
Abstract
Two series of (0–4 wt%) potassium doped oxide catalysts based on iron and manganese spinel were prepared. The synthesized materials were characterized in terms of their structure (XRD, Raman spectroscopy) and surface electronic properties (work function measurements). The catalytic activity towards soot combustion was determined by temperature programmed oxidation of a physical mixture of soot and catalyst in tight contact in gas oxygen mixtures with and without NO addition. For iron spinel based materials, where potassium is localized at the surface, the catalytic activity correlates with the work function lowering upon K doping, while for manganese spinel based materials, where potassium is incorporated into the bulk (formation of KMn4O8 or KMn8O16), the correlation was not found. The presence of NO in the gas mixture leads to a systematic decrease of soot ignition temperature for all samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Soot is a product of the incomplete combustion of hydrocarbons, coal or other such materials, comprised of various arrangements of carbon molecules. The composition of soot depends highly on the conditions and substance being burned. Typical size of soot particles/aggregates between 50 and 500 nm, is large enough to be harmful for human health. Soot has also been proven to have carcinogenic and mutagenic properties [1]. Jacobson [2], from Stanford University, claims in “Journal of Geophysical Research” that soot pollution is one of the main negative influences causing climate change (global warming). Soot being emitted by factories can stay aloft in the air for as long as a dozen weeks, creating an aerosol which absorbs solar radiation, which can have a direct effect on the climate. According to Jacobson’s calculations, the lowering of soot emissions into the atmosphere would cause the lowering of yearly temperatures by 1 °C in 15 years. Soot, which has settled on the surface of ice, lowers the ability of the ice to reflect solar light—part of the solar radiation is absorbed by the soot and converted into thermal energy. An atmosphere content of a few parts per billion is enough for the settling soot to change the snows reflecting ability by 1 %. This phenomenon has negative consequences for the climate, since such interference into the global energy balance could result in a rise in temperature.
To prevent human illness and climate change soot must be removed or its emissions stopped. Several ways have been developed for this purpose. Reactors or engines may be designed to prevent the formation of soot, yet this method has technological hindrances. On the other hand soot may be removed by use of filters. A disadvantage of this method is that the filters must be replaced and the soot is not permanently eliminated. A third method is to use a catalyst which would lower the temperature of ignition of soot, triggering its combustion in the reactor [3].
Existing catalysts are either ineffective or based on expensive noble metals. One of such catalysts is based on platinum applied to a cerium–zirconium support. The catalyst is based on technology which places a catalytically active material on a diesel particulate filter (DPF) [3]. Promising substitutes are catalysts based on transition metals, which are relatively cheap and harmless to the environment. Oxides containing transition metals such as perovskite-type [4] and spinel-type [5] are being investigated intensively. Moreover, it is widely accepted that the catalytic activity of the material in the soot combustion process can be enhanced by alkali doping [6, 7]. Nitrogen oxides are present in diesel exhaust fumes and some papers report that NO in particular has a positive effect on catalyst activity in soot combustion, as it forms NO2, which is a stronger oxidant for carbon particles than oxygen [8]. The aim of this study is to investigate how alkali doping influences the catalytic activity of iron and manganese oxides with a spinel structure in soot combustion. Moreover, the effect of NO addition will also be addressed.
2 Experimental
2.1 Catalyst Preparation
The catalysts were prepared by weighing 1 g of commercial spinel (Mn3O4 and Fe3O4, Aldrich) and adding an appropriate amount of potassium carbonate solution to achieve the desired alkali loading (0–4 wt%). Afterwards, the catalysts were dried for 1 h at 100 °C and subsequently calcined at 400 °C for 4 h.
