
Abstract
A series of potassium-promoted spinels (Mn, Fe, Co) were prepared with various K+ promoter locations: on the surface (surface promotion) or in the bulk (formation of new layered and tunneled nanostructures via solid state reaction). All prepared samples were characterized by means of X-ray diffraction, Raman spectroscopy, X-ray fluorescence and N2-BET specific surface area analysis. Catalytic activity in soot combustion in different reaction conditions was investigated (tight contact, loose contact, loose contact with NO addition). It was shown that in all cases the nanostructuration is more effective than the surface promotion, with the layered structures of KCo4O8, KMn4O8 being the most catalytically active phases, lowering the soot combustion down to 250 °C. The difference in activity between tight and loose contacts can be bridged in the presence of NO due to its transformation into NO2, which acts as the oxygen carrier from the catalyst surface into soot particles, eliminating the soot-catalyst contact difference.
Similar content being viewed by others

1 Introduction
During pyrolysis and the incomplete combustion of fuels (such as wood, coal, lignite or different fractions of petroleum) agglomerated soot particles of different carbon forms are produced. They are one of the main components of particulate matter (PM). Long exposition of the human organism to PM leads to many illnesses like cardiovascular and lung diseases, heart attacks or arrhythmia. The nanometric size of the soot particles allows for their high penetration into the human body causing DNA code modifications that result in mutations and possible cancerogenic changes. Moreover, PM accumulation leads to atherosclerosis, antenatal complications, and respiratory system diseases. Deposition of PM, especially soot, on snow and ice, lowers its ability to reflect sunlight. The ice or snow, absorbing solar energy more readily, melts and in further effect has an impact on global warming [1].
The most important anthropogenic sources are coal combustion in electric plants and home heating systems, waste incineration, and car emissions—soot from fuel combustion and dust from tires wear. Since the main part of anthropogenic particulate matter is soot, it is an important task to decrease its emissions from diesel engines [1]. Many governments have introduced strict norms for pollutant emission [2]. These have led to the development of new technologies for soot emission reduction. It seems that catalytic diesel particulate filters (CDPF) are the most promising technology. DPFs can lower soot emission by up to 99 % but soot accumulates inside them and regeneration is needed. Catalysts can regenerate DPFs in the temperature of exhaust gasses while a temperature of up to 600 °C is needed for regeneration without any catalyst [3, 4].
A lot of different groups of catalysts were investigated for soot combustion—noble metal-based, perovskites, spinels, ceria catalysts, chlorides, low melting point catalysts. The catalytically active phases were recently reviewed in [3, 5–8].
The alkali-containing systems form an interesting group of soot combustion catalysts. Alkali metals (Li, Na, K, Rb, Cs) can be used as surface or bulk dopants in the metal oxide matrix. Surface promotion of CeO2 by a series of alkali metals showed that the catalytic activity depends both on the dopant nature and its concentration [9, 10]. In [11] and [12] structural and surface alkali promotion of SrTiO3 was compared. The results showed that all dopants increased the activity with potassium promotion being the most effective. It was also shown that structural promotion was more effective than the surface one. Details concerning alkali promotion can be found in [13]. Systematic investigations of perovskite KxLa1-xFeO3, spinels A2xCu1-xFe2O4 and A2xCo1-xFe2O4 (where A is an alkali metal) led to the conclusion that the most effective promoter is potassium and the activity depends on the promoter concentration. More comprehensive studies on the surface alkali promotion of d- and f-electron metal oxides such as cobalt, lanthanum, iron, chromium, manganese, niobium, vanadium, and cerium are presented in [14]. Examples of the use of mixed oxides (potassium bulk promotion) as catalysts include K6Ti4O11, K2Ti2O5, K2Ti4O9 [15], KNbO3 [16], KFeO2 and K2Fe22O34 [17] or KVO3 and K4V2O7 [18]. Potassium containing glasses [19, 20] have also been found recently as a component of the most active soot combustion catalysts [21].
Since nitrogen oxides are present in diesel exhaust, their influence on catalytic activity in the soot oxidation process has been investigated. It was found that NO has a positive effect on catalyst activity in soot combustion as it forms NO2, which is a stronger oxidant for carbon particles than the oxygen [22]. A systematic shift of the temperature window of soot combustion on potassium-promoted spinels in the presence of NO was also reported [23]. It was found that potassium-containing catalysts can be used for the simultaneous removal of NOx and soot [24–26], so similar catalytic systems were also investigated in bench-scale experiments [26–28].
