
Abstract
Iron-based Fischer–Tropsch synthesis (FTS) catalysts evolve in situ on exposure to synthesis gas (CO & H2) forming a mixture of iron oxides, iron carbides and carbonaceous deposits. Recently, the application of inelastic neutron scattering has shown the progressive formation of a hydrocarbonaceous overlayer during this catalyst conditioning period. The evolving nature of the catalyst alters the proportion of phases present within the catalyst, which may influence the transport of hydrogen within the reaction system. Preliminary quasi-elastic neutron scattering (QENS) measurements are used to investigate hydrogen diffusion within an un-promoted iron FTS catalyst that has experienced varying levels of time-on-stream (0, 12 and 24 h) of ambient pressure CO hydrogenation at 623 K. Measurements on the catalyst samples in the absence of hydrogen show the unreacted sample (t = 0 h) to exhibit little increase in motion over the temperature range studied, whereas the t = 12 and 24 h samples exhibit a pronounced change in motion with temperature. The contrast is attributed to the presence of the afore-mentioned hydrocarbonaceous overlayer. Measurements on the samples in the presence of liquid hydrogen show hydrogen diffusional characteristics to be modified as a function of the catalyst conditioning process but, due to the complexity of the evolving catalyst matrix, the hydrogen motion cannot be attributed to a particular phase or component of the catalyst. Problems in the use of hydrogen as a probe molecule in this instance are briefly considered. Coincident neutron diffraction studies undertaken alongside the QENS measurements confirm the transition from hematite pre-catalyst to that of Hägg carbide during the course of extended times-on-stream.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Fischer–Tropsch synthesis (FTS) reaction is a well-established, industrially significant, catalytic reaction involving the conversion of synthesis gas (CO and H2) into a wide range of hydrocarbon products, which can be further processed to produce high value chemical intermediates utilised in the chemical manufacturing industry [1,2,3]. Co and Fe based catalysts dominate the industrial FTS scene; this work concentrates solely on iron-based FTS systems. Despite wide industrial application of iron FTS, there remains uncertainty in the mechanistic operation of these catalysts. This may be due in part to the ‘evolutionary’ phase in the initial stages of reaction that can result in a variety of molecular entities including iron oxides, iron carbides, metallic Fe, carbon, and a hydrocarbonaceous overlayer; with the final composition dependent on the reaction conditions [2, 4,5,6,7,8].
Previous work has utilised neutron spectroscopic techniques, in particular inelastic neutron scattering (INS) for the investigation of iron FTS systems, allowing for identification of hydrogenous moieties in post-reaction iron FTS catalysts [9, 10]. Alongside complimentary analytical techniques such as XRD, Raman, temperature-programmed oxidation (TPO) and TEM, INS has been used to investigate both the evolutionary profile of the active phase of the catalyst as well as the development of a hydrocarbonaceous overlayer [8, 11,12,13,14]. These investigations have utilised a promoter-free iron-based catalyst (α-Fe2O3) and have examined its evolutionary profile over a 24 h period using ambient pressure CO hydrogenation conditions as a representative FTS reaction [8]. Consequently, a qualitative model has been proposed indicating a role for the hydrocarbonaceous overlayer in the process chemistry, where the overlayer ‘templates’ access to two active sites: one primarily supporting dissociative hydrogen adsorption and the other supporting dissociative CO adsorption and, sequentially, product formation. A component of this model requires adsorbed hydrogen to diffuse through the overlayer, with the expectation that the rate of diffusion will change as the overlayer evolves [8]. More recently, the period of reaction under examination by INS has been extended from 24 h to 10 days (240 h) time-on-stream [15], with that work revealing further complexity within the catalyst conditioning period. Specifically, catalytic performance was observed to be decoupled from certain aspects of the evolving catalyst matrix: whereas CO hydrogenation activity had stabilised by 9 h time-on-stream, carbide formation and maturing of the hydrocarbonaecous overlayer, comprising sp3- and sp2-hybridized C-H components, exhibited different temporal profiles [15].
In order to test the hypothesis of moderated hydrogen diffusion throughout the iron based FTS catalyst conditioning period, here we report a preliminary investigation employing the technique of quasi-elastic neutron scattering (QENS) to examine the diffusion characteristics of an iron based FTS catalyst that has experienced different times-on-stream. Hydrogen was selected as a probe molecule because it is directly linked with the process chemistry under consideration. In this role, the hydrogen molecule is viewed as existing as a physisorbed entity that can access porosity within the various phases of the catalyst matrix, viz. iron oxides, iron carbides, amorphous carbon and hydrocarbonaceous overlayer [16].
