported previously by our research group using a homogeneous V–Cu catalyst system under similar reaction conditions was approximately 3.5 wt%, albeit with an initial LB concentration of 25 g/L [30]. The initial LB concentration used in the present study was lower (10 g/L), which may have influenced the yield. However, based on the encouraging results, the VCu1:2Zr catalyst was selected for the investigation of catalyst recovery and reuse in subsequent experiments.
The reusability of the VCu1:2Zr catalyst was investigated by using the spent catalyst in two subsequent oxidation reactions to observe the effect on the monomer yield and vanillin selectivity. As can be seen from Fig. 2, the vanillin yield was 4.3 wt% in both recycling experiments (VCu1:2Zr-RU and VCu1:2Zr-RU II), similar to the yield obtained in the control reaction. The total monomer yields obtained in the two recycling experiments were 7.7 wt% and 7.8 wt%, which is only 0.2–0.3 wt% higher than in the control reaction, and slightly lower than that with pure ZrO2. These results indicate that the catalyst might not be stable under the reaction conditions tested; this was further investigated and confirmed through catalyst characterization (see Sect. 3.2).
2D 1H–13C HSQC NMR was used to estimate the relative amounts of the interunit linkages present in the LB substrate and in the residual lignin that was recovered after oxidative depolymerization reactions catalyzed by the VCu1:0Zr, VCu1:2Zr, VCu0:1Zr catalysts, and the control reaction. The results for the fractions of the main linkages, β-O-4, β–5, and β–β, are given in Table 2. It should be emphasized that such determinations are semiquantitative, through comparison to aromatic signal as internal reference, assuming that virtually all aromatic rings have three protons and that the gamma signals from the linkages are integrated due to their favorable NMR relaxation behavior. The fact that two protons contribute to the linkage signal in the HSQC and three protons to the aromatic signals was accounted for.
As can be seen from Table 2, both the ether bonds and the more robust C–C bonds were cleaved in the oxidative depolymerization reactions. No significant differences were seen between the control and the catalyzed reactions, which may be due to the relatively low metal loading of the catalysts. However, the fact that the control reaction resulted in the cleavage of lignin interunit bonds indicates that the reaction conditions were favorable for oxidative depolymerization. Interestingly, the residual lignin obtained from the reaction catalyzed by the VCu1:2Zr catalyst showed the highest cleavage of all three interunit bonds, implying that this is a promising catalyst for the oxidative depolymerization of technical lignin.
The 2D 1H–13C HSQC NMR spectra from the bio-oils obtained in the reactions employing the VCu1:0Zr, VCu1:2Zr, and VCu0:1Zr catalysts are shown in Fig. 3, together with the results from the control reaction. Each spectrum is plotted together with the NMR spectrum obtained from the LB substrate.

Comparison of the 1H–13C HSQC NMR spectra obtained from bio-oils resulting from the reactions catalyzed by VCu1:0Zr, VCu1:2Zr, VCu0:1Zr, and the control reaction, compared with the NMR spectrum obtained from the LB substrate (gray). The signals in the yellow-highlighted boxes correspond to the signals from the β-O-4, β–5, and β–β linkages.
It can be seen that most of the signal from the β-O-4 linkages disappeared in the spectrum obtained from the bio-oil using the VCu1:2Zr catalyst, confirming that they were successfully cleaved during this reaction. Furthermore, the signals from the β–β linkages also decreased significantly, compared to the experiments with the other two catalysts. A smaller decrease in these two signals was observed in the control reaction, indicating that less cleavage of the β-O-4 and β–β bonds occurred in the absence of the metal catalyst. The signals from these bonds observed with the VCu1:0Zr catalyst were only slightly lower than those in the control reaction, which indicates that V alone was not very selective for the cleavage of these types of bonds in the lignin substrate. The VCu0:1Zr catalyst induced more cleavage than the VCu1:0Zr catalyst, however, the signals from both the β-O-4 linkages and β–β linkages were low. This further indicates that the combination of V and Cu rendered the catalyst more selective to linkage cleavage.