The aim of the study is to show how the doping of transition metal (Mn, Fe, Co) oxides by alkali facilitates the soot combustion process, by shifting the combustion temperature window below the target 400 °C. This issue was addressed by systematic studies of catalytic activity in the soot combustion of a series of surface and structural potassium-promoted transition metal spinels in tight, loose contacts and various gas environments (with and without NO).
2 Experimental
2.1 Catalyst Preparation
The surface doped catalysts were prepared by the wet impregnation of 1 g of the iron (Aldrich) and cobalt spinels (Fluka) with a potassium carbonate solution, to achieve the optimal alkali loading corresponding to the theoretical 0.5 monolayer, as described in [23]. The impregnated spinel was dried at 100 °C for 1 h and subsequently calcined at 400 °C for 4 h. These samples were further designated as K/Fe3O4 and K/Co3O4, respectively.
The samples of K2Fe22O34 ferrite were prepared by the solid-state reaction of a stoichiometric mixture of K2CO3 (POCh) with α-Fe2O3 (Merck) at 1200 °C as described previously [29]. The KFeO2 sample was synthesized from K2CO3 with α-Fe2O3 according to the procedure described in [30]. In brief, the mixtures of finely ground powders were placed in a porcelain crucible and heated at the rate of 5 °C/min in the static air up to the final temperatures of 800 °C for KFeO2 and 1200 °C for K2Fe22O34.
The cryptomelane phase was prepared as described in [31]. The mass of 11 g of manganese(II) acetate was dissolved in 40 ml of distilled water and 5 ml of glacial acetic acid was added to achieve a pH = 5. The solution was heated and stirred under reflux for 30 min. 6.5 g of potassium permanganate was dissolved in 150 ml of distilled water, then this solution was added to the previously prepared solution of Mn2+. The mixture was heated and stirred for 24 h under reflux. The solid residue was washed with water (for neutral pH), dried overnight at 110 °C, and calcined at 450 °C for 2 h.
Birnessite was synthesized as described in [32]. A glucose solution (1.4 mol/dm3) and KMnO4 solution (0.27 mol/dm3) were mixed, with a glucose to KMnO4 molar ratio equal 1.5. The solution was stirred at room temperature until a xerogel was formed. The xerogel was washed with water and dried at 110 °C overnight. The powder was calcined at 450 °C for 2 h.
The cobalt oxide nanostructured by potassium (KCo4O8) was prepared by grinding 2.13 g of the cobalt spinel with 1.1 g of potassium hydroxide (POCh). The produced powder was placed in a preheated oven and calcined at 700 °C for 12 h. The pre-calcined bulk materials were placed in preheated ovens to avoid alkali loss due to thermal desorption, further compensated by the use of excessive amounts of alkali substrate (three times excess of potassium hydroxide). The synthesized bulk-doped sample was thoroughly washed with distilled water to remove excess potassium and ground in an agate mortar into a fine powder for further studies [33].
2.2 Catalyst Characterization
XRD patterns were recorded on a Rigaku MiniFlex powder diffractometer with Cu Kα radiation, 2θ step scans of 0.02° and a counting time of 1 s per step. The micro-Raman spectra were recorded under ambient conditions using a Renishaw InVia spectrometer equipped with a Leica DMLM confocal microscope and a CCD detector, with an excitation wavelength of 785 nm. The laser power at the sample position was 1.5 mW (0.5 % of total power) with a magnification of 20×. The Raman scattered light was collected in the spectral range of 150–800 cm−1. At least 6 scans, 10 s each, were accumulated to ensure a sufficient signal to noise ratio. For XRF analysis, ARL Quant’X spectrometer with Uniquant software, calibrated with metal standards series, was used. Specific surface area (SSA) was determined by three-point nitrogen adsorption/desorption with BET isotherm method with Quantasorb Junior (Quantachrome). The X-ray photoelectron spectra (XPS) of the catalyst samples were measured with a Prevac photoelectron spectrometer equipped with a hemispherical VG SCIENTA R3000 analyzer. The photoelectron spectra were measured using a monochromatized aluminum AlKα source (E = 1486.6 eV). The base pressure in the analysis chamber during the measurements was 5 × 10−9 mbar. Spectra were recorded with a constant pass energy of 100 eV for the survey and narrow scan spectra. The binding energy scale was aligned with the main C1 s band set to 295 eV.