Within the context of this application of QENS, it is informative to consider how neutrons interact with matter. Due to their lack of charge, neutrons generally interact only weakly with matter. When they do interact, they may either be scattered or absorbed. If we assume absorption to be negligible in our system, we can limit our interpretation to four types of scattering. Neutrons may be scattered elastically or inelastically, and coherently or incoherently. Whilst the theoretical basis for the separation of these components is well understood, we can only measure the neutrons that are scattered and infer from our knowledge of the system the most likely source [17]. Coherent elastic scattering is the most common and familiar event, where there is no change in the energy of the neutron, which interacts as a wave interfering with the extended lattice and leads to diffraction. Coherent inelastic scattering occurs where a neutron scatters from a correlated lattice motion (phonon). Incoherent inelastic scattering provides information about uncorrelated atomic motion. At higher energies this may represent molecular vibrations, but around the elastic peak this represents the stochastic diffusion processes occurring within a sample. Incoherent elastic scattering also occurs, but does not carry useful information.
The probability of a neutron scattering from an atom is given by its scattering cross section. This can be split into coherent and incoherent cross sections, which determine the relative strengths of these two modes of scattering and is isotope dependent. 1H has such a uniquely high incoherent cross that in hydrogenous samples all incoherent scattering is generally treated as scattering from hydrogen. In a system with a solid phase and a hydrogen-containing liquid the incoherent inelastic scattering will represent the stochastic motions of the liquid, whilst the coherent elastic scattering represents the structural order of the solid. This simplification allows us to neglect the effects of correlated motions in the solid, and structure in the liquid, although we should remember that this is an approximation that becomes decreasingly accurate with as the percentage of hydrogen in our sample reduces.
In this study, QENS has been used for the first time to investigate the diffusion of the hydrogen reactant in FTS catalysts that have been exposed to CO hydrogenation conditions at 623 K for 0, 12 and 24 h. The three samples have previously been analysed by INS by Warringham et al. [8]. Specifically, this preliminary QENS study seeks to investigate whether this technique may provide any insight towards how the evolving hydrocarbonaceous overlayer affects reactant diffusion.
2 Experimental
2.1 Catalyst Preparation
The iron oxide catalyst sample used for this investigation was prepared using the co-precipitation of iron nitrate (Sigma Aldrich, 99.99%) and sodium carbonate (Sigma-Aldrich, 99.99%). The preparative procedure utilises a batch reactor apparatus for reproducible sample synthesis and is described elsewhere [14]. The resulting slurry is filtered and washed with warm deionised water followed by a calcination step involving a step wise heating programme, with a maximum temperature of 623 K [14]. The procedure produces hematite (α-Fe2O3) and an absence of promoters/modifiers. The surface area of the samples examined in this work was 70.8 m2 g−1. All samples were ground and sieved to a particle size range of 250–500 µm. Catalyst characterisation has been carried out in previous investigations [8, 14].
2.2 Quasielastic Neutron Measurements
In previous studies, approximately 10 g of iron oxide catalyst was exposed to CO hydrogenation conditions (heated to 623 K at 5 K min−1 under a flow of H2, 150 sccm, CK Gas, 99.9%, and CO, 75 sccm, CK Gas, 99.9%, in a carrier gas He, 600 sccm, CK Gas, 99.9%, total WHSV of 1.47 h−1) in a custom built sample preparation rig [8, 9]. Each sample was exposed to reaction conditions for varying lengths of time on stream: 0, 12 and 24 h; labelled as FT0, FT12 and FT24 respectively. Following exposure to reaction conditions, each of the reacted samples were subjected to a passivation procedure involving the introduction of small amounts of O2 to the reactor, gradually increasing to atmospheric levels (i.e. 20% O2 in the gas feed) [18]. Each sample was sealed into cylindrical aluminium sample cans that had an annular sample space of 2 mm and allowed connection to gas handling facilities. The three samples were first dried in a vacuum furnace at 363 K overnight. They were then mounted onto a gas handling sample stick to allow control of the gas atmosphere in the sample space whilst the sample was cooled in a closed cycle refrigerator (CCR) in the neutron beam. For the hydrogen-containing samples the gas was admitted to the cell and an attached gas buffer of 500 cm3 to approximately 1 bar at 300 K and sealed. The pressure drop upon cooling was significant due to adsorption/condensation of the hydrogen gas. Data was collected using the IRIS spectrometer at ISIS using the 002 reflection of the pyrolitic graphite analyser bank [19, 20]. This gives an energy transfer range of − 0.5 to 0.5 meV with an energy resolution of 17.5 µeV. IRIS has 51 spectroscopy detectors viewing the graphite analyser that cover a 2θ range of 27.07–158.4 degrees. In the PG002 configuration, this corresponds to 0.442–1.854 Å−1. It is common to group these detectors to improve the signal to noise ratio at the expense of Q resolution. The choice of groups was optimised for quasielastic signal, with detectors coincident with strong Bragg scattering peaks from the reacted catalysts omitted from the analysis.