3.2 Catalyst Properties
The catalysts prepared were characterized using various techniques to obtain information on their textural and structural properties. The VCu1:2Zr catalyst was the main focus of characterization, as this afforded the highest yield of vanillin, and was used in the recycling experiments.
The specific surface area and pore volume determined by N2 physisorption, together with the calculated average pore size are given in Table 3 for all the catalysts prepared. The table also includes data for the spent VCu1:2Zr catalyst after one oxidation reaction with and without subsequent recalcination, and after being reused twice (Entries 5, 6, and 7, respectively).
Pristine zirconia (Entry 1) had a specific surface area of 85.8 m2/g, a pore volume of 0.20 cm3/g, and an average pore size of 90.4 Å (after suspension in deionized water and calcination). When the support was loaded with the V and Cu metal oxides, all of the three values increased (Entries 2–4 and 8–10), suggesting some agglomeration of the metal oxides with interparticle pores.
After the VCu1:2Zr catalyst had been used once, the reaction mixture removed, and the catalyst washed with water and acetone (Entry 5), all three textural properties were lower than those in the fresh catalyst (Entry 4), possibly due to heavier organic compounds blocking some of the catalyst pores. Moreover, the specific surface area of the catalyst was similar to that of the pristine ZrO2 after recalcination (Entry 6), suggesting that some of the carbon deposits on the surface were removed during calcination, and that metal oxide clusters on the surface were also lost. After the VCu1:2Zr catalyst had been reused twice, the specific surface area and pore volume barely changed, while the pore size decreased slightly (Entry 7), indicating that the active phase of the catalyst remained largely unmodified between the two consecutive reactions.
Powder XRD patterns were obtained for the pristine ZrO2 support and selected VCux:yZr catalysts, and are shown in Fig. 4, together with the reference diffraction pattern of monoclinic ZrO2 (Inorganic Crystal Structure Database [41]). The most representative peaks of monoclinic ZrO2 (reference), tetragonal ZrO2, monoclinic CuO, and orthorhombic V2O5 are also indicated [42,43,44].

XRD patterns of fresh VCux:yZr catalysts and the pristine ZrO2 support, together with the reference diffraction pattern of monoclinic ZrO2. The vertical dotted lines indicate the most prominent peaks of monoclinic ZrO2, tetragonal ZrO2, monoclinic CuO, and orthorhombic V2O5.
The most prominent peaks of the ZrO2 support (28.1° and 31.4°) corresponded well with the peaks observed for the monoclinic ZrO2, and these peaks were also observed in the diffraction patterns of the VCu0:1Zr, VCu1:2Zr, and VCu1:0Zr catalysts (Fig. 4), indicating that the monoclinic phase of ZrO2 was retained after impregnation with the metals. However, an additional small shoulder peak appeared in the diffraction pattern at the most prominent peak (30.2°) of the tetragonal phase of ZrO2 for all the metal-loaded catalysts. This suggests that metal impregnation induced a minor transformation of the support to the tetragonal crystalline phase of ZrO2.
The most prominent peaks in the diffraction pattern of monoclinic CuO (35.6° and 38.8°) are close to peaks observed for monoclinic ZrO2, thus making it difficult to distinguish them. However, the peak intensity was unchanged in the diffraction pattern of the VCu0:1Zr catalyst, which was richest in Cu (dark blue line), and in that of the VCu1:0Zr catalyst containing no Cu (light blue line). This observation suggests that Cu was not detected in the XRD pattern. Likewise, no peaks were observed in the diffraction patterns of the analyzed catalysts at the positions corresponding to V2O5 (20.3° and 26.2°), implying that the diffraction patterns obtained arose only from the ZrO2 support, likely due to the low V and Cu loadings, in combination with good metal oxide dispersion.