2.3 Soot Combustion Tests
The activity in soot combustion of the catalysts was measured by means of TPO (temperature-programmed oxidation). The quartz fixed-bed reactor was heated (10 °C/min) from RT to 750 °C while the concentrations of the released gases were measured by a quadrupole mass spectrometer (SRS RGA 200). Two gas mixtures were used in the tests: 5 % O2 in He, as well as 3.3 % O2, 0.3 % NO in He, both at a 60 ml/min flow. The samples for the combustion test were prepared by weighing 0.05 g of soot (Degussa–Printex80, with the SSA of 230 m2 g−1 and average composition of 98 % carbon and 1 % oxygen, 1 % of hydrogen, and other mineral components (of which 80 % sulfur, 10 % phosphorus and 10 % potassium), examined by XRF and XPS) and the catalyst with an 8:1 ratio and then grinding them in a mortar for 10 min following the tight contact method or gently mixing in an Eppendorf tube following the loose contact protocol. The shifts of the temperature of 50 % soot conversion (ΔT 50) was defined as T 50 for unpromoted spinel oxides minus T 50 for potassium-promoted samples. We also assessed the value of the reaction onset. For this purpose we used of the Arrhenius plot, assuming that CO2 concentration is proportional to the reaction rate constant (at the beginning of the reaction), and plot log (pCO2/Pa) as a function of T −1. On such Arrhenius-like plot the intercept between the line associated with the p CO2 background and the line corresponding to CO2 evolution due to soot combustion reaction is taken as an indicator of the ignition temperature. We found this value to be independent on the amount of soot, thus more reliable than T 10, and take it as a measure of the reaction onset.
3 Result and Discussion
3.1 Characterization
The stoichiometry of all the investigated catalysts was assessed by XRF analysis (Table 1). An excess of potassium (observed also as potassium carbonate segregation on XRD results, see below) was confirmed in the birnessite and β-ferrite samples. The specific surface area of the investigated samples varied from 4 to 70 m2/g. Separate catalytic experiments with the same phase composition (birnessite) with different specific surface areas showed no significant difference in their reactivity.
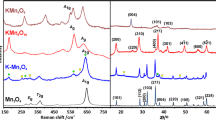
Diffraction patterns obtained for the investigated samples are shown in Fig. 1—manganese-based in Fig. 1a, iron-based in Fig. 1b, and cobalt-based in Fig. 1c. For all samples, the diffraction peaks were labeled with Miller indexes. The manganese catalysts, i.e., spinel, cryptomelane (tunneled structure), and birnessite (layered structure), were indexed in the Pmab (ICSD-30005 [34]), I4/m (ICSD-59159 [35]) and \(R\overline{3} m\) (ICSD-152290 [36]) space groups, respectively. Some reflections originating from an excess of potassium carbonate are only detectable in the case of the birnessite phase (marked with violet diamonds). Likewise, the iron catalysts were indexed, with the spinel (Pmc2 1 (ICSD-35002 [37]), β-ferrite K2Fe22O34 (tunneled structure) (P6 3 /mmc (ICSD-83285 [29]) and monoferrite KFeO2 (layered structure), (Pbca (ICSD-94467 [38]) space groups. Some minor reflections from excess hematite α-Fe2O3 are visible for the surface promoted and β-ferrite samples (marked with orange squares). The cobalt spinel Co3O4 was indexed in the \(Fd\overline{3} m\) (ICSD-150805 [39]) space group. Potassium cobaltate KCo4O8 (tunneled structure) was indexed in the R3 m space group (ICSD-89454 [40]).

Diffraction patterns of manganese-based (a), iron-based (b) and cobalt-based (c) catalysts. Manganese spinel Mn3O4 (a), cryptomelane KMn8O16 (b), birnessite KMn4O8 (c), iron spinel Fe3O4 (d), potassium-doped Fe3O4 (e), β-ferrite K2Fe22O34 (f), monoferrite KFeO2 (g), cobalt spinel Co3O4 (h), potassium-doped Co3O4 (i), potassium cobaltate KCo4O8 (j). Hematite Fe2O3 phase is marked by orange squares
Raman spectra obtained for investigated samples were shown in Fig. 2—manganese-based in Fig. 2a, iron-based in Fig. 2b, and cobalt-based in Fig. 2c. Where possible the symmetry was assigned based on the supporting literature. The Mn3O4 spectra showed six visible bands: 175, 289, 318, 374, 477, and 658 cm−1 [41]. Bands at 186, 285, 393, 479, 514, 582, 639 and 750 cm−1 were visible on the cryptomelane spectra [42]. On the birnessite spectra, bands at 280, 409, 477, 506, 577, 634 and 728 cm−1 are present. The same bands were observed in [43].