2.2 Physicochemical Characterization
The Raman spectra were recorded at room temperature in ambient conditions using a Renishaw InVia spectrometer equipped with a Leica DMLM confocal microscope and a CCD detector, with an excitation wavelength of 785 nm. The laser power at the sample position was 1.5 or 3 mW (0.5 or 1 % of total power) with a magnification of 20×. The Raman scattered light was collected in the spectral range of 150–850 cm−1. At least 6 scans, 10 s each, were accumulated to ensure a sufficient signal to noise ratio. XRD patterns were recorded by a Philips X’Pert APD powder diffractometer with Cu Kα radiation at 10 mA and 10 kV, 2θ step scans of 0.02° and a counting time of 1 s per step. Electron donor properties (work function) of the samples were determined by Kelvin Probe (McAllister, KP6500). For each measurement 0.1 g of sample was pressed under 8 MPa for 1 min to form a pellet. The measurement was performed in a vacuum (10−6 mbar) at 150 °C after heating the sample to 400 °C to standardize the catalyst surface. A stainless steel electrode (d = 3 mm, Φ = 4.1 eV) was used as a reference.
2.3 Soot Combustion Tests
The activity of the catalysts in soot combustion was measured by means of temperature programmed reaction (TPR). The quartz fixed-bed reactor was heated (10 °C/min) from RT to 900 °C and two gas mixtures were used during the tests: 5 % O2 in He, as well as 3.3 % O2, 0.3 % NO in He, both at a 60 ml/min flow. The samples for the soot combustion tests were prepared by weighing 0.05 g of soot (Degussa–Printex80) and the catalyst in an 1:8 ratio and then grinding them in a mortar for 10 min following the tight contact method. The changes in the gas composition upon the soot oxidation reaction were monitored by a quadrupole mass spectrometer (SRS RGA200, lines for m/z = 46 (NO2), 44 (CO2), 32 (O2), 30 (NO), 28 (CO), 18 (H2O)).
3 Results and Discussion
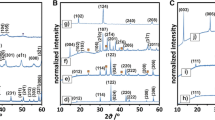
The X-ray diffractograms prove the spinel structure of both iron [9] and manganese spinel materials [10], as shown in Fig. 1. The diffractograms were indexed in the Fd3m and Pmab space groups, respectively. In the case of iron oxide, both magnetite (Fe3O4) and maghemite (γ-Fe2O3) crystalize with a spinel structure, thus, based on the XRD data, we cannot unambiguously determine the definite phase composition. Raman spectra of the investigated iron spinel based materials are shown in Fig. 2. The peak at 640 cm−1 originates from magnetite (curve spinel) [11]. Wide peaks at around 190, 350, 500 and 700 cm−1 are characteristic for maghemite (circles), while narrow peaks at 225, 247, 299, 412, 498 and 613 cm−1 belong to hematite (diamonds). Pop-up and a evanescent narrow peak at 690 cm−1 may be assigned to a bipyramidal FeO5 [12]. The presence of maghemite is not surprising since fine powdered magnetite easily undergoes oxidation, which may be induced by laser radiation during the measurement or due to calcination of potassium doped materials [13]. In the case of manganese spinel we observe peaks characteristic for Mn3O4 [14] (curve a, Fig. 3). Upon the potassium doping and calcination the structure of the sample changes and peaks characteristic of cryptomelane-type manganese dioxide (circles) [15], as well as γ-MnO2 (diamonds) [16] and pyrolusite β-MnO2 (squares) [16] appear. Other manganese compounds, such as birnessite (KMn4O8) exhibit peaks close to cryptomelane (KMn4O16) and spinel type compounds and thus it is difficult to confirm or exclude the presence of this phase.


Raman spectra of potassium doped, iron oxide based samples: Fe3O4 (a), potassium doped with 0.01 theoretical monolayers of K2CO3 (b), 0.2 (c), 0.3 (d), 0.6 (e). Maghemite phase–circles, hematite phase–diamonds

Raman spectra of potassium doped, manganese oxide based samples: Mn3O4 (a), potassium doped with 0.5 theoretical monolayers of K2CO3 (b), 3 (c), 4.5 (d), 6.5 (e), 11 (f)
The influence of potassium addition on the catalytic activity in soot oxidation for the iron spinel is shown in Fig. 4 (diamonds). The character of this dependence is similar to a Morse curve (steep slope, minimum and asymptote), which brings to mind the Topping model of surface alkali promotion [17]. It is reported that catalytic activity changes (especially in catalytic processes where the electronic factor plays a key role) are excused by this model [18]. This is also a typical dependence for systems where alkalis are located on the catalyst surface, forming surface dipoles [19].

