
Abstract
Oxidation catalysts are modeled by oxide single crystals, thin oxide films, as well as supported oxide nanoparticles. We characterize the surface of those materials using a variety of surface sensitive techniques including scanning tunneling microscopy and spectroscopy, photoelectron spectroscopy, infrared spectroscopy, and thermal desorption spectroscopy. We find temperature dependent structural transformations from V2O5(001) to V2O3(0001) via V6O13(001). V2O3(0001) is found to be vanadyl terminated in an oxygen ambient and it loses the vanadyl termination after electron bombardment. It is shown that the concentration of vanadyl groups controls the selectivity of the methanol oxy-dehydrogenation towards formaldehyde. A proposal for the mechanism is made. The results on single crystalline thin films are compared with similar measurements on deposited vanadia nanoparticles. The experimental results are correlated with theoretical calculations and models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vanadium oxides supported by a second oxide such as TiO2, SiO2, or Al2O3 represent an important class of active catalysts industrially applied to a variety of reactions. These include oxidation as well as reduction reactions, e.g., the oxidation of o-xylene to phthalic anhydride, the oxidation of sulfur dioxide to sulfur trioxide, the ammoxidation of aromatic hydrocarbons, and the selective catalytic reduction of NO x with ammonia [1, 2]. Typically, submonolayer to monolayer quantities of vanadium oxides are dispersed on the substrate using impregnation, grafting, or chemical vapor deposition techniques followed by calcination cycles. Numerous investigations have been dedicated to the role of the support oxide and to the identification of active species. It was found that the choice of the support oxide can influence the activity of a catalyst system by several orders of magnitude [1, 2]. The reducibility of the support oxide and the coupling between the support and vanadia, mediated via V–O support bonds, have been proposed to play a decisive role [3–6]. However, many questions remain unanswered, such as the role of vanadyl groups or of vanadium oxides in an oxidation state lower than +5 [1, 2].
Theoretical studies by density functional theory (DFT) for vanadium oxide species supported on silica revealed the key steps of the oxidation of methanol to formaldehyde [7]. First methanol binds on the surface by heterolytically splitting the V–O–Si interface bond (Scheme 1).

Binding of methanol on isolated vanadia sites on silica
The second step is the redox step, a hydrogen atom is transferred from the methyl group to the vanadyl oxygen atom which creates a kind of biradical structure with an unpaired electron at the •CH2O– group and one electron in the vanadium d-states. The reaction is completed when the V–O(OCH2 •) bond splits releasing formaldehyde and a second electron goes into the vanadium d-states (Scheme 2).