TGA was carried out on the fresh VCu1:2Zr catalyst and the catalyst after being reused twice (VCu1:2Zr-RU II) without preceding calcination (Fig. 5). The two profiles obtained were very different at temperatures below 120 °C, where the fresh catalyst lost more adsorbed water (~ 2 wt%) than the reused catalyst (~ 0.3 wt%). This was identified as being due to storage of the fresh catalyst for several months under ambient conditions before analysis.

TGA profiles of the VCu1:2Zr and VCu1:2Zr-RU II catalysts.
Upon heating to 600 °C, the fresh and reused catalysts showed a total weight loss of ∼1.7 and ∼2.2 wt%, respectively (disregarding the adsorbed water), implying that they were reasonably thermally stable, and thus suggesting that catalyst degradation was not responsible for the reduced catalytic effect of the VCu1:2Zr catalyst observed during reuse (Sect. 3.1). In addition, the reused catalyst had lost ∼0.5 wt% at a temperature of about 260 °C, likely corresponding to the release of organic compounds retained in the pores of the catalyst after reaction, which is consistent with our previous observations on catalytic lignin depolymerization under oxidative conditions [27]. Clogging of pores could reduce the activity of the catalyst, demonstrating the need for recalcination during catalyst regeneration.
SEM images were obtained for the fresh VCu1:2Zr and reused VCu1:2Zr-RU II catalysts to examine their morphological features (Fig. 6). When comparing the images of the catalysts on the 20 μm scale, it was clear that the fresh catalyst (Fig. 6a) had a smoother surface than the reused catalyst (Fig. 6b), which had an uneven layer with several protrusions on the support surface.

SEM images of the fresh VCu1:2Zr (a) and reused VCu1:2Zr-RU II (b) catalysts.
SEM-EDS elemental mapping of the catalysts (Fig. 7) showed that Cu and V were evenly distributed on the surface of the fresh catalyst (Fig. 7b and c), whereas the amounts of both metals were considerably decreased in the reused catalyst (Fig. 7e and f). This indicates that leaching of the active metals could have occurred during oxidative depolymerization.

SEM-EDS elemental mapping of the fresh VCu1:2Zr (a-c) and reused VCu1:2Zr-RU II (d-f) catalysts.
The relative amounts of metals and sulfur in the VCu1:2Zr and VCu1:2Zr-RU II catalysts determined by SEM-EDS (bulk) and XPS (surfaces) are given in Table 4. The results show that V and Cu had indeed been leached from the surface of the catalyst by use, which could explain the lower monomer yields obtained in the recycling experiments (Sect. 3.1). In addition, sulfur had accumulated on the catalyst surface after being reused twice, probably due to the presence of sulfur-containing functionalities in pristine LB. This could also cause deactivation of the catalyst.
Since the results of TGA analysis showed that the VCu1:2Zr catalyst was stable at temperatures up to 600 °C in air, the harsh alkaline reaction medium or the combination of elevated temperature and alkaline solution could have caused leaching of the active metals from the catalyst (e.g., the formation of soluble copper complex ions and vanadates). Further investigations are therefore required to elucidate the main cause of metal leaching and to devise strategies for catalyst stabilization.
4 Conclusions
The oxidative depolymerization of LB into high-value aromatic monomers over heterogeneous V–Cu catalysts with O2 as oxidant has been demonstrated with promising results. Vanillin, vanillic acid, acetovanillone, 4-hydroxybenzoic acid, and 4-hydroxybenzaldehyde were identified as the main monomers in the bio-oil fraction obtained. V and Cu had a combined effect on the cleavage of various lignin interunit linkages, and the VCu1:2Zr catalyst system exhibited an improvement in the total monomer yield of about 20% compared to the control reaction. Different V–Cu molar ratios were investigated, showing the superiority of VCu1:2Zr, which exhibited the highest selectivity for vanillin. However, the performance of the VCu1:2Zr catalyst decreased with reuse as a result of leaching of V and Cu species from the catalyst surface. Leaching was confirmed by SEM-EDS and XPS analysis and led to the conclusion that the catalyst system was not stable under the reaction conditions investigated. The accumulation of sulfur on the surface of the used catalyst was also observed. Thus, further optimization is required to identify the optimal operating conditions and to ensure the long-term stability of this catalyst system.