Raman spectra of manganese-based (a), iron-based (b) and cobalt-based (c) catalysts. Manganese spinel Mn3O4 (a), cryptomelane KMn8O16 (b), birnessite KMn4O8 (c), iron spinel Fe3O4 (d), potassium-doped Fe3O4 (e), β-ferrite K2Fe22O34 (f), monoferrite KFeO2 (g), cobalt spinel Co3O4 (h), potassium-doped Co3O4 (i), potassium cobaltate KCo4O8 (j). Fingerprints without assigned symmetry are marked by blue stars (for KFeO2, K2Fe22O34 Raman spectra are reported for the first time in the literature). Hematite α-Fe2O3 phase is marked by orange squares
The iron spinel Fe3O4 spectrum shows three visible bands: 304, 541 and 665 cm−1 [44]. In the potassium doped Fe3O4 spectrum, some additional peaks (marked with orange squares) were assigned to hematite, formed during calcination. On the other hand potassium β-ferrite displays rich Raman spectrum. There is no literature concerning this material, but using detailed studies on BaFe12O19 spectrum, based on the vibrations of β-alumina (isostructural with β-ferrite) and other ferrites described in [45], allowed for some interpretation. The most intensive band, observed at 414 cm−1, was assigned to the vibration of the octahedral group FeO6 in the A 1g symmetry, as were the intensive bands at 517 and 618 cm−1 and the less intensive bands at 445 and 489 cm−1. Relatively intensive peaks at 680 and 719 cm−1 were assigned to the vibrations of the FeO5 bipyramid and FeO4 tetrahedral group, respectively, both in the A 1g symmetry. The less intensive band at 221 cm−1 was assigned to vibrations of the O–Fe–O bridge in the A 1g symmetry. Relatively intensive and less intensive bands at 166 and 185 cm−1 were assigned to vibrations of spinel blocks in E 1g. The rest of the bands were not assigned to specific vibrations but symmetry can be assigned to–206 cm−1 (E 2g), 245 cm−1 (E 1g), 288 cm−1 (E 1g) and 373 cm−1 (E 2g) Potassium monoferrite spectra had fewer peaks than the β-ferrite spectra. It was related to a higher symmetry of the KFeO2 primitive cell than that of K2Fe22O34. This spectrum was different from other simple and mixed iron oxides so it can be used for identification but the lack of literature on this material prevented vibration assignment.
The Co3O4 spectrum displayed five bands: 193, 476, 518, 617 and 685 cm−1 [46]. No additional peaks were visible on potassium-doped Co3O4 Raman spectrum. On the KCo4O8 spectrum, three broad bands were visible: 464, 575, and 695 cm−1.
Binding energies for K 2p3/2, O 1s and 2p3/2 maxima for transition metals are summarized in Table 2. In the case of birnessite and cryptomelane peaks assigned to both Mn(III) and Mn(IV) were clearly visible and shifted to higher energies due to potassium promotion. In the case of Fe-based samples, potassium promotion led to an increase in the iron binding energy. As described in [48] it is connected with the increase of Fe(III) content. The shift in Co 2p3/2 peak position to a higher energy can be interpreted as an increase in the Co(IV) content, as discussed in [33]. The highest differences between the binding energies of K 2p3/2 for surface and bulk promotion were seen for Fe-based samples 1.1–1.3 eV, whereas for Co-based catalysts it was only 0.3 eV. It showed a remarkable difference of the potassium state on the surface and in the bulk of the iron oxide matrix, whereas the difference in the case of the cobalt-oxide matrix is much lower. The difference of potassium binding energy between the tunneled cryptomelane and layered birnessite phases is 0.7 eV, whereas for the tunneled monoferrite and layered β-ferrite it is only 0.1–0.3 eV. Oxygen 1 s peak energy was slightly increased in the case of Mn- and Co-based samples, whereas for Fe-based samples the observed shift was much higher.
In summary, the performed characterization of the obtained materials revealed that the catalysts stoichiometry was close to the assumed. The samples are essentially monophasic—surface promotion did not alter the diffraction patterns—whereas for bulk promoted samples the presence of minor spurious phases undetectable by XRD was also excluded by the locally more sensitive Raman probe. For all surface promoted materials the XPS analysis confirmed the presence of potassium. The surface or bulk (nano-structuration) location of the promoter was confirmed in the case of iron and cobalt. For the manganese spinel, due to a very low temperature of intercalation, nanostructured phases alone were observed [49].
