
Abstract
Cobalt(II) tri-tert-butoxysilanethiolates with 2,5-dimethylpyridine, 3,4-dimethylpyridine and 3,5- dimethylpyridine co-ligands have been synthesized by reaction of bimetallic [Co{μ-SSi(OtBu)3}{SSi(OtBu)3}(NH3)]2 with the appropriate pyridines. The complexes were characterized by elemental analysis, single-crystal X-ray structure determination, IR and UV–Vis spectroscopy. These complexes are tetra- or penta-coordinated with CoN2S2 and CoNO2S2 cores, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transition metal silanethiolates (MSSiR3) have recently attracted much attention owing to their potential applications as precursors in sulfido cluster-forming reactions, thin layer fabrication and catalysis [1–5]. Generally, silanethiols are sensitive to atmospheric conditions and hydrolyze easily with the evolution of H2S. In our syntheses, we use tri-tert-butoxysilanethiol as a source of S-ligand. This sterically hindered compound is resistant to hydrolysis and enables the preparation of monomeric and relatively stable complexes for synthesis conducted under normal atmospheric conditions [6–10].
Our goal is to design complexes that can act as molecular models for biological systems, both in the solid state and in the solution. Previous results indicate that Co(II) and Zn(II) tri-tert-butoxysilanethiolates show strong correlations with biological systems and imitate the coordination environment of metal-containing proteins like alcohol dehydrogenase ADH, transcription factor TFIIIA or farnesyltransferase FTase, where the metal atom is coordinated by histidines and cysteines [6–14].
Owing to the structural and chemical similarity of Co(II) and Zn(II) silanethiolates, we plan to synthesize a series of cobalt(II) tri-tert-butoxysilanethiolates, with additional N donor ligands, in order to elaborate their spectroscopic and structural properties. Relying on our earlier UV–Vis analysis of cobalt(II) silanethiolates with imidazoles as a co-ligand [10], we suppose that such models will be useful for the spectral studies of the active centers of other metalloproteins [10, 15–18], and this paper is the continuation of our research. Here, we describe the syntheses, structures and spectral analysis of three monomeric new cobalt(II) tri-tert-butoxysilanethiolates with dimethylpyridine co-ligands.
Experimental
The elemental analyses (C, H, S and N contents) were performed on an Elemental Analyser EA 1108 (Carlo Erba Instruments). Solution electronic spectra were recorded on a Unicam SP300 spectrometer in n-hexane in the range 200–800 nm. Quartz cuvettes and n-hexane as solvent were used. The IR spectra were measured for crystalline compounds in the range of 4,000–700 cm−1 with a Momentμm microscope (IR detector) attached to a Mattson Genesis II Gold spectrometer (IR source).
Syntheses
Tri-tert-butoxysilanethiol and [Co{μ-SSi(tBuO)3}{SSi(tBuO)3}(NH3)]2 were obtained according to procedures described previously [13, 19]. All other reagents were obtained commercially. Amines were dried by standard methods and distilled prior to use.
All three complexes were obtained using the same synthesis procedure. To a solution of [Co{μ-SSi(tBuO)3}{SSi(tBuO)3}(NH3)]2 (0.25 g, 0.2 mmol) in n-hexane (10 mL), freshly prepared base was added dropwise (2,5-dimethylpyridine: 46.3 μL, 0.4 mmol 1, 3,5-dimethylpyridine: 116 μL, 1 mmol 2, 3,4-dimethylpyridine: 116 μL, 1 mmol 3). After gentle stirring for a few minutes, the mixtures were allowed to stand at 4 °C to yield crystals of the complexes.
[Co{SSi(OtBu) 3 } 2 (2,5-dimethylpyridine)]1 Crystals are very stable in atmospheric conditions. Anal. Found for C31H63NO6S2Si2Co: C 51.4, H 8.8, N 2.1, S 9.0%. Calc. C 51.3, H 8.8, N 2.0, S 8.8%. M. p. at 172–173 °C.
[Co{SSi(OtBu) 3 } 2 (3,5-dimethylpyridine)(NH 3 )]2 is unstable at room temperature and after some hours changes color from blue to pink-violet. Anal. Found for C31H66N2O6S2Si2Co: C 50.8, H 8.6, N 3.7, S 8.8%. Calc. C 50.2, H 8.9, N 3.8, S 8.6%. M. p. at 132–134 °C.
[Co{SSi(OtBu) 3 } 2 (3,4-dimethylpyridine) 2 ]3 is also unstable just as complex 2. Anal. Found for C38H72N2O6S2Si2Co: C 54.1, H 8.8, N 2.9, S 8.0%. Calc. C 54.8, H 8.7, N 3.4, S 7.7%. M. p. at 93–96 °C.
X-ray crystallography
Diffraction data were recorded on a KUMA KM4 diffractometer with graphite-monochromated Mo-Kα radiation using a Sapphire-2 CCD detector (Oxford Diffraction Ltd). The apparatus was equipped with an open flow thermostat (Oxford Cryosystems), which enabled experiments at 120 K. The structures were solved with direct methods and refined with the SHELX98 program package [20] with the full-matrix least-squares refinement based on F 2.
Basic crystal data, description of the diffraction experiment, and details of the structure refinement are given in Table 1. Crystallographic data (without structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre with reference numbers CCDC 772024 and 772025 and can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
Results and discussion
Previously, we described the structures of several mononuclear complexes with different CoNxOySz and CoNxSy cores, most of which were molecular and neutral [21, 22]. As a starting compound for all the syntheses, we use bimetallic [Co{μ-SSi(tBuO)3}{SSi(tBuO)3}(NH3)]2 with Co(II) coordinated by ammonia plus two bridging and two terminal silanethiolato ligands. The addition of a heterocyclic N−ligand to a solution of this complex causes breakage of the (Co–μS)2 ring with simultaneous elimination of ammonia [21, 22].
The currently presented complexes (Scheme 1) have been obtained with dimethylpyridine derivatives that introduce similar steric hindrance on the aromatic ring but with different positioning of the methyl groups. Two equivalents of 2,5-dimethylpyridine added to an n-hexane solution of the Co(II) complex gave pink-violet crystals of penta-coordinated [Co{SSi(OtBu)3}2(2,5-dimethylpyridine)] 1 (Fig. 1) which is stable in atmospheric conditions. We did not observe any other products in the reaction mixture, which may suggest that the presence of methyl groups in positions 2 and 5 on the pyridine ring of the amine forces the formation of complex 1 that contains one amine ligand coordinated to the metal.