References
Schutyser W, Renders T, Van den Bosch S et al (2018) Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem Soc Rev 47:852–908. https://doi.org/10.1039/C7CS00566K
Zhu P, Abdelaziz OY, Hulteberg CP, Riisager A (2020) New synthetic approaches to biofuels from lignocellulosic biomass. Curr Opin Green Sustain Chem 21:16–21. https://doi.org/10.1016/j.cogsc.2019.08.005
Li T, Takkellapati S (2018) The current and emerging sources of technical lignins and their applications. Biofuels Bioprod Biorefining 12:756–787. https://doi.org/10.1002/bbb.1913
Abdelaziz OY, Hulteberg CP (2020) Lignin depolymerization under Continuous-Flow Conditions: highlights of recent developments. Chemsuschem 13:4382–4384. https://doi.org/10.1002/cssc.202001225
Ragauskas AJ, Beckham GT, Biddy MJ et al (2014) Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843. https://doi.org/10.1126/science.1246843
Abdelaziz OY, Al-Rabiah AA, El-Halwagi MM, Hulteberg CP (2020) Conceptual design of a Kraft Lignin Biorefinery for the production of Valuable Chemicals via oxidative depolymerization. ACS Sustain Chem Eng 8:8823–8829. https://doi.org/10.1021/acssuschemeng.0c02945
Sun Z, Fridrich B, De Santi A et al (2018) Bright side of Lignin depolymerization: toward new platform chemicals. Chem Rev 118:614–678. https://doi.org/10.1021/acs.chemrev.7b00588
Sudarsanam P, Duolikun T, Babu PS et al (2020) Recent developments in selective catalytic conversion of lignin into aromatics and their derivatives. Biomass Convers Biorefinery 10:873–883. https://doi.org/10.1007/s13399-019-00530-1
Gale M, Cai CM, Gilliard-Abdul-Aziz KL (2020) Heterogeneous Catalyst Design Principles for the Conversion of Lignin into High-Value Commodity. Fuels and Chemicals ChemSusChem 13:1947–1966. https://doi.org/10.1002/cssc.202000002
Guadix-Montero S, Sankar M (2018) Review on Catalytic cleavage of C–C inter-unit linkages in Lignin Model Compounds: towards Lignin Depolymerisation. Top Catal 61:183–198. https://doi.org/10.1007/s11244-018-0909-2
Xu J, Zhou P, Zhang C et al (2022) Striding the threshold of photocatalytic lignin-first biorefinery via a bottom-up approach: from model compounds to realistic lignin. Green Chem 24:5351–5378. https://doi.org/10.1039/d2gc01409b
Behling R, Valange S, Chatel G (2016) Heterogeneous catalytic oxidation for lignin valorization into valuable chemicals: what results? What limitations? What trends? Green Chem 18:1839–1854. https://doi.org/10.1039/c5gc03061g
Rinaldi R, Jastrzebski R, Clough MT et al (2016) Paving the way for Lignin Valorisation: recent advances in Bioengineering, Biorefining and Catalysis. Angew Chemie Int Ed 55:8164–8215. https://doi.org/10.1002/anie.201510351
Ren T, Qi W, Su R, He Z (2019) Promising techniques for depolymerization of Lignin into Value-added chemicals. ChemCatChem 11:639–654. https://doi.org/10.1002/cctc.201801428
Abdelaziz OY (2021) Lignin Conversion to Value-Added small-molecule chemicals: towards Integrated Forest Biorefineries. Lund University, Sweden. PhD Thesis
Hatti-Kaul R, Nilsson LJ, Zhang B et al (2020) Designing Biobased Recyclable Polymers for Plastics. Trends Biotechnol 38:50–67. https://doi.org/10.1016/j.tibtech.2019.04.011