Oxidation of surface bound methanol to formaldehyde
To obtain a more direct insight into structure–reactivity relations supported vanadia systems have been prepared under ultrahigh vacuum conditions on silica, alumina, and ceria thin film supports [8–16]. Scanning tunneling microscopy has been used to show that under such conditions the vanadia does not wet the surfaces of silica and alumina even if those surfaces had been exposed to water beforehand. Rather the vanadia particles grow three dimensionally. The vibrational and electronic properties of such model systems have been investigated with infrared spectroscopy and X-ray photoelectron spectroscopy and the analysis has been compared with the equivalent information from real catalysts [11]. It was found, in particular, that the vanadium oxidation state is +3 in the bulk but +5 at the surface where the vanadium is fourfold coordinated to three oxygen atoms in the surface plane and to one oxygen atom via a vanadium–oxygen double bond (vanadyl) perpendicular to the surface plane. It is expected that the vanadyl oxygen is a decisive factor for the surface chemistry.
The role of vanadyl species will be investigated in this paper with respect to alcohol oxidation even though in this case the process used commercially involves silver and ironmolybdate catalysts [17, 18]. In this publication we present detailed catalytic studies on alcohol oxidation for supported vanadia and for ordered V2O3(0001) thin films grown on Au(111). Similar to the case of the supported vanadia particles the ordered films exhibit vanadium in a +3 oxidation state in the bulk and +5 at the surface which is terminated by a layer of vanadyl groups [19, 20].
Also, we address the interconversion of vanadium oxides. Specifically, we will report changes of the surface structure of a V2O5(001) single crystal surface upon heat treatment. The experimental studies are paralleled by theoretical investigations using electronic structure calculations and Monte Carlo simulations [21].
Experimental
The experiments have been performed in a number of different ultrahigh vacuum chambers. The setups are described in the literature. The V2O5(001) single crystal surfaces were prepared by cleavage in UHV using adhesive tape [21].
The preparation of vanadia clusters on silica follows the recipes described in ref. [8–11] and the synthesis of the thin vanadia films has been detailed in Ref. [16, 19]. The V2O3 films have been either used as prepared or they have been reduced by electron bombardment before exposure to the alcohol. Methanol has been dosed from a capillary doser typically at low temperature (~80 K) before annealing to given temperature.
Theoretical Calculations
Calculations are based on spin-DFT and employ a plane-wave basis set as implemented in the Vienna Ab initio Simulation Package (VASP). We use gradient corrected exchange-correlation (GGA) functionals (PW91 or PBE) [22, 23]. The electron–core interaction is described by the projector augmented wave (PAW) method [24]. The clean and defective V2O5(001) surfaces are modeled using a fully relaxed two-layer slab and an energy cutoff of 800 eV. We have considered a wide range of defect concentrations (1/6 ≤ Θ ≤ 1). In order to rationalize the scanning tunneling spectra we also perform GGA + U calculations with U = 3 eV [25]. Furthermore, we employ the Monte Carlo method with pair interaction energies between reduced sites fitted to the DFT results to simulate the reduced V2O5(001) surface at a given temperature and defect concentration. The network consists of 50 double rows of 2 × 150 sites, which corresponds to an area of 57.75 × 53.7 nm2. Ten trajectories, starting from random states, are simulated using the Metropolis algorithm [26] for a varying fraction of reduced sites (0–100%, every 2.5%).
For the study of the oxidation of methanol to formaldehyde on V2O3(0001) surfaces we employed a four-layer slab with a (2 × 2) periodicity and a 400 eV cutoff; the (V–O3–V) bottom layer is kept fixed at its position in the fully relaxed slab representing the clean surface.
Results and Discussion
To analyze the surface properties of vanadia based catalysts we have studied single crystalline systems as in Ref. [21]. Figure 1a shows a scanning tunneling microscopy image of a V2O5(001) single crystal surface revealing the double rows of vanadyl–oxygen atoms protruding out of the surface of the V2O5(001) sheets which are held together by van der Waals forces along the (001) direction as expected from the bulk structure schematically represented in Fig. 1b. This surface undergoes a transformation via V6O13 towards V2O3(0001) keeping its single crystallinity over large areas of the macroscopic surface. The final result of a V2O3(0001) surface shows in Fig. 1c the typical hexagonal lattice of protrusions of a depolarized corundum type (0001) surface, which is expected to be terminated by half a metal ion layer with exactly the exposed surface unit cell, if the surface structure is directly derived from a cut through the bulk structure in between the puckered vanadium ion layers [16]. This situation is depicted in Fig. 1d. It will become clear further below in this paper that the surface is, in fact, not terminated in this way but rather by a layer of V=O (vanadyl) bonds with the same registry [16].