Obtained complexes 1 – 3

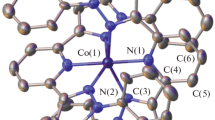
Molecular structure of {[Co{SSi(tBuO)3}2]2(2,5-dimethylpyridine)} 1 with atom labeling scheme (hydrogen atoms of tBuO groups omitted). Thermal ellipsoids are drawn at 30% probability
The result is different when 3,5-dimethylpyridine is used in the synthesis. First of all, an excess of the amine is necessary to obtain the blue crystals of tetrahedral [Co{SSi(OtBu)3}2(3,5-dimethylpyridine)(NH3)] 2. This complex is coordinated by four ligands: two silanethiolates, one pyridine and one ammonia, giving a CoN2S2 kernel to the complex. Previously, we have obtained a similar compound with 2-methylpyridine [23], which was however significantly less stable than 2. We have also published the penta-coordinated [Co{SSi(OtBu)3}2(3,5-dimethylpyridine)] [21], which is a close analog of (3,5-dimethylpyridine)bis(tri-tert-butoxysilanethiolato) Zn(II) and Cd(II) [24, 25]. However, the Co(II) complex was obtained in the synthesis with the solution of 2,4,6-trimethylpyridine, in which 3,5-dimethylpyridine was present as a contaminant [21]. We did not obtain this complex in the direct synthesis of the amine with [Co{μ-SSi(tBuO)3}{SSi(tBuO)3}(NH3)]2 in n-hexane.
The above results prompted us to explore the synthesis with dimethylpyridine, with the methyl groups located in positions of 3 and 4; the basicity of 3,4-dimethylpyridine (pKb = 7.54) is very similar to that of 3,5-dimetylpyridine (pKb = 7.85). This synthesis also leads to a tetrahedral product, namely [Co{SSi(OtBu)3}2(3,4-dimethylpyridine)2] 3; however, this time Co(II) is coordinated by two silanethiolate residues and two dimethylpyridine ligands. Complex 3 is very soluble in different solvents and precipitates as a blue hair-like forms, and up to now we have not received any crystals of this compound, despite the use of a variety of crystallization methods. Meanwhile, the composition of 3 was determined by elemental and spectral analysis in the solid state and in solution.
The results obtained, together with the comparison with previous results [21–23], strongly suggest that the steric hindrance exerted by the methyl groups on pyridine has the main influence on the products formation.
Crystal structures
The structures of complexes 1 and 2 with atom labeling schemes and crystal packing are illustrated in Figs. 1, 2, 3 and 4, and selected bonds and angles are listed in Tables 2 and 3.