Influence of potassium promotion and NO addition (lower curve) on ignition temperature of iron spinel
The influence of NO addition on the catalytic activity is shown in Fig. 4, (squares). The effect is beneficial and exhibits a systematic character (~30 °C), but disappears near the 1.5 monolayer coverage, which could be the result of full coverage of the catalyst surface by potassium carbonate. The influence of potassium addition on the catalytic activity of the manganese spinel is shown in Fig. 5 (diamonds). This dependence is similar to that of the iron spinel, however, less pronounced. The influence of NO addition (squares) measures between 5 °C (measurement error) and 25 °C. It is worth mentioning that during the catalytic tests for both series, m/z = 46 was traced with mass spectrometer but no signal was detected, which indicates that NO2 was not produced during the reaction. It indicates that NO acts directly as an oxidizing agent. This behavior is different from that observed for other oxide based catalysts during soot oxidation, where the formation of NO2 is often postulated [20, 21].

Influence of potassium promotion and NO addition (lower curve) on ignition temperature of manganese spinel
The minor effect of addition of NO for the catalysts with high potassium loading is related to the formation of surface potassium carbonate. Indeed, we found, that if pure K2CO3 is used as a catalyst for soot oxidation the addition of NO does not induce any changes in its reactivity. This is in contrast to the redox and noble metal based catalysts where the NO addition usually enhances the catalytic activity [22].
The non-systematic effect for K doped Mn3O4, in contrast to Fe3O4 may be brought about by structural changes of the material as indicated by Raman spectroscopy (Fig. 3). This nanostructurization of manganese oxide based materials by potassium, leading to the formation of a layered structure of KMn4O8 and KMn4O16, is found to be the main factor responsible for the very high activity of these catalysts. The ignition temperature of potassium promoted manganese oxide catalysts can be as low as 250 °C. In Fig. 6 the dependence between activity and electro-donor properties of iron (diamonds) and manganese (squares) spinels is shown. The work function of the iron spinel changes in a wide range upon K addition and the highest activity is observed for a potassium coverage of 0.5, which corresponds to the lowest work function value. This type of dependence of reactivity on the loading of alkali promoter is typical for a surface promotion [18]. The fact that mainly surface promotion is observed is caused by the high temperatures required for the formation of bulk potassium ferrite phases, which exceed 600 °C [23]. The minimum of ignition temperature, shown in Fig. 4, is related to the optimal dispersion of the surface potassium, which brings its highest promotional effect. This is associated with the formation of surface dipoles and resultant changes in the work function of the catalytic material (∆Φ ≈ 0.6 eV), much higher than for Mn based samples, (Fig. 6). For manganese spinel the work function is rather independent on potassium addition (∆Φ ≈ 0.2 eV, Fig. 6), suggesting that the promoter is incorporated into the bulk of the manganese oxide phase, as revealed by Raman spectroscopy (Fig. 3). In this case, at the calcination temperature of 400 °C, mixed potassium-manganese phases can be already formed [24]. Thus, much higher potassium loading (per unit surface area) in comparison with the K/Fe3O4 samples is necessary for formation of the surface K2CO3 phase, (Fig. 5).