(a) Scanning tunneling microscopy image of a V2O5(001) single crystal surface revealing the double rows of vanadyl-oxygen atoms protruding out of the surface. (b) Schematic representation of Fig. 1a. (c) V2O3 resulting from V2O5 reduction. (d) Schematic view of oxygen and vanadium layer stacking in V2O3(0001)
Before we discuss the evidence for this we present details for the first steps of the processes involved in the transformation from V2O5(001) towards V6O13 and V2O3 respectively. Figure 2a and b shows color coded gap maps of a V2O5 surface after heating to 350 and 400 K, respectively. To construct these maps we measured scanning tunneling spectra for every pixel, i.e. the derivative of the I–V-curve as a function of the tip voltage. The blue, red and yellow areas correspond to regions of different gap width. The corresponding scanning tunneling spectra are revealed in Fig. 2c. The blue trace has been taken on a fully oxidized V2O5(001) surface at 300 K exhibiting a 1.8 eV gap and well ordered structure, while the yellow and red pixels stem from disordered and already reduced parts in which we observe a smaller gap (yellow) and a closed gap (red). Those changes have been modeled using DFT + U and Monte Carlo simulations. The DFT + U calculated density of states (DOS) of the clean V2O5(001) surface yields a band gap of ~2.3 eV (Fig. 2f), which is similar to the measured value (~1.8 eV). The formation of isolated vanadyl oxygen defects results in the reduction of the band gap down to ~1.1 eV, whereas the formation of defect pairs along the [010] direction leads to a band gap of ~0.5 eV, and a trench-like structure with missing rows of vanadyl–oxygen atoms exhibits a metallic character. Earlier plain DFT-PW91 calculations of reduced V2O5(001) surfaces predicted the facile reduction along the direction of the vanadyl rows [27]. Therefore, we ascribe the red and yellow areas in Fig. 2a and b to randomly distributed isolated vanadyl oxygen defects and to the missing-row phase, respectively. Monte Carlo simulations at 300 K and UHV conditions (10−10 mbar of O2) for 3% (Fig. 2d) and 7% (Fig. 2e) of reduced sites combined with the DFT + U band gaps result in simulated band gap maps which exhibit a disordered pattern with the same amount of red and yellow areas as the experimental ones. Hence, the anisotropic growth of vanadyl oxygen vacancies is concomitant with an insulator-to-metal transition at the surface.

(a, b) Band gap maps of the V2O5(001) surface recorded at 350 K (a) and 400 K (b). (c) Normalized conductance spectra over areas A and B at 350 K, and over V2O5(001) at 300 K. (d, e) Simulated gap maps at 300 K for 3% (d) and 7% (e) of reduced sites. The local band gap value was taken equal to the smallest gap within a radius of 2.78 nm. The band gap scale (in eV) is indicated. (f) Calculated density of states of the clean V2O5(001) surface (blue), of the defective surface containing isolated vanadyl–oxygen vacancies (yellow), and of the missing-row structure (red). The curves are smoothed by a Gaussian level broadening of 0.2 eV
We now return to the V2O3(0001) surface as imaged in Fig. 1c. We have shown earlier that this surface is terminated by a layer of vanadyl groups, increasing the formal oxidation state from V3+ in the bulk to V5+ at the surface [14, 15]. Synchrotron based photoemission studies were employed to prove this. Such a surface is completely unreactive with respect to methanol. Only physisorbed species have been observed and characterized by TDS and spectroscopic techniques. However, if the surface is reduced the situation changes considerably. We note that, different from Cr2O3(0001) [28], where we encountered a similar structural situation, the vanadyl groups cannot be reduced thermally without destroying the surface structure [16, 19]. In fact, it is necessary to electron bombard the surface to selectively remove the vanadyl oxygen atoms as shown in Fig. 3 and discussed in a previous paper [16]. Ultimately, all vanadyl oxygen atoms may be removed by this procedure and we are left with vanadium terminated surfaces just as it was indicated in Fig. 1d. This surface is very reactive with respect to a variety of different molecules, including CO, CO2, H2O and alcohols [29]. There is no specific selectivity observed under those circumstances. The situation changes when we consider intermediate vanadyl coverages, i.e. situations where vanadyl groups as well as surface vanadium ion sites are exposed simultaneously. The results presented in the following have been obtained on thin V2O3 films grown on Au(111). Figure 4a shows an area at 5 K where a few vanadyl oxygen atoms have been removed (dark spots) [30]. Figure 4b shows scanning tunneling spectra taken within an area where full vanadyl coverage is observed as opposed to a position where apparently an individual vanadyl oxygen atom is missing. Obviously, this very small variation in vanadyl oxygen coverage leads to a change in the local electronic structure, i.e. the surface band gap starts to close! Note that the band gap referred to here is the band gap in the surface region which may be different from the bulk band gap.