Crystal packing of 1. All hydrogen atoms of aromatic ring have been omitted for clarity

Two molecules of {[Co{SSi(OtBu)3}2(3,5-dimethylpyridine)(NH3)} 2 with atom labeling and hydrogen bonds scheme. All tBu groups and hydrogen atoms of aromatic ring are omitted for clarity. Thermal ellipsoids are drawn at 30% probability

Crystal packing of 2
Complex 1 consists of discrete molecules and crystallizes in space group P–1 with two molecules in a unit cell (Figs. 1 and 2). The central Co(II) atom is coordinated by two S atoms and two O atoms derived from silanethiolate residues and one N atom of 2,5-dimethylpyridine. The geometry of the complex can be described as distorted trigonal bipyramidal with basal plane S1, S2 and N1 and Co1 lying in this plane. The Co1–O1 and Co1–O4 distances (2.3548(16) and 2.3145(15) Ǻ, respectively) distinctly exceed the sum of covalent radii for Co and O, which is equal to 1.89 Å, but are nevertheless shorter than those found in similar metal silanethiolates with methyl and dimethyl pyridine derivatives. The S1–Co1–S2 angle is noticeably one of the narrowest angles among tri-tert-butoxysilanethiolate complexes with O,S-chelating bonds of silanethiolate residues [21, 22].
The overall coordination pattern is typical for penta-coordinated Co(II), Mn(II), Zn(II) or Cd(II) tri-tert-butoxysilanethiolates with pyridines [6, 21, 22, 24, 25], where metal–oxygen distances are longer than the typical covalent bonds. Therefore, considering these observations, we think that the formation of such interactions in silanethiolate complexes is forced by the spatial arrangement of the bulky tert-butoxy groups.
Complex 2 crystallizes in space group P21/c with four molecules in a unit cell (Fig. 4). The Co(II) atom is coordinated by two S atoms from the silanethiolate ligands and two N atoms from different ligands, namely ammonia and 3,5-dimethylpyridine. This gives the complex with a tetrahedral structure with CoN2S2 core (Fig. 3). Additionally, each ammonia ligand is engaged in two hydrogen bonds: one intermolecular N–H···S of the silanethiolate residue and one intramolecular N–H···O of the tert-butoxy group. These additional interactions enable the formation of two hydrogen-bonded dimers in a unit cell, with the pyridine rings lying in the same plane, approached in a head to tail mode (Fig. 4). In comparison, the congruent complex [Co{SSi(OtBu)3}2(2-methylpyridine)(NH3)] contains two intramolecular N–H···O hydrogen bonds (distances 3.041(6) and 3.092(6) Å) [23]. The remaining bond lengths and angles in complex 2 are comparable to these found in other Co(II) tri-tert-butoxysilanethiolates.
Spectral analysis
The structures of complexes 1–3 were confirmed with vibrational spectra recorded in the solid state in the range of 3,500–700 cm−1 as presented in Figs. 5 and 6. In metal silanethiolates, where oxygen from the Si–O–tBu ligand interacts with the metal center, we can observe a broad band at about 980 cm−1, as seen in the spectrum of complex 1 (Fig. 5) [22]. This intensity is absent in the spectra of tetrahedral complexes 2 and 3, and only the strong asymmetric ν (Si–O–C) appears at about 1,010 cm−1 (Fig. 5). In contradistinction to complexes 1 and 3, complex 2 shows additional sharp bands in the region 3,400–3,100 cm−1, ascribed as the antisymmetric and symmetric NH3 stretching vibrations, as shown in Fig. 6 [26].

Solid-state FT-IR spectra of complexes 1 – 3 in the range 3,400–3,100 cm−1

Solid-state FT-IR spectra of complexes 1 – 3 in the range 1,700–700 cm−1
During the preparation of the n-hexane solutions for spectral measurements, we noticed that in the case of dilute solutions, their blue color changes and some indefinite precipitate appears. Therefore, we performed the UV–Vis analysis in order to determine the composition of the complexes in the solutions with diverse concentrations. The results are presented in Fig. 7.