3 Results and Discussion
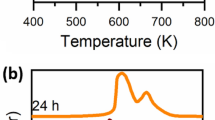
An initial assessment of the interaction between hydrogen and the sample utilised elastic window scans under varying temperature [17]. The total integrated elastic peak intensity was obtained by integration between − 0.0175 and + 0.0175 meV energy transfer (the instrumental resolution) for the FT24 sample with and without hydrogen. Results are shown in Fig. 1. At temperatures of 100 K and above the samples with and without hydrogen show little difference, indicating that hydrogen is only present in the sample space as a rarefied gas, which scatters too weakly to be detected. Below 100 K, however a condensed phase forms on the catalyst, presumably within pores present in the sample indicated by additional scattering from the hydrogen atoms. As the liquid range of hydrogen is 14–20 K at one atmosphere the difference in the elastic peak intensities in the range 20–100 K suggests that there is a favourable interaction between the catalyst and the hydrogen leading to an absorptive process. Any information that may be obtained about the nature of this sorbed phase and its interaction with the catalyst could lead to increased understanding of the catalytic reaction mechanism.

Elastic peak intensity as a function of temperature for FT24 without (a, grey) and with (b, red) an overpressure of hydrogen. Decreasing elastic intensity indicates increasing molecular motion
Figure 1 therefore allows the temperature region of interest to be defined (20–100 K); longer data accumulation times were then used for all subsequent measurements to allow line-shape analysis. The data summed across Q for all three samples (FT0, FT12 and FT24) and conditions (30, 50, 70 K with and without H2) is presented in Fig. 2.

Variation in quasielastic peak maxima summed across Q for catalyst samples before and during hydrogen exposure: a FT0, b FT12, c FT24, d FT0 + H2, e FT12 + H2, f FT24 + H2
Figure 2 presents the peak maxima for the six samples considered. For the samples without hydrogen dosing (Fig. 2a–c), the differing intensity between catalyst samples is attributed to differing sample masses. Figure 2a shows the unreacted sample (FT0) to exhibit little increase in motion over the temperature range studied, as would be expected for a rigid, coherently-scattering structure. However, for the FT12 and FT24 samples (Figs. 2b and c), there is a more pronounced change in motion with temperature, indicating that there is increasing incoherent scattering with temperature from hydrogen contained within these samples. Moreover, as previous characterisation of the FT12 and FT24 samples has shown the only hydrogeneous entities present are encompassed with the hydrocarbonaceous overlayer [8], the motion observed in Fig. 2b and c is uniquely assigned to the presence of the hydrocarbonaceous overlayer [8].
The addition of hydrogen was performed in situ and the increased intensity of the quasielastic peak reflects the increased number of scatters in the neutron beam. This is visible in the increased intensity for the lower temperatures (Fig. 2d–f). The increase in quasielastic peak intensity on hydrogen adsorption across all samples is remarkably similar in magnitude, suggesting that the sorption capacity of the α-Fe2O3 pre-catalyst does not significantly change during the transformation to the active carbided form. It should be remembered however that the quality of the data recorded was limited by the weak interaction with the sample. At lower temperatures the dynamics were too slow to be observed. At higher temperatures, equilibrium shifted towards the gas phase, resulting in decreased signal, preventing determination of the activation energy of diffusion.