Correlation of temperature of ignition and work function for K-doped iron (diamonds) and manganese (squares) oxide based catalysts
4 Conclusions
Two series of (0–4 wt%) potassium promoted iron and manganese spinel based catalysts were investigated in model soot oxidation processes. The temperatures of soot ignition for K-promoted iron spinels were found to be in the range of 320–360 °C, while for K-promoted manganese spinels, in the range of 250–300 °C. We found that presence of NO in the gas feed leads to a decrease of the soot ignition temperature independently of the base metal oxide or potassium loading, suggesting that NO acts as better oxidizing agent than O2. The lowest determined ignition temperature with the presence of NO was found to be ca. 290 °C for K–Fe3O4 and ca. 240 °C for K–Mn3O4. In the case of iron spinel based materials the changes of the observed work function upon K addition correlate with soot oxidation reactivity, while for manganese spinel based materials such correlation is not observed. Whereas, for iron oxide catalyst the addition of potassium leads to the surface promotional effect, for manganese oxides potassium invokes more severe structural changes (formation of KMn4O8 and KMn4O16).
References
van Setten BAAL, Makkee M, Moulijn JA (2001) Cat Rev 43:489–564
Jacobson MZ (2010) J Geophys Res D 115:24–31
Fino D (2007) Sci Technol Adv Mater 8:93–100
Ura B, Trawczyński J, Kotarba A, Bieniasz W, Illan-Gomez MJ, Bueno-López A, López-Suárez FE (2011) Appl Catal 101:169–175
Fino D, Russo N, Saracco G, Specchia V (2006) J Catal 242:38–47
An H, McGinn PJ (2006) Appl Catal 62:46–56
Legutko P, Stelmachowski P, Trębala M, Sojka Z, Kotarba A (2013) Top Catal 56:489–492. doi:10.1007/s11244-013-0003-8
Kumar PA, Tanwar MD, Russo N, Pirone R, Fino D (2012) Catal Today 184:279–287
Haavik C, Stolen S, Fjellvag H, Hanfland M, Hausermann D (2000) Am Minerol 85:514–523
Baron V, Gutzmer J, Rundlof H, Tellgren R (1998) Am Minerol 83:739–786
de Faria DLA, Venancio Silva S, de Oliveira MT (1997) J Raman Spectrosc 28:873–878
Kreisel J, Lucazeau G, Vincent H (1998) J Solid State Chem 137:127–137
Shebanova ON, Lazor P (2003) J Raman Spectrosc 34:845–852
Han Y-F, Chen F, Zhong Z, Ramesh K, Chen L, Widjaja E (2006) J Phys Chem B 110:24450–24456
Gao T, Glerup M, Krumeich F, Nesper R, Fjellvåg H, Norby P (2008) J Phys Chem C 112:13134–13140
Julien C, Massot M, Rangan S, Lemal M, Guyomard D (2002) J Raman Spectrosc 33:223–228
Brison J, Mine N (2007) Surf Sci 601:1467–1472
Zasada F, Stelmachowski P, Maniak G, Paul J-F, Kotarba A, Sojka Z (2009) Catal Lett 127:126–131
Verhoef RW, Asscher M (1997) Surf Sci 391:11–18
Fino D, Russo N, Saracco G, Specchia V (2008) Powder Technol 180:74–78
Fino D, Russo N, Saracco G, Specchia V (2006) J Catal 242:38–47
Cauda E, Fino D, Saracco G, Specchia V (2004) Top Catal 30–31:299–304
Joseph Y, Ketteler G, Kuhrs C, Ranke W, Weiss W, Schlogl R (2001) Phys Chem Chem Phys 3:4141–4153
Atribak I, Bueno-López A, García-García A, Navarro P, Frías D, Montes M (2010) Appl Catal 93:267–273
Acknowledgments
The project was financed by the Polish National Science Center awarded by decision number DEC-2011/01/B/ST4/00574
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Legutko, P., Kaspera, W., Jakubek, T. et al. Influence of Potassium and NO Addition on Catalytic Activity in Soot Combustion and Surface Properties of Iron and Manganese Spinels. Top Catal 56, 745–749 (2013). https://doi.org/10.1007/s11244-013-0026-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-013-0026-1