STM images illustrating the reduction of vanadyl terminated V2O3(0001) towards vanadium terminated V2O3(0001) via electron irradiation (500 eV). Images of the vanadyl terminated surface, the vanadium terminated surface (after irradiation with an electron dose of 80 mC), and two intermediated reduction states (electron doses of 1 and 10 mC) are shown

(a) STM image of V2O3/Au(111) taken at 5 K (4.0 × 4.0 nm, 1.4 V, 100 pA). (b) dI/dV spectra taken at a defect site (violet line) and above a V=O group (blue line). The additional dI/dV intensity above the V=O lattice is attributed to a V=O related electronic state
There are indications that even defects within the material influence the band gap at the surface, and that this band gap varies across the surface [30]. The band gap is believed to be important in oxidation reactions where redox processes involve electron transfer and intermediate storage [4]. It is therefore a working hypothesis to assume that a combination of the ensemble and the local band gap governs the reactivity [4–6].
In Fig. 5a we plot the reactivity of a V2O3(0001) surface at two different degrees of reduction with respect to the formation of various reaction products as measured via TDS after exposing the surface to methanol at ~90 K. The black and the red traces correspond to mass 31 (methanol) and mass 29 (formaldehyde plus a methanol fragment which results from cracking in the mass-spectrometer), respectively. In addition we show water (mass 18), hydrogen (mass 2) and methane (mass 16). Below 200 K the desorption signals are basically determined by desorption of the methanol multilayer. A prominent peak at ~275 K is assigned to the desorption of the methanol monolayer [29], essentially up to 500 K the desorption traces of 29 amu (formaldehyde) and 18 amu (water) follow the methanol signal (31 amu). The water signal at ~250 K which does not follow methanol desorption is probably to be assigned to the reaction of hydroxyl groups resulting from the fission of methanol into methoxy and hydrogen [29]. An interesting point to note is the dependence of the selectivity for formaldehyde formation on the presence of vanadyl groups on the surface: their presence leads to an increased amount of produced formaldehyde and decreases the amount of formed methane indicating that the surface vanadyl groups are an important ingredient for the control of the selectivity of the V2O3(0001) surface.

(a) Thermal desorption spectra of a methanol multilayer on a partially reduced V2O3(0001) surface (top, electron dose: 2 mC), and a fully reduced surface (bottom, electron dose: 80 mC, electron energy: 500 eV). Spectra for mass 2 (molecular hydrogen), 16 (methane), 18 (water), 29 (formaldehyde + methanol), and 31 (methanol) are shown. Methanol was adsorbed at 95 K. (b) Thermal desorption spectra of a methanol multilayer on V2O3(0001) recorded at mass 29 (methanol + formaldehyde) as a function of the degree of surface reduction. The originally vanadyl terminated V2O3(0001) surface was reduced with different doses of electron irradiation (500 eV) as given in the figure. At the bottom a spectrum of a methanol (90%) plus oxygen (10%) gas mixture is shown. The adsorbates were dosed at 90 K
Above 500 K small traces of H2O are observed and a considerable signal at mass 29 which largely stems from formed formaldehyde as judged by the small intensity of the signal at mass 31. After the onset of the formaldehyde signal we see desorption of methane as well as traces of H2O and hydrogen. All of the signals above 500 K are absent for a fully vanadyl covered surface (not shown here).
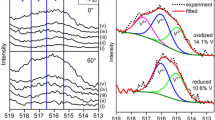
Figure 5b shows a series of traces for mass 29 as a function of the degree of reduction of the V2O3(0001) surface. There is a clear maximum of formaldehyde production at a moderate state of reduction corresponding to a 4 mC dose of electrons reducing the surface (full reduction needs about 80 mC). Figure 6a shows STM images of this intermediate reduction stage before and after exposure to methanol and heating to 400 K. The XPS data in Fig. 6b as well as infrared data (Fig. 7 to be discussed below) indicate the presence of methoxy groups on this surface. Figure 6b contains the C1s XPS data for methanol dosed at 100 K and consecutively heated to three different temperatures. The lowest trace is for methanol exposure at 100 K revealing spectra characteristic of methanol. At 550 K the states characteristic of methanol have entirely disappeared and a species with a C1s binding energy of 286.5 eV remains. Indeed, the XPS binding energy of 286.5 eV is typical for methoxy. Figure 7 shows temperature dependent IR spectra after exposure of the partially reduced surface (second trace from bottom) to methanol at 88 K and subsequent stepwise heating to 660 K. In comparison, the spectrum of the fully vanadyl terminated surface is shown. The vanadyl groups had been partially labeled with 18O isotopes giving rise to the doublet at 998 and 1032 cm−1 respectively. It is interesting to note that this doublet shifts by more than 10 cm−1 to lower energies (982 and 1021 cm−1, see the right panel) upon reduction indicating the influence of the modified electronic structure discussed above in connection with the low temperature STM data. Upon exposure to methanol even at the lowest temperature the vanadyl vibrations disappear and methoxy bands appear in the spectra. This indicates that methanol first loses the hydrogen atom from the hydroxyl group and a V–OH species must be formed.