3.1 Quasielastic Fitting
Fitting of the data was carried out using the DAVE software package [21] with a two component fit. The experimentally measured resolution function was convoluted with a delta function to represent the elastic scattering, and a single Lorentzian was used to represent the quasielastic broadening. Many fitting models were attempted, with the most consistent results obtained using un-subtracted data. The elastic component in this model represents both the coherent and incoherent elastic scattering, which is the sum of the static and dynamic structure factors, and reduces the number of variables in our interpretation. Attempts to fit the subtracted data, where the static structure from the immobile phase has been removed, resulted in negative peaks at higher Q values.
Even with this restricted model, it was not possible to achieve reliable fitting for the H-dosed sample at 30 K or for the evacuated samples. An example fit for a subset of the hydrogen loaded 50 K data is shown in Fig. 3. Various diffusion models were considered, with little to recommend one over another. For illustration purposes, the simple Fickian model is selected for discussion. In this case the width at half-maximum of the Lorentzian component, FWHM = DsQ2, where Ds is the self-diffusion coefficient. Figure 4 shows plots of the FWHM as a function of Q2 along with linear fits to the data for the hydrogen dosed samples analysed at 50 and 70 K. The error bars shown correspond to the standard error obtained through least squares data fitting. The calculated diffusion coefficients are presented in Table 1 and are comparable to previous reports of hydrogen diffusing on carbon [22]. Little difference is apparent between 50 and 70 K for FT0 in Fig. 4a, and the rate is similar at 50 K for all three catalysts. The 70 K data for the FT12 and FT24 catalysts (Fig. 4b and c) show an increase in diffusion rate that is more pronounced with increasing reaction time. The significant result is that the difference in gradient increases with time on stream, with the FT24 sample showing a pronounced difference. Importantly, Fig. 4 and Table 1 show that hydrogen diffusion within the overlayer is physically feasible and that the more lengthy the catalyst conditioning period, the greater the diffusion rate, whether or not this diffusion follows the Fickian model. These results provide additional evidence for a role of hydrogen diffusion within the evolving catalyst matrix. However, whereas the experimental constraints associated with Fig. 2a–c (absence of probe molecule) permitted deductions to be made concerning an exclusive contribution from the hydrocarbonaceous overlayer, such distinction in not possible when examining the mobility of the liquid hydrogen data. The hydrogen is present in various components of the catalyst matrix, e.g. iron carbide, amorphous carbon or hydrocarbonaceous overlayer, with the hydrogen in each environment having different dynamics. Diffusion for adsorbed hydrogen was observed over the molecular length scales accessed by the QENS measurement (< 2.7 nm), suggesting a bulk-like liquid in contact with the solid below 70 K. Though not directly comparable with the FTS reaction conditions, the differences observed suggest that the changing nature of the evolving catalyst is apparent using cryogenic hydrogen as a surface probe.

Example fits for FT24 loaded with hydrogen at 50 K and Q = 0.48 (a), 0.96 (b), 1.27 (c) and 1.77 Å− 1 (d); limited Q values shown for clarity to illustrate broadening of Lorentzian with increasing Q

FWHM variation with Q2; linear fit corresponding to Fickian long-range diffusion of hydrogen dosed onto a FT0, b FT12 and c FT24 catalysts at 50 (grey) and 70 K (red)
The FTS model catalyst is able to sorb hydrogen throughout its life cycle at cryogenic temperatures, either through chemisorption or encapsulation. As the condensed phase only forms within a limited temperature range, complete characterisation of the hydrogen interaction with QENS remains challenging. Use of a sorption probe with a wider liquid range is recommended for future studies. The use of methane as a probe molecule is proposed as an alternative to hydrogen [16].
3.2 Diffraction
The IRIS spectrometer has the additional capability to collect time of flight neutron diffraction data, across a limited Q range in parallel with normal spectroscopic measurement. Diffraction data collected from the three evacuated samples at base temperature is shown in Fig. 5. All patterns show weak peaks from the Al sample holder corresponding to d-spacings of 3.37 and 3.65 Å. The FT0 sample displays a single reflection at 3.68 from the hematite (012) plane. The FT12 and FT24 catalysts show (111) reflections at d-spacings of 3.16 and 3.32 Å. This is consistent with the transformation of the hematite FT0 pre-catalyst into Hägg carbide in samples FT12 and FT24 [8]. Phases were assigned using neutron diffraction patterns computed by the PowderCell software [23] from literature CIF files [24, 25]. No significant differences in diffraction were seen due to temperature or hydrogen loading.

