
Abstract
A controlled substitution reaction of the chlorine atoms of 1,3,5-benzenetricarbonyl trichloride by the organoiron fragment (CpFe(CO)2S) has been achieved. The complexes CpFe(CO)2SCO-3,5-C6H3(COCl)2 (1), 1,3-[CpFe(CO)2SCO]2-5-C6H3COCl (2) and 1,3,5-[CpFe(CO)2SCO]3C6H3 (3) were prepared from the reaction of (μ-S x )[CpFe(CO)2]2 (x = 3, 4) with 1,3,5-C6H3(COCl)3 in a 1:1, 2:1, or 3:1 metal to ligand molar ratio. The reactions of (1) with amines, thiols, and carboxylic acids produce the trifunctional mono-iron complexes CpFe(CO)2SCO-3,5-C6H3(COY)2 [Y = NR2 (4), SR (5), OCOR (4)]. The X-ray structure determination of (1) is reported.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chemistry of ligands containing sulfur, selenium, or tellurium has been actively pursued [1–3]. These ligands have high affinity to coordinate with transition metals in inorganic and organometallic complexes [2, 3]. The interest in such ligands is motivated by their bonding diversity, perspectives of reactivity, and relevance to bioinorganic chemistry [4–7].
The iron sulfide complexes (μ-S x )[CpFe(CO)2]2 (x = 3, 4), which are accessible through the insertion of elemental sulfur into the iron–iron bond of the dimer [CpFe(CO)2]2 [8], were found to be reactive toward electrophiles. Their reactions with mono-functional electrophiles such as acid chlorides [9, 10], sulfonyl chlorides [11], chloroformates [12], chlorothioformates [13], chlorodithioformates [14], and O-alkyloxalyl chlorides [15] produced the complexes CpFe(CO)2SZ (Z = COR, SO2R, CO2R, C(S)OR, CS2R, COCO2R) which contain one iron centre. The reaction of these iron sulfides with bi-functional acid chlorides, such as oxalyl chloride, gave [CpFe(CO)2SCO]2 which contains two iron atoms [15]. On the other hand, their reaction with terephthaloyl chloride gave either the di-iron thioterephthalates [CpFe(CO)2SCO]2(4-C6H4) or the mono-iron complex CpFe(CO)2SCO-4-C6H4COCl, depending on the molar ratio of the reactants [16]. The latter complex was found to be reactive toward amines, phenols, carboxylic acids, and thiols to produce the bi-functional products CpFe(CO)2ECO-4-C6H4COY (Y = NR2, OR, OCOR, SR) [16–19]. The dimeric complexes containing two iron atoms with either homo- or hetero-bis(chalcogeno)-ligands were prepared by the reactions of CpFe(CO)2SCO-4-C6H4COCl with the iron chalcogenides (μ-E x )[CpFe(CO)2]2 (E = S, Se) [18, 19].
In this article, we describe the reaction of the iron sulfides (μ-S x )[CpFe(CO)2]2 with 1,3,5-benzenetricarbonyl trichloride, which contains three acid chloride groups as an extension to our efforts in this area. The reactions of the mono-iron complex CpFe(CO)2SCO-3,5-C6H3(COCl)2 with amines, thiols, or carboxylic acids are discussed.
Experimental
General considerations
All reactions and manipulations were carried out under a dinitrogon atmosphere by using standard Schlenk line techniques. The use of dry solvents is necessary. The starting materials (μ-S x )[CpFe(CO)2]2 (x = 3, 4) were prepared according to the literature method [8], while 1,3,5-C6H3(COCl)3, amines, thiols, and carboxylic acids were obtained commercially (Acros) and were used without further purification. Infrared (i.r.) spectra were recorded on a Nicolet-Impact 410 FT-IR spectrometer. 1H-NMR spectra were recorded on a Bruker-Avance-400 MHz spectrometer. Chemical shifts are given in ppm relative to TMS at 0 ppm. Elemental analyses were carried out on a Perkin-Elmer 2400 Series II CHN-analyzer.
Preparation of CpFe(CO)2SCO-3,5-C6H3(COCl)2 (1)
A solution of (μ-S x )[CpFe(CO)2]2 (x = 3, 4) (2.00 mmol) in diethyl ether (50 cm3) was dropwise added, over a period of 30 min, to a solution of 1,3,5-C6H3(COCl)3 (0.67 g, 2.5 mmol) in diethyl ether (10 cm3). The resulting mixture was stirred for 2 h at room temperature. The volatiles were removed under vacuum and re-dissolved in a minimum amount of CH2Cl2. This solution was introduced to a silica gel column made up in hexane. Elution with a mixture of diethyl ether and hexane (7:3 volume ratio) gave an orange band which was collected and identified as CpFe(CO)2SCO-3,5-C6H3(COCl)2, followed by a red band which was identified as CpFe(CO)2Cl. The title complex was re-crystallized from CH2Cl2/hexane. Yield = 0.54 g, 62%. M.p = 193–195 °C. I.r. (CH2Cl2, cm−1): νC≡O = 2047 (vs); 2003 (vs); νC=O = 1764 (s); νSC=O = 1612 (m). 1H-NMR (CDCl3): δ 9.10 (d, 2H, Ha); 8.84 (t, 1H, Hb); 5.09 (s, 5H, Cp). C16H8FeO5SCl2 calcd.: C, 43.8; H, 1.8; S, 7.3%. Found: C, 43.4; H, 2.1; S, 6.9%.