(a) STM image of a weakly reduced surface (electron dose: 2 mC, electron energy: 500 eV). (b) STM image of the weakly reduced surface after exposure to methanol in multilayer amounts at 95 K and annealing at 400 K. (b) C1s-XPS spectra of reduced V2O3(0001) after annealing at 400 K. Methanol (three different doses: 0.2, 0.75, 2.25 L) was dosed at 100 K. The spectra were recorded with Al Kα radiation at an electron exit angle of 70° with respect to the surface normal

Infrared spectra of methanol adsorbed on fully reduced [panel (a), electron dose: 80 mC] and partially reduced [panel (b), electron dose: 8 mC] V2O3(0001) as a function of temperature. Methanol was adsorbed at 88 K. Before recording a spectrum, the samples were shortly flashed to the temperature given at the respective spectrum and the spectra were recorded after the sample had cooled down to below 100 K. The V2O3(0001) substrate contains a mixture of 18O and 16O. All spectra are referenced to the spectra of the respective reduced surfaces except the spectrum of the partially reduced and the vanadyl terminated surfaces in panel (b) which were referenced to the spectrum of the fully reduced surface of panel (a)
The vanadyl groups are actively involved in the formaldehyde production as the formaldehyde produced contains both 16O and 18O if CH3 16OH is dosed onto the surface that contains vanadyl groups formed from both isotopes as shown in Fig. 8.

Thermal desorption spectra (mass 29 and 31) of methanol on a weakly reduced V2O3(0001) layer (electron dose: 8 mC) containing a mixture of 16O and 18O. Methanol was adsorbed at 88 K
It is clear that the mechanism involves both vanadyl and open vanadium surface ion sites, as schematically depicted in Scheme 3, see also Fig. 9. Methanol binds to the vanadium ion sites transferring the hydroxyl hydrogen to a vanadyl group with low activation energy. The methyl hydrogen is then transferred to another vanadyl group or directly to a V–OH group leading to the formation of water. Alternatively, the methyl group could take up a hydrogen atom to form methane.

Tentative reaction scheme for formaldehyde formation from methanol on V2O3(0001) surfaces