Neutron diffraction data for FT0 (a, yellow), FT12 (b, green) and FT24 (c, orange). Peaks labelled ‘hem’ are assigned to hematite; ‘car’ refers to the Hägg carbide. Vertical lines indicate the position of background from the aluminium sample holder
4 Conclusions
With reference to an iron based un-promoted FTS catalyst, samples that have previously experienced 12 and 24 h time on stream in ambient pressure CO hydrogenation at 623 K were investigated for hydrogen diffusion by QENS. The following conclusions can be drawn.
-
An elastic peak intensity scan as a function of temperature for the 24 h sample shows hydrogen incorporation into the catalyst surface structure over the range 20–100 K (Fig. 1).
-
The structure of the unreacted catalyst is rigid over the temperature range 30–70 K but catalysts possessing a hydrocarbonaceous overlayer show increased incoherent scattering (Fig. 2b, c).
-
The analysis of samples loaded with hydrogen at 50 and 70 K suggests diffusion within a condensed hydrogen phase. That the diffusion rate qualitatively varies with catalyst time-on-stream is consistent with the hydrogen associating with the evolving catalyst matrix.
-
QENS has the potential to probe diffusion issues connected with iron-based FTS catalysts however, better correspondence between molecular interactions, superior signal:noise ratio spectra, and spectrometer temporal range is desirable.
Data Availability
The neutron scattering data is available at https://doi.org/10.5286/ISIS.E.87841524 [26]. Other datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
de Smit E, Weckhuysen BM (2008) The renaissance of iron-based Fischer-Tropsch synthesis: on the multifaceted catalyst deactivation behaviour. Chem Soc Rev 37:2758–2781
Van de Loosdrecht J, Botes FG, Ciobica IM, Ferreira A, Gibson P, Moodley DJ, Saib AM, Visage JL, Weststrate CJ, Niemantsverdriet JW (2013) Fischer-Tropsch synthesis: catalysts and chemistry. In: Reedijk J, Poeppelmeier KR (eds) Comprehensive inorganic chemistry II. Eslvier, Oxford, pp 525–557
Steynberg AP (2004) Studies in surface science and catalysis. In: Steynberg AP, Dry M (eds) Fischer-Tropsch technology, vol 152. Elsevier, Amsterdam, pp 1–63
Niemantsverdriet JW, Van der Kraan AM, Van Dijk WL, Van der Baan HS (1980) Behaviour of metallic iron catalysts during Fischer-Tropsch synthesis studied with Mössbauer spectroscopy, X-ray diffraction, carbon content determination, and reaction kinetic measurements. J Phys Chem 84:3363–3370
Dry ME (1981) The Fischer-Tropsch Synthesis Catalysis Science and Technology. Springer, New York, pp 160–255
de Smit E, Cinquini F, Beale AM, Safonova OV, van Beek W, Sautet P, Weckhuysen BM (2010) Stability and reactivity of ε-χ-θ Iron carbide catalyst phases in Fischer-Tropsch synthesis: controlling μc. J Am Chem Soc 132:14928–14941
Ning W, Koizumi N, Chang H, Mochizuki T, Itoh T, Yamada M (2006) Phase Transformation of unpromoted and promoted Fe catalysts and the formation of carbonaceous compounds during Fischer-Tropsch synthesis reaction. App Catal A 312:35–44
Warringham R, Davidson AL, Webb PB, Tooze RP, Ewings RA, Parker SF, Lennon D (2019) Examining the temporal behaviour of the hydrocarbonaceous overlayer on an iron based Fischer-Tropsch catalyst. RSC Adv 9:2608–2617
Warringham R, Bellaire D, Parker SF, Taylor J, Ewings RA, Goodway CM, Kibble M, Wakefield SR, Jura M, Dudman MP, Tooze RP, Webb PB, Lennon D (2014) Sample environment issues relevant to the acquisition of inelastic neutron scattering measurements of heterogeneous catalyst samples. J Phys Conf Ser 554:012005
Parker SF, Lennon D, Albers PW (2011) Vibrational spectroscopy with neutrons: a review of new directions. Appl Spectrosc 65:1325–1341
Hamilton NG, Silverwood IP, Warringham R, Kapitán J, Hecht L, Webb PB, Tooze RP, Parker SF, Lennon D (2013) Vibrational analysis of an industrial Fe-based Fischer-Tropsch Catalyst employing inelastic neutron scattering. Angew Chem Int Ed 52:5608–5611