3.2 Catalytic Activity
Detailed investigations of catalytic activity in different reaction conditions of the investigated samples were performed and the results are shown in Fig. 3 for tight contact, loose contact and loose contact with NO addition. As a reference, a conversion curve of the non-catalytic soot combustion is also presented (dotted line). The arrows represent the promotional effect as a shift of the conversion curves towards lower temperature around the T 50. The results clearly reveal that the soot combustion progress depends strongly on both the contact mode and also the type of transition metal and potassium location (bulk or surface), as well as the presence of NO.

Soot combustion tests on manganese, iron and cobalt spinels unpromoted and promoted surficial and structural by potassium in different reaction conditions-tight contact, loose contact, loose contact with NO addition
In the tight contact mode, the most active phases were potassium cobaltate (KCo4O8), birnessite (KMn4O8) and monoferrite (KFeO2)—the temperature of 50 % conversion (T 50) of soot were shifted by about 45, 90, and 130 °C, respectively, in comparison to the unpromoted spinels.
Surface potassium promotion is beneficial for soot combustion, however, only in the case of iron (ΔT 50 ~ 105 °C), while for cobalt it has a slightly negative effect (ΔT 50 ~ −20 °C). A proposed explanation for these observations is the different interaction with oxygen. Whereas magnetite is readily oxidized into the corresponding sesquioxide (γ-Fe2O3/α-Fe2O3) in the reaction conditions, cobalt spinel is oxidation resistant [50]. The interaction of the unpromoted Co3O4 with oxygen leads to the formation of surface oxygen species, which participate in the soot oxidation. However, the presence of potassium leads to facile recombination of the surface oxygen species reducing their availability in the course of soot combustion [33]. The highest activity is exhibited by KCo4O8, and KMn4O8 catalysts which lower down the reaction onset (determined from Arrhenius-like plot) to 255 and 250 °C, respectively, in comparison to non-catalytic soot combustion, beginning above 550 °C.
Summarizing, for all the investigated catalyst potassium promotion has a dramatic effect on catalytic activity, and the best results were observed for bulk promotion in each case. The highest activity is exhibited by KCo4O8, and KMn4O8 catalysts, which lower the onset of the soot oxidation down to the spectacular value of ~250 °C.
In the loose contact mode the reaction, as expected, shifts to much higher temperatures (by ~150 °C), but the observed changes are not that regular as those in the tight contact mode. The most active phases were the same as in tight contact mode: KMn4O8, KFeO2, and KCo4O8. In the case of the manganese-based samples, the largest activity increase for the KMn4O8 catalyst was revealed (ΔT 50 ~ 150 °C). Surprisingly, the second structurally promoted sample (KMn8O16) was less active than the unpromoted manganese spinel (ΔT 50 ~ −10 °C), in contrast to the tight contact mode. Only for iron catalyst the activity sequence KFeO2 > K/Fe3O4 > K2Fe22O34 > Fe3O4/Fe2O3 is univocally preserved, though the differences are much smaller.
In the loose contact mode, the presence of NO in the gas mixture leads to the substantial decrease in the reaction temperature, making the situation quite similar to that observed for the tight contact mode measurements. In these conditions the characteristics of the conversion curves are different from those of tight and loose contact conditions—they exhibit a smaller slope, implying a lower activation energy for the reaction. The most striking observation was that for all the samples potassium promotion of bare spinel oxides decreased the T 50 for the same values as in the tight contact conditions. The only exception was the surface-promoted cobalt spinel, which had a lower activity than the unpromoted spinel in tight contact conditions, and in loose contact with NO addition became the most active.
The shifts of the temperature of 50 % of soot conversion for all investigated samples are collected in Fig. 4. Inspection of the presented results clearly shows that the potassium promotional effects (shift in ΔT 50 values with respect to the parent spinels) are strikingly similar to the tight contact conditions and the loose contact with NO addition. At the same time, they are dramatically different from the results obtained in the loose contact mode. This indicates that the loss of the mutual contact between the catalyst surface and the soot particles upon passing from tight to loose contact can be to a large extent compensated in the presence of NO. It may be accounted for by the crucial role of surface mobility of oxygen species in soot oxidation [3, 7].