Electronic spectrum in n-hexane solution of complexes 1 – 3. Concentration of the solutions: 0.00012 M (UV) and 0.005 M (Vis) 1; 0.00056 M (UV) and 0.0078 M (Vis) 2; 0.00012 M (UV) and 0.012 M (Vis) 3
Normally, Co(II) tri-tert-butoxysilanethiolates with additional aromatic N-ligands show an absorption band at about 270 nm, arising from N → Co LMCT, and a second band at about 350 nm, attributed to S → Co charge transfer transition of the terminal sulfur (St) of the silanethiolato residues [22, 27, 28]. Co(II) complexes also show specific d–d transitions in the visible region: penta-coordinated complexes show two broad bands at about 520 and 680 nm, whereas three bands in the range of 520–720 nm are characteristic for the 4A2 → 4T1(P) d–d transitions of high-spin pseudotetrahedral compounds [15, 23, 27, 28].
The spectra of complexes 1–3 differ from the spectra of other cobalt(II) silanethiolates with N-ligands and in the LMCT region contain only the N → Co LMCT absorption band, shifted insignificantly (264 nm for 2, 256 nm for 3) with a visible shoulder. The St → Co band is present in the spectrum of complex 2, but very weak, whereas in the case of complexes 1 and 3, there is a definite lack of this band. It suggests that in dilute solutions, these complexes dissociate and the number of the silanethiolate ligands on the cobalt atom decreases rapidly.
Changes are also observed in the visible region. The diluted solutions of complexes 1–3 show no absorption bands in this region. The spectra presented in Fig. 7 are obtained only for concentrated solutions, and only the spectrum of complex 1 contains separated bands, similar to those of penta-coordinated complexes, while the spectra of complexes 2 and 3 reveal some bands, which are not symmetric. These suggest that the examined solutions contain mixtures of the compounds, where Co(II) is four- and five-coordinated, respectively.
Conclusions
Three cobalt(II) tri-tert-butoxysilanethiolates with additional dimethylpyridines have been synthesized. The crystal structures have been determined for two of them. The spectral analysis using UV–Vis and IR shows the relative stability of the complexes in the solid state but their instability in dilute solutions. We suppose that the presence of hydrogen bonds stabilizes the structures in the crystals.
References
Cai L, Holm RH (1994) J Am Chem Soc 116:7177–7188
Tran DTT, Taylor NJ, Corrigan JF (2000) Angew Chem Int Ed 39:935–937
Shapley PA, Liang HC, Dopke NC (2001) Organometallics 20:4700–4704
Komuro T, Matsuo T, Kawaguchi H, Tatsumi K (2002) Chem Commun 9:988–989
Kropidłowska A, Rotaru A, Strankowski M, Becker B, Segal E (2008) J Therm Anal Calor 91:903–909
Kropidłowska A, Chojnacki J, Becker B (2007) Inorg Biochem 101:578–584
Dołęga A, Pladzyk A, Baranowska K, Wieczerzak M (2008) Inorg Chem Commun 11:847–850
Dołęga A, Baranowska K, Pladzyk A, Majcher K (2008) Acta Cryst C 64:m259–m263
Dołęga A, Farmas A, Baranowska K, Herman A (2009) Inorg Chem Comm 12:823–827
Dołęga A, Pladzyk A, Baranowska K, Jezierska J (2009) Inorg Chim Acta 362:5085–5096
Corwin DT, Fikar R, Koch SA (1987) Inorg Chem 26:3079–3080
Corwin DT, Gruff ES, Koch SA (1987) J Chem Soc Chem Commun 13:966–967
Piękoś R, Wojnowski W (1962) Z Anorg Allg Chem 318:212–216
Hightower KE, Fierke CA (1999) Curr Opin Chem Biol 3:176–181
Eklund H, Nordström B, Zeppezauer E, Söderlund G, Ohlsson I, Boiwe T, Söderberg BO, Tapia O, Brändén CI, Åkeson Å (1976) J Mol Biol 102:27–59
Kimblin C, Bridgewater BM, Churchill DG, Hascall T, Parkin G (2000) Inorg Chem 39:4240–4243
Maret W (1989) Biochemistry 28:9944–9949
Chang S, Karambelkar VV, Sommer RD, Rheingold AL, Goldberg DP (2002) Inorg Chem 41:239–248
Becker B, Zalewska A, Konitz A, Wojnowski W (2001) Z Anorg Allg Chem 627:271–279
Sheldrick GM (2008) Acta Crystallogr Sect A Found Crystallogr 64:112
Becker B, Zalewska A, Konitz A, Wojnowski W (2001) Polyhedron 30:2567–2576
Pladzyk A, Baranowska K, Hapter P (2010) Trans Met Chem 35:373–379
Becker B, Pladzyk A, Konitz A, Wojnowski W (2002) Appl Organometal Chem 16:517–524
Dołęga A, Konitz A, Baum E, Wojnowski W (2005) Acta Cyst E61:m2582–m2584
Dołęga A, Chojnacki J, Konitz A, Komuda W, Wojnowski W (2006) Acta Cyst E62:m636–m639
Nakamoto K (1997) Infrared and raman spectra of inorganic and coordination compounds. Wiley, New York
Vašák M, Kägi JHR (1981) Proc Natl Acad Sci USA 78: 6709–6713
Vašák M, Kägi JHR, Holmquist B, Vallee BL (1981) Biochemistry 20:6659–6664
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pladzyk, A., Olszewska, J., Baranowska, K. et al. Cobalt(II) silanethiolato complexes with dimethylpyridines: crystal structures and spectroscopic studies. Transition Met Chem 35, 821–827 (2010). https://doi.org/10.1007/s11243-010-9399-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-010-9399-1