Preparation of 1,3-[CpFe(CO)2SCO]2-5-C6H3COCl (2)
A solution of 1,3,5-C6H3(COCl)3 (0.26 g, 1 mmol) in diethyl ether (150 cm3) was added to a solution (2.00 mmol) of (μ-S x )[CpFe(CO)2]2 (x = 3, 4) (2.00 mmol) in diethyl ether (50 cm3) under dinitrogen atmosphere. The resulting mixture was stirred for 3 h at room temperature. The volatiles were removed under vacuum and re-dissolved in a minimum amount of CH2Cl2. This solution was introduced to a silica gel column made up in hexane. Elution with THF/hexane gave an orange band which was discarded. Elution with THF gave an orange band which was collected and identified as 1,3-[CpFe(CO)2SCO]2-5-C6H3COCl and was re-crystallized from CH2Cl2/hexane. Yield = 0.17 g, 29%. M.p = 213–214 °C. I.r. (CH2Cl2, cm−1): νC≡O = 2046 (vs); 2001 (vs), νC=O = 1761 (w); νSC=O = 1610 (m). 1H-NMR (CDCl3): δ 9.18 (bs, 1H, Ha); 8.86 (bs, 2H, Hb); 5.06 (s, 10H, Cp). C23H13Fe2O7S2Cl calcd.: C, 45.1; H, 2.1; S, 10.5%. Found: C, 44.9; H, 2.0; S, 9.8%.
Preparation of 1,3,5-[CpFe(CO)2SCO]3C6H3 (3)
A solution of 1,3,5-C6H3(COCl)3 (0.18 g, 0.67 mmol) in diethyl ether (100 cm3) was added slowly over a period of 1 h to a solution of (μ-S x )[CpFe(CO)2]2 (2.00 mmol) (x = 3, 4) in diethyl ether (50 cm3) under dinitrogen atmosphere. The resulting mixture was stirred for 3 h at room temperature. The volatiles were removed under vacuum and re-dissolved in a minimum amount of CH2Cl2. This solution was introduced to a silica gel column made up in hexane. Elution with 1:1 volume ratio of THF/hexane gave an orange band, which was discarded. Elution with THF gave an orange band which was collected and recrystallized from CH2Cl2/hexane. Yield = 0.24 g, 48%. M.p = 227–228 °C. I.r. (CH2Cl2, cm−1): νC≡O = 2044 (vs); 2000 (vs), νSC=O = 1606 (m). 1H-NMR (CDCl3): δ 8.91 (s, 3H, Ar-H); 5.03 (s, 15H, Cp). C30H18Fe3O9S3 calcd.: C, 45.8; H, 2.3; S, 12.2%. Found: C, 45.3; H, 2.3; S, 12.4%.
Preparation of CpFe(CO)2SCO-3,5-C6H3(CONR1R2)2 (4)
A THF solution (60 cm3) containing the amine (2.3 mmol) and (1) (0.5 g, 1.14 mmol) was refluxed. The volatiles were removed under vacuum and the residue was re-dissolved in CH2Cl2 and transferred to a chromatographic column made up in hexane. An orange band was elueted with CH2Cl2/diethyl ether (9:1 volume ratio). This band was collected and identified as CpFe(CO)2SCO-3,5-C6H3(CONR1R2)2 and re-crystallized from CH2Cl2/hexane.
CpFe(CO)2SCO-3,5-C6H3(CONHPh)2 (4a)
Yield = 0.42 g, 67%. M.p = 220–221 °C. Reflux time = 2 h. I.r. (KBr, cm−1): νC≡O = 2036 (vs); 1987 (v), νNC=O = 1647 (s); νSC=O = 1601 (m). 1H-NMR (CDCl3): δ 8.63 (s, 2H, Ha); 8.39 (s, 1H, Hb); 8.29 (bs, 2H, NH); 7.70 (d, 4H, Ar-H); 7.37 (t, 4H, Ar-H); 7.16 (t, 2H, Ar-H), 5.00 (s, 5H, Cp). C28H21FeO5SN2.2CH2Cl2 calcd.: C, 54.6; H, 3.5; S, 5.0; N, 4.4%. Found: C, 54.2; H, 3.5; S, 4.7; N, 4.3%.