Temperature shifts in soot combustion process caused by structural or surface potassium promotion of manganese, iron and cobalt spinels in tight contact (a), loose contact (b), loose contact and NO conditions (c)
In the case of mobile surface oxygen species, the number of direct soot-catalyst contact is of less importance that for rigid oxygen. Indeed, spillover of mobile reactive oxygen species allows for oxidation of remote soot particles, whereas for immobile species their direct contact with the soot particles is required. This effect implies a large difference between tight and loose contact as observed in the case of e.g., Mn3O4 (immobile surface oxygen) and a smaller effect for KMn4O8 (mobile surface oxygen). As reported in detailed investigations, in the catalytic cycle of soot combustion NO can be oxidized to NO2, which in turn oxidizes soot more effectively [51, 52]. In such cases, it can be supposed that NO is preferentially oxidized on the investigated catalysts, transferring effectively the catalyst generated oxygen species into the soot particles. Thus, the difference in contact between soot and the catalyst resulting from the different mixing of the solids is substantially attenuated since the NO/NO2 couple acts as an oxygen carrier. Such a role of NO2 as an oxygen carrier has also been reported in the case of deNOx reactions over metalozeolites [53].
Summarizing, for Mn, Fe spinels the potassium promotion leads to pronounced enhancement in soot combustion activity in tight and loose contacts, however, for Co spinel the bulk promotion is positive, whereas surface promotion is slightly inhibitive. In all the investigated cases the nanostructuration of the parent spinel into the layered or tunneled structures results in the best catalytic performance. The difference in activity between tight and loose contacts can be bridged in the presence of NO in the feed due to its transformation into NO2, which acts as the oxygen carrier from the catalyst surface (NO + Osurf → NO2) into soot particles (NO2 + C → CO2 + N2).
4 Conclusions
The paper presents systematic studies of the promotional effect of potassium on the activity of transition metal (Mn, Fe, Co) spinels in catalytic soot combustion. The research presented in the paper reveals, that potassium addition can substantially improve the spinel activity in soot combustion, however, the effect strongly depends in each case on the promoter concentration but also on its location (bulk or surface). It was shown that the bulk promotion is more effective than the surface, with the layered structures of KCo4O8, KMn4O8 being the most active phases, which lower the onset of the soot oxidation down to the spectacular value of ~250 °C. The difference in activity between tight and loose contacts can be bridged in the presence of NO due to its transformation into NO2, which acts as the oxygen carrier from the catalyst surface into soot particles, eliminating the soot-catalyst contact difference.
References
EEA (2014) Air quality in Europe—2014 report. European Environment Agency, Copenhagen
Johnson T (2008) Diesel emission control in review. SAE Tech Pap 1:69–81
Fino D (2007) Diesel emission control: catalytic filters for particulate removal. Sci Tech Adv Mater 8:93–100
Caroca J, Villata G, Fino D, Russo N (2009) Comparison of different diesel particulate filters. Top Catal 52:2076–2082
Van Setten BAAL, Makkee M, Moulijn JA (2001) Science and technology of catalytic diesel particulate filters. Catal Rev 43:489–564
Bueno-López A (2014) Diesel soot combustion ceria catalysts. Appl Catal B 146:1–11
Stanmore BR, Brilhac JF, Gilot P (2001) The oxidation of soot: a review of experiments, mechanisms and models. Carbon 39:2247–2268
Hernández-Giménez AM, Castelló DL, Bueno-López A (2014) Diesel soot combustion catalysts: review of active phases. Chem Pap 68:1154–1168
Aneggi E, de Leitenburg C, Dolcetti G, Trovarelli A (2008) Diesel soot combustion activity of ceria promoted with alkali metals. Catal Today 136:3–10
Legutko P, Kaspera W, Stelmachowski P, Sojka Z, Kotarba A (2014) Boosting the catalytic activity of magnetite in soot oxidation by surface alkali promotion. Catal Commun 56:139–142