Calculated structures of (a) molecular adsorption of methanol on V2O3(0001), (b) dissociative adsorption, and (c) surface after methanol oxidation
It is the hydrogen attachment to the vanadyl group that goes hand in hand with a change in the electron count and could be influenced by the ability of the vanadium oxide to accept and release electrons, which in turn is controlled by the vanadium oxide surface band gap.
The mechanism suggested by Scheme 3 is supported by DFT calculations using the PBE functional. The energy for molecular adsorption of methanol on the fully oxidized V2O3(0001) surface is only −16 kJ/mol. This step differs substantially from adsorption on silica supported vanadia shown in Scheme 1, for V2O3 forms a closely packed lattice and insertion of a methoxy group in a V–O bond is not possible. On the reduced surface with a vanadyl defect concentration of 1/4 we find stronger adsorption with energies of −122 and −193 kJ/mol for non-dissociative (Fig. 9a) and dissociative adsorption (Fig. 9b). The latter involves hydrogen transfer to a neighboring vanadyl group. Hydrogen transfer to a three-fold coordinated surface oxygen atom is much less favorable and results in an adsorption energy of only −103 kJ/mol. The large energy difference underlines the importance of having both reduced and oxidized surface ion sites for dissociative methanol adsorption creating a hydroxyl and a methoxy site, respectively. In a second step, the methoxy group formed is oxidized to formaldehyde by hydrogen transfer to a neighboring vanadyl group (Fig. 9c). This redox step is similar to silica supported vanadia (Scheme 2). For this reaction we find a reaction energy of +141 kJ/mol with respect to the dissociative adsorption state. This reaction is only possible if two adjacent vanadyl sites are available on the surface, one for the initial adsorption and a second one for the oxidation step. For higher defect concentrations (1/2), dissociative adsorption is 20–30 kJ/mol less exothermic. However, the main difference is that after adsorption vanadyl groups are no longer available. Hence, in the redox step, hydrogen has to be transferred to a three-fold coordinated surface oxygen atom, which is far more endothermic (244 kJ/mol with respect to the dissociative adsorption state). Hence, the effect of the defect concentration on these reactions energies is sizable. This result is supported by the TDS data presented in Fig. 5b which show a formaldehyde desorption peak at 550 K which grows at the expense of the state at ~515 K when the degree of surface reduction is increased. In the light of the results of the calculation presented above, the peak at 515 K could be attributed to formaldehyde formed using vanadyl oxygen while the peak at higher temperature might be assigned to formaldehyde formed involving three-fold coordinated surface oxygen atoms.
So far we have discussed the reactivity of a well ordered surface. Realistically the reactions happen on systems where the vanadium oxide is supported on another oxide. We have studied in the past vanadium oxide (VO x ) deposits on alumina and on silica in order to clarify spectroscopic assignments previously proposed for powder samples using Raman spectroscopy [11]. The typical sample shows an STM image as shown in Fig. 10 where the vanadia is prepared by physical vapor deposition of vanadium in an oxygen ambient onto the surface of a silica film. The morphology of the film is granular indicating three-dimensional growth of the vanadium oxide particles, which have been proven to be mainly V2O3 particles exposing vanadyl groups at their surface [8–11]. In this sense they are similar to the single crystal V2O3(0001) surface. The morphology of the vanadium oxide deposits can only be modified within a very narrow range. Attempts have been made to deposit the vanadium within an oxygen ambient into a pre-deposited water (ice) layer [31] on the thin silica film surface. While this procedure does not lead to hydroxylation of the silica, the vanadium oxide particles expose OH and partially V=O groups at 300 K. Upon heating to 500 K, V2O3 particles are formed which mainly expose V sites [12]. Note that water assisted preparation slightly smoothes the vanadium oxide layer. When oxidized at 500 K in 10−6 mbar of O2, the vibrational properties are very similar to that of the fully oxidized V2O3(0001) surface. If such a system, fully vanadyl covered, is exposed to methanol, it is as unreactive as a fully V=O covered V2O3(0001) surface. For fully reduced vanadia particles (i.e. before oxidation treatment) we find some amount of formaldehyde in the desorption spectra. However, if the reduced particles prepared using water assisted preparation are exposed to oxygen at 300 K which leads to the partially reduced (or partially oxidized) surface (note that quantifying the state of reduction is even more difficult here than in the case of single crystal surface), the amount of formaldehyde produced is much greater than for fully reduced or fully oxidized particles. Figure 11 shows the TDS traces equivalent to the ones shown for the single crystal in the relevant temperature range. It is clear that the deposited particles behave qualitatively similar, and it is tempting to assume the same mechanistic steps as more clearly underpinned by the single crystal data.

Typical STM image of vanadia prepared by physical vapor deposition of vanadium in an oxygen ambient on silica and alumina films. Size: 100 × 100 nm2