Cywar RM, Rorrer NA, Hoyt CB et al (2022) Bio-based polymers with performance-advantaged properties. Nat Rev Mater 7:83–103. https://doi.org/10.1038/s41578-021-00363-3
Cabral Almada C, Kazachenko A, Fongarland P et al (2021) Supported-metal catalysts in upgrading lignin to Aromatics by oxidative depolymerization. Catalysts 11:467. https://doi.org/10.3390/catal11040467
Levec J, Pintar A (2007) Catalytic wet-air oxidation processes: a review. Catal Today 124:172–184. https://doi.org/10.1016/j.cattod.2007.03.035
Liu X, Bouxin FP, Fan J et al (2020) Recent advances in the Catalytic depolymerization of Lignin towards Phenolic Chemicals: a review. Chemsuschem 13:4296–4317. https://doi.org/10.1002/cssc.202001213
Ma R, Guo M, Zhang X (2018) Recent advances in oxidative valorization of lignin. Catal Today 302:50–60. https://doi.org/10.1039/9781788010351-00128
Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM (2010) The Catalytic valorization of lignin for the production of renewable chemicals. Chem Rev 110:3552–3599. https://doi.org/10.1021/cr900354u
Vangeel T, Schutyser W, Renders T, Sels BF (2018) Perspective on Lignin Oxidation: advances, Challenges, and future directions. Top Curr Chem 376:30. https://doi.org/10.1007/s41061-018-0207-2
Abdelaziz OY, Clemmensen I, Meier S et al (2022) On the oxidative valorization of Lignin to High-Value Chemicals: a critical review of Opportunities and Challenges. Chemsuschem 15:e202201232. https://doi.org/10.1002/cssc.202201232
Deng W, Zhang H, Wu X et al (2015) Oxidative conversion of lignin and lignin model compounds catalyzed by CeO2-supported pd nanoparticles. Green Chem 17:5009–5018. https://doi.org/10.1039/c5gc01473e
Song W-L, Dong Q, Hong L et al (2019) Activating molecular oxygen with Au/CeO2 for the conversion of lignin model compounds and organosolv lignin. RSC Adv 9:31070–31077. https://doi.org/10.1039/c9ra04838c
Abdelaziz OY, Meier S, Prothmann J et al (2019) Oxidative depolymerisation of Lignosulphonate Lignin into Low-Molecular-Weight Products with Cu–Mn/δ-Al2O3. Top Catal 62:639–648. https://doi.org/10.1007/s11244-019-01146-5
Deng H, Lin L, Sun Y et al (2008) Perovskite-type oxide LaMnO3: an efficient and recyclable heterogeneous Catalyst for the Wet Aerobic oxidation of lignin to aromatic aldehydes. Catal Lett 126:106–111. https://doi.org/10.1007/s10562-008-9588-0
Deng H, Lin L, Liu S (2010) Catalysis of Cu-Doped Co-Based Perovskite-Type Oxide in Wet Oxidation of Lignin to produce aromatic aldehydes. Energy Fuels 24:4797–4802. https://doi.org/10.1021/ef100768e
Walch F, Abdelaziz OY, Meier S et al (2021) Oxidative depolymerization of Kraft lignin to high-value aromatics using a homogeneous vanadium–copper catalyst. Catal Sci Technol 11:1843–1853. https://doi.org/10.1039/d0cy02158j
Basahel SN, Mokhtar M, Alsharaeh EH et al (2016) Physico-Chemical and Catalytic Properties of Mesoporous CuO-ZrO2 catalysts. Catalysts 6:57. https://doi.org/10.3390/catal6040057
Kumar A, Biswas B, Bhaskar T (2020) Effect of cobalt on titania, ceria and zirconia oxide supported catalysts on the oxidative depolymerization of prot and alkali lignin. Bioresour Technol 299:122589. https://doi.org/10.1016/j.biortech.2019.122589
Brunauer S, Emmett PH, Teller E (1938) Adsorption of gases in Multimolecular Layers. J Am Chem Soc 60:309–319. https://doi.org/10.1021/ja01269a023