Ura B, Trawczyński J, Kotarba A, Bieniasz W, Illan-Gomez MJ, Bueno-López A, López-Suárez FE (2011) Effect of potassium addition on catalytic activity of SrTiO3 catalyst for diesel soot combustion. Appl Catal B 101:169–175
López-Suárez FE, Bueno-López A, Illán-Gómez MJ, Ura B, Trawczynski J (2009) Potassium stability in soot combustion perovskite catalysts. Top Catal 52:2097–2100
An H, Kilroy C, McGinn PJ (2004) Combinatorial synthesis and characterization of alkali metal doped oxides for diesel soot combustion. Catal Today 98:423–429
An HM, McGinn PJ (2006) Catalytic behavior of potassium containing compounds for diesel soot combustion. Appl Catal B 62:46–56
Meng X, Wang Q, Duan L, Chung J-S (2012) Catalytic combustion of diesel soot over perovskite-type catalyst: Potassium titanates. Kinet Catal 53:560–564
Pecchi G, Cabrera B, Delgado EJ, García X, Jimenez R (2013) Activity of KNbO3 as catalyst for soot combustion: Effect of the preparation method. Appl Catal A 453:341–348
Legutko P, Stelmachowski P, Trębala M, Sojka Z, Kotarba A (2013) Role of electronic factor in soot oxidation process over tunnelled and layered potassium iron oxide catalysts. Top Catal 56:489–492
Saracco G, Badini C, Russo N, Specchia V (1999) Development of catalysts based on pyrovanadates for diesel soot combustion. Appl Catal B 21:233–242
An H, Su C, McGinn PJ (2009) Application of potash glass as a catalyst for diesel soot oxidation. Catal Commun 10:509–512
Su C, McGinn PJ (2013) The effect of Ca2+ and Al3+ additions on the stability of potassium disilicate glass as a soot oxidation catalyst. Appl Catal B 138–139:70–78
Su C, McGinn PJ (2014) Application of glass soot catalysts on metal supports to achieve low soot oxidation temperature. Catal Commun 43:1–5
Kumar PA, Tanwar MD, Russo N, Pirone R, Fino D (2012) Synthesis and catalytic properties of CeO2 and Co/CeO2 nanofibres for diesel soot combustion. Catal Today 184:279–287
Legutko P, Kaspera W, Jakubek T, Stelmachowski P, Kotarba A (2013) Influence of potassium and NO addition on catalytic activity in soot combustion and surface properties of iron and manganese spinels. Top Catal 56:745–749
Matarrese R, Castoldi L, Lietti L, Forzatti P (2007) High performances of Pt–K/Al2O3 versus Pt–Ba/Al2O3 LNT catalysts in the simultaneous removal of NOx and soot. Top Catal 42:293–297
Matarrese R, Castoldi L, Lietti L, Forzatti P (2009) Simultaneous Removal of NOx and Soot Over Pt–Ba/Al2O3 and Pt–K/Al2O3 DPNR Catalysts. Top Catal 52:2041–2046
Gálvez ME, Ascaso S, Moliner R, Lázaro MJ (2013) Influence of the alkali promoter on the activity and stability of transition metal (Cu Co, Fe) based structured catalysts for the simultaneous removal of soot and NOx. Top Catal 56:493–498
Doggali P, Kusaba H, Rayalu S, Teraoka Y, Labhsetwar N (2013) Bench scale experiments of diesel soot oxidation using Pr0.7Sr0.2K0.1MnO3 perovskite type catalyst coated on ceramic foam filters. Top Catal 56:457–461
Mizutani K, Takizawa K, Shimokawa H, Suzawa T, Ohyama N (2013) A Novel α-Al2O3 diesel particulate filter with alkali metal-based catalyst for diesel soot oxidation. Top Catal 56:473–476
Ito S, Kurosawa H, Akashi K, Michiue Y, Watanabe M (1996) Crystal structure and electric conductivity of K+-β-ferrite with ideal composition KFe11O17. Solid State Ionics 86:745–750
Kotarba A, Barański A, Hodorowicz S, Sokołowski J, Szytuła A, Holmlid L (2000) Stability and excitation of potassium promoter in iron catalysts–the role of KFeO2 and KAlO2 phases. Catal Lett 67:129–134
Luo J, Zhang Q, Huang A, Suib SL (2000) Total oxidation of volatile organic compounds with hydrophobic cryptomelane-type octahedral molecular sieves. Microporous Mesoporous Mater 35:209–216
Ching S, Landrigan JA, Jorgensen ML, Duan NG, Suib SL, OYoung CL (1995) Sol-Gel synthesis of birnessite from KMnO4 and simple sugars. Chem Mater 7:1604–1606
Jakubek T, Kaspera W, Legutko P, Stelmachowski P, Kotarba A (2015) Surface versus bulk alkali promotion of cobalt-oxide catalyst in soot oxidation. Catal Commun 71:37–41
Ross CR, Rubie DC, Paris E (1990) Rietveld refinement of the high-pressure polymorph of Mn3O4. Am Mineral 75:1249–1252