TDS of methanol on vanadia particles (different degrees of reduction). About 1 L of methanol was adsorbed at ~140 K in each case
Conclusions
We have shown how vanadium oxide surfaces of different composition may be transformed into each other. The final reduction stage of V2O5 is V2O3 and it is possible to collect solid experimental information on reaction steps and mechanism. The results revealed for the single crystal surfaces may be transferred to the situation of vanadium oxide particles on a supporting oxide, i.e. silica.
The oxidation of methanol to formaldehyde was investigated for V2O3(0001) thin films and deposited vanadia clusters. It is shown that the vanadyl oxygen atoms are involved in the reaction and that they influence its selectivity. Indications were found that the band gap plays a prominent role for the reactivity.
This study indicates that through a combination of experimental work on well defined model systems in combination with theoretical modeling useful insight into catalytic active systems may be gained that are otherwise difficult or impossible to obtain.
References
Bond GC, Tahir SF (1991) Appl Catal 71:1
Deo G, Wachs IE, Haber J (1994) Crit Rev Surf Chem 4:141
Wachs IE (2005) Catal Today 100:79–94
Argyle MD, Chen K, Resini C, Krebs C, Bell AT, Iglesia E (2004) J Phys Chem B 108:2345–2353
Argyle MD, Chen K, Iglesia E, Bell AT (2005) J Phys Chem. B 109:2414–2420
Chen K, Bell AT, Iglesia E (2002) J Catal 209:35–42
Döbler J, Pritzsche M, Sauer J (2005) J Am Chem Soc 127:10861–10868
Magg N, Giorgi JB, Schroeder T, Bäumer M, Freund HJ (2002) J Phys Chem B 106:8756–8761
Magg N, Giorgi JB, Hammoudeh A, Schroeder T, Bäumer M, Freund HJ (2003) J Phys Chem. B 107:9003–9010
Magg N, Giorgi J, Frank M, Immaraporn B, Schroeder T, Bäumer M, Freund H-J (2004) J Amer Chem Soc 126:3613
Magg N, Immaraporn B, Giorgi J, Schroeder T, Bäumer M, Döbler J, Wu Z, Kondratenko E, Cherian M, Baerns M, Stair PC, Sauer J, Freund H-J (2004) J Catal 226:88–100
Kaya S, Sun YN, Weissenrieder J, Stacchiola D, Shaikhutdinov S, Freund HJ (2007) J Phys Chem. C 111:5337–5344
Lu JL, Gao HJ, Shaikhutdinov S, Freund HJ (2006) Surf Sci 600:5004–5010
Dupuis A-C, Abu Haija M, Richter B, Kuhlenbeck H, Freund H-J (2003) Surf Sci 539:99–112
Abu Haija M, Guimond S, Romanyshyn Y, Uhl A, Kuhlenbeck H, Todorova TK, Ganduglia-Pirovano MV, Döbler J, Sauer J, Freund HJ (2006) Surf Sci 600:1497–1503
Guimond S, Abu Haija M, Kaya S, Lu J, Weissenrieder J, Shaikhutdinov S, Kuhlenbeck H, Freund H-J, Döbler J, Sauer J (2006) Top Catal 38:117–125
Gerberich HR, Seaman GC (1994) Kirk-Othmer encyclopedia of technical technology, vol 11. Wiley, New York, pp 929–951
Tatibouet JM (1997) Appl Catal A 148:213–252
Dupuis A-C, Abu Haija M, Richter B, Kuhlenbeck H, Freund HJ (2003) Surf Sci 539:99–112
Todorova TK, Ganduglia-Pirovano MV, Sauer J (2005) J Phys Chem. B 109:23523–23531
Blum RP, Niehus H, Hucho C, Ganduglia-Pirovano MV, Fortrie R, Sauer J, Shaikhutdinov S, Freund H-J (2007) Phys Rev Lett 99:226103
Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C (1992) Phys Rev B 46:6671
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Blöchl PE (1994) Phys Rev B 50:17953
Dudarev SL, Botton GA, Savrasov SY, Humphreys CJ, Sutton AP (1998) Phys Rev B 57:1505
Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E (1953) J Chem Phys 21:1087–1092
Ganduglia-Pirovano MV, Sauer J (2004) Phys Rev B 70:045422
Dillmann B, Rohr F, Seiferth O, Klivenyi G, Bender M, Homann K, Yakovkin IN, Ehrlich D, Bäumer M, Kuhlenbeck H, Freund H-J (1996) Farad Disc 105:295–315
Romanyshyn Y, PhD Thesis HU Berlin (to be published)
Simic-Milosevic V, Nilius N, Rust HP, Freund H-J (2008) Phys Rev B 77:125112-1–125112-5
Kaya S, Weissenrieder J, Stacchiola D, Shaikhutdinov S, Freund HJ, Phys J (2007) Chem C 111:759–764
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 546) and the “Fonds der chemischen Industrie”. The calculations were carried out on the IBM pSeries 690 system of the Norddeutscher Verbund für Hoch- und Höchstleistungsrechnen (HLRN).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Romanyshyn, Y., Guimond, S., Kuhlenbeck, H. et al. Selectivity in Methanol Oxidation as Studied on Model Systems Involving Vanadium Oxides. Top Catal 50, 106–115 (2008). https://doi.org/10.1007/s11244-008-9114-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-008-9114-z