Hamilton NG, Warringham R, Silverwood IP, Kapitán J, Hecht L, Webb PB, Tooze RP, Zhou W, Frost CD, Parker SF, Lennon D (2014) The application of inelatsic neutron scattering to investigate CO hydrogenation over an iron Fischer-Tropsch synthesis catalyst. J Catal 312:221–231
Warringham R, Hamilton NG, Silverwood IP, How C, Webb PB, Tooze RP, Zhou W, Frost CD, Parker SF, Lennon D (2015) The application of inelastic neutron scattering to investigate a hydrogen pre-treatment stage of an iron Fischer-Tropsch catalyst. Appl Catal A 489:209–217
Warringham R, McFarlane AR, Webb PB, Tooze RP, Taylor J, MacLaren DA, Ewings R, Parker SF, Lennon D (2015) The application of inelastic neutron scattering to explore the significance of a magnetic transition in an iron based Fischer-Tropsch catalyst that is active for the hydrogenation of CO. J Chem Phys 143:174703
Davidson AL, Webb PB, Parker SF, Lennon D (2020) Hydrogen partitioning as a function of time-on-stream for an unpromoted iron-based Fischer-Tropsch synthesis catalyst applied to CO hydrogenation. Ind Eng Chem Res 59:52–60
Silverwood IP, Agote Arán M, Lezcano González I, Kroner A, Beale AM (2018) QENS study of methane diffusion in Mo/H-ZSM-5 used for the methane dehydroaromatisation reaction. AIP Conf Proc 1969:030002
Embs JP, Juryani F, Hempelmann R (2010) Introduction to quasielastic neutron scattering. Z Phys Chem 224:5–32
Shroff MD, Dayte AK (1996) The importance of passivation in the study of iron Fischer-Tropsch catalysts. Catal Lett 37:101–106
IRIS Technical Information, https://www.isis.stfc.ac.uk/Pages/Iris-technical-information.aspx. Accessed Sep 2019
IRIS Publications, https://www.isis.stfc.ac.uk/Pages/Iris-publications.aspx. Accessed Sep 2019
Azuah RT, Kneller LR, Qiu Y, Tregenna-Piggott PLW, Brown CM, Copley JRD, Dimeo RM (2009) DAVE: a comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data. J Res Nat Inst Stan Technol 114:341
Bahn E, Czakkel O, NagyB LK, Villar-Rodil S, Tascón JMD, Demmel F, Telling MTF, Fouquet P (2016) Diffusion of molecular hydrogen in carbon aerogel. Carbon 98:572–581
Federal Institute for Materials and Research and Testing, PowederCell 2.3 https://www.ccp14.ac.uk/ccp/web-mirrors/powdcell/a_v/v_1/powder/e_cell.html. Accessed Sep 2019
Fruchart D, Chaudouet P, Fruchart R, Rouault A, Senateur JP (1984) Structural studies of cementite type compounds—effect of hydrogen on Fe3C followed by neutron diffraction-Mössbauer spectrometry of Fe57 doped FeCo2B and Co3B. J Solid State Chem 51:246–252
Maslen EN, Streltsov VA, Streltsova NR, Ishizawa N (1994) Synchrotron X-ray study of the electron density in α-Fe2O3. Acta Cryst B 50:435–441
Silverwood I, Davidson A, Parker S, Lennon D (2017) Hydrogen diffusion in the reactive carbon overlayer on a Fischer-Tropsch catalyst. STFC ISIS Neutron and Muon Source. https://doi.org/10.5286/ISIS.E.87841524
Acknowledgements
Sasol Ltd., (PGRS-3) the University of Glasgow and the Engineering and Physical Sciences Research Council (EP/P505534/1) are thanked for the provision of postgraduate studentship (ALD). The STFC Rutherford Appleton Laboratory is thanked for access to neutron beam facilities (RB1720188) [26]. The Royal Society are thanked for the provision of an Industrial Fellowship (PBW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
Research Involving Human Participants and/or Animals
There were no human or animal subjects involved in this research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davidson, A.L., Webb, P.B., Silverwood, I.P. et al. The Application of Quasi-Elastic Neutron Scattering to Investigate Hydrogen Diffusion in an Iron-Based Fischer–Tropsch Synthesis Catalyst. Top Catal 63, 378–385 (2020). https://doi.org/10.1007/s11244-020-01259-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-020-01259-2