Vicat J, Fanchon E, Strobel P, Tran Qui D (1986) The structure of K1.33Mn8O16 and cation ordering in hollandite-type structures. Acta Crystallogr B 42:162–167
Gaillot AC, Lanson B, Drits VA (2005) Structure of birnessite obtained from decomposition of permanganate under soft hydrothermal conditions. 1. Chemical and structural evolution as a function of temperature. Chem Mater 17:2959–2975
Iizumi M, Koetzle TF, Shirane G, Chikazumi S, Matsui M, Todo S (1982) Structure of magnetite (Fe3O4) below the Verwey transition temperature. Acta Crystallogr B 38:2121–2133
Tomkowicz Z, Szytuła A (1977) Crystal and magnetic structure of KFeO2. Phys Chem Solids 38:1117–1123
Shaheen WM, Ali AA (2001) Characterization of solid–solid interactions and physico–chemical properties of copper–cobalt mixed oxides and CuxCo3–xO4 spinels. Mater Res Bull 36:1703–1716
Butel M, Gautier L, Delmas C (1999) Cobalt oxyhydroxides obtained by ‘chimie douce’ reactions: structure and electronic conductivity properties. Solid State Ionics 122:271–284
Ren L, Zhou W, Wang Y, Meng M, Wu S, Li S (2014) Magnetic properties of Mn3O4 film with a coexistence of two preferential orientations. J Appl Phys 116:023906
Iyer A, Del-Pilar J, King’ondu CK, Kissel E, Garces HF, Huang H, El-Sawy AM, Dutta PK, Suib SL (2012) Water oxidation catalysis using amorphous manganese oxides, octahedral molecular sieves (OMS-2), and octahedral layered (OL-1) manganese oxide structures. J Phys Chem C 116:6474–6483
Julien C, Massot M, Hadjean R, Franger S, Bach S, Pereira-Ramos J-P (2003) Raman spectra of birnessite manganese dioxides. Solid State Ionics 159:345–356
Lin J-F, Wu J, Zhu J, Mao Z, Said AH, Leu BM, Cheng J, Uwatoko Y, Jin C, Zhou J (2014) Abnormal elastic and vibrational behaviors of magnetite at high pressures. Sci Rep 4:6282
Kreisel J, Lucazeau G, Vincent H (1998) Raman spectra and vibrational analysis of BaFe12O19 hexagonal ferrite. J Solid State Chem 137:127–137
Hadjiev VG, Iliev MN, Vergilov IV (1988) The Raman spectra of Co3O4. J Phys C 21:L199–L201
Gnana Sundara Raj B, Asiri AM, Wu JJ, Anandan S (2015) Synthesis of Mn3O4 nanoparticles via chemical precipitation approach for supercapacitor application. J Alloy Compd 636:234–240
Iyengar SJ, Joy M, Maity T, Chakraborty J, Kotnala RK, Ghosh S (2016) Colloidal properties of water dispersible magnetite nanoparticles by photon correlation spectroscopy. RSC Adv 6:14393–14402
Legutko P, Jakubek T, Kaspera W, Stelmachowski P, Sojka Z, Kotarba A (2014) Soot oxidation over K-doped manganese and iron spinels—How potassium precursor nature and doping level change the catalyst activity. Catal Commun 43:34–37
Kaczmarczyk J, Zasada F, Janas J, Indyka P, Piskorz W, Kotarba A, Sojka Z (2016) Thermodynamic Stability, Redox Properties and Reactivity of Mn3O4, Fe3O4, and Co3O4 Model Catalysts for N2O Decomposition: Resolving the Origins of Steady Turnover. ACS Catal. doi:10.1021/acscatal.5b02642
Fino D, Bensaid S, Piumetti M, Russo N (2016) A review on the catalytic combustion of soot in Diesel particulate filters for automotive applications: from powder catalysts to structured reactors. Appl Catal A 509:75–96
Rico Pérez V, Bueno-López A (2015) Catalytic regeneration of diesel particulate filters: comparison of Pt and CePr active phases. Chem Eng J 279:79–85
Modén B, Da Costa P, Ki Lee D, Iglesia E (2002) Transient studies of oxygen removal pathways and catalytic redox cycles during NO decomposition on Cu–ZSM5. J Phys Chem B 106:9633–9641
Acknowledgments
The project was financed by the Polish National Science Center awarded by decision number DEC-2011/01/B/ST4/00574. Piotr Legutko received funding for the preparation of his doctoral dissertation from the Polish National Science Center under the doctoral scholarship funding program based on the decision number DEC-2014/12/T/ST4/00687. This research was partially carried out using the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Legutko, P., Jakubek, T., Kaspera, W. et al. Strong Enhancement of deSoot Activity of Transition Metal Oxides by Alkali Doping: Additive Effects of Potassium and Nitric Oxide. Top Catal 60, 162–170 (2017). https://doi.org/10.1007/s11244-016-0727-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-016-0727-3