Barrett EP, Joyner LG, Halenda PP (1951) The determination of pore volume and area distributions in porous substances. I. computations from Nitrogen Isotherms. J Am Chem Soc 73:373–380. https://doi.org/10.1021/ja01145a126
Pinto PCR, da Silva EAB, Rodrigues AE (2011) Insights into oxidative Conversion of Lignin to High-Added-value phenolic aldehydes. Ind Eng Chem Res 50:741–748. https://doi.org/10.1021/ie102132a
Pinto PCR, Costa CE, Rodrigues AE (2013) Oxidation of lignin from Eucalyptus globulus pulping liquors to produce syringaldehyde and vanillin. Ind Eng Chem Res 52:4421–4428. https://doi.org/10.1021/ie303349j
Patankar SC, Liu L-Y, Ji L et al (2019) Isolation of phenolic monomers from kraft lignin using a magnetically recyclable TEMPO nanocatalyst. Green Chem 21:785–791. https://doi.org/10.1039/c8gc03304h
Liu S, Das L, Blauch DN et al (2020) Statistical design of experiments for production and purification of vanillin and aminophenols from commercial lignin. Green Chem 22:3917–3926. https://doi.org/10.1039/d0gc01234c
Zirbes M, Quadri LL, Breiner M et al (2020) High-temperature electrolysis of Kraft Lignin for selective Vanillin formation. ACS Sustain Chem Eng 8:7300–7307. https://doi.org/10.1021/acssuschemeng.0c00162
Abdelaziz OY, Ravi K, Mittermeier F et al (2019) Oxidative depolymerization of Kraft Lignin for Microbial Conversion. ACS Sustain Chem Eng 7:11640–11652. https://doi.org/10.1021/acssuschemeng.9b01605
McCullough JD, Trueblood KN (1959) The Crystal structure of Baddeleyite (Monoclinic ZrO2). Acta Crystallogr 12:507–511. https://doi.org/10.1107/s0365110x59001530
Bondars B, Heidemane G, Grabis J et al (1995) Powder diffraction investigations of plasma sprayed zirconia. J Mater Sci 30:1621–1625. https://doi.org/10.1007/BF00375275
Åsbrink S, Norrby L-J (1970) A refinement of the Crystal structure of copper(II) oxide with a discussion of some exceptional E.s.d.’s. Acta Crystallogr Sect B Struct Crystallogr Cryst Chem 26:8–15. https://doi.org/10.1107/s0567740870001838
Haberkorn R, Bauer J, Kickelbick G (2014) Chemical Sodiation of V2O5 by Na2S. Z für Anorg und Allg Chemie 640:3197–3202. https://doi.org/10.1002/zaac.201400381
Acknowledgements
This work was supported by the Technical University of Denmark, Lund University, the Swedish Foundation for Strategic Environmental Research MISTRA (F2019/1822) within the framework of the research program STEPS – Sustainable Plastics and Transition Pathways at Lund University, the Swedish Energy Agency (P2021-00137), and the Research Council of Norway through the project L2BA – Lignin to BioAromatics (321427). We thank Dr. Leonhard Schill, Ping Zhu, and Bodil Fliis Holten for technical support with catalyst characterization. The NMR spectra were recorded using the 800 MHz spectrometer at the NMR Center DTU, supported by the Villum Foundation.
Funding
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdelaziz, O.Y., Clemmensen, I., Meier, S. et al. Oxidative Depolymerization of Kraft Lignin to Aromatics Over Bimetallic V–Cu/ZrO2 Catalysts. Top Catal 66, 1369–1380 (2023). https://doi.org/10.1007/s11244-023-01826-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-023-01826-3