CpFe(CO)2SCO-3,5-C6H3(CON(CH3)Ph)2 (4b)
Yield = 0.37 g, 56%. M.p = 180–181 °C. Reflux time = 3 h. I.r. (KBr, cm−1): νC≡O = 2041 (vs); 1991 (vs), νNC=O = 1648 (s); νSC=O = 1601 (m). 1H-NMR (CDCl3): δ 7.91 (bs, 2H, Ha); 7.29 (bs, 1H, Hb); 7.19 (t, 4H, Ph); 7.11 (t, 2H, Ph); 6.88 (d, 4H, Ph); 4.98 (s, 5H, Cp); 3.38 (s, 6H, CH3). C28H24FeO5SN2.0.5CH2Cl2 calcd.: C, 58.8; H, 4.1; S, 5.2; N, 4.5%. Found: C, 58.4; H, 3.9; S, 4.9; N, 4.4%.
CpFe(CO)2SCO-3,5-C6H3(CON(CH3)CH2Ph)2 (4c)
Yield = 0.52 g, 75%. M.p = 118–120 °C. Reflux time = 3 h. I.r. (KBr, cm−1): νC≡O = 2038 (vs); 1985 (vs), νNC=O = 1648 (s); νSC=O = 1590 (m). 1H-NMR (CDCl3): δ 8.25 (s, 2H, Ha); 7.65 (s, 1H, Hb); 7.36 (m, 8H, Ph); 7.14 (m, 2H, Ph); 5.05 (s, 5H, Cp); 4.51 (s, 4H, CH2); 3.03 (s, 6H, CH3). C32H28FeO5SN2.0.5CH2Cl2 calcd.: C, 60.0; H, 4.5; S, 4.9; N, 4.3%. Found: C, 59.6; H, 4.3; S, 4.7; N, 3.9%.
Preparation of CpFe(CO)2SCO-3,5-C6H3(COSR)2 (5)
A THF solution (50 cm3) containing 2.50 mmol of thiol and (1) (0.50 g, 1.14 mmol) was refluxed for 6 h. The solvent was removed under vacuum and the residue was re-dissolved in a minimum amount of CH2Cl2 and transferred to a silica gel column made up in hexane. An orange band was eluted by a mixture of CH2Cl2/diethylether (6:4 volume ratio). The collected products were recrystallized from CH2Cl2/hexane.
CpFe(CO)2SCO-3,5-C6H3(COSR)2 (5a)
Yield = 0.53 g, 77%. M.p = 152–153 °C. I.r. (KBr, cm−1): νC≡O = 2040 (vs); 1991 (vs), νC=O = 1661 (s); νSC=O = 1611 (m). 1H-NMR (CDCl3): δ 9.01 (d, 2H, Ha); 8.67 (t, 1H, Hb); 7.56 (m, 4H, Ph); 7.45 (m, 6H, Ph); 5.13 (s, 5H, Cp). C28H18FeO5S3.0.5CH2Cl2 calcd.: C, 54.4; H, 3.0; S, 15.3%. Found: C, 54.4; H, 2.9; S, 14.6%.
CpFe(CO)2SCO-3,5-C6H3(COSCH2Ph)2 (5b)
Yield = 0.62 g, 89%. M.p = 139–141 °C. I.r. (KBr, cm−1): νC≡O = 2038 (vs); 1987 (vs), νC=O = 1658 (s); νSC=O = 1608 (m). 1H-NMR (CDCl3): δ 8.91 (d, 2H, Ha); 8.72 (t, 1H, Hb); 7.37–7.53 (m, 10H, Ph); 5.08 (s, 5H, Cp); 4.36 (s, 4H, CH2). C30H22FeO5S3.0.5CH2Cl2 calcd.: C, 55.7; H, 3.5; S, 14.6%. Found: C, 55.2; H, 3.4; S, 13.8%.
Preparation of CpFe(CO)2SCO-3,5-C6H3(C2O3R)2 (6)
A THF solution (50 cm3) containing 2.60 mmol of carboxylic acid and (1) (0.50 g, 1.14 mmol) was refluxed for 12 h. The solvent was removed under vacuum and the residue was re-dissolved in a minimum amount of THF and transferred to a silica gel column made up in hexane. An orange band was eluted by a mixture of THF/hexane (8:2 volume ratio), the collected products were re-crystallized from THF/hexane.
CpFe(CO)2SCO-3,5-C6H3(C2O3CF3)2 (6a)
Yield = 0.38 g, 56%. M.p = 176–178 °C. I.r. (KBr, cm−1): νC≡O = 2037 (vs); 1996 (vs), νC=O = 1728 (s); 1696 (s); νSC=O = 1608 (m). 1H-NMR (Acetone-d 6): δ 8.95 (d, 2H, Ha); 8.77 (t, 1H, Hb); 5.34 (s, 5H, Cp). C20H8FeO9SF6.2THF calcd.: C, 45.0; H, 3.3; S, 4.3%. Found: C, 44.9; H, 3.2; S, 4.1%.
CpFe(CO)2SCO-3,5-C6H3(C2O3CCl3)2 (6b)
Yield = 0.31 g, 39%. M.p = 187–189 °C. I.r. (KBr, cm−1): νC≡O = 2036 (vs); 1995 (vs), νC=O = 1728 (s); 1697 (s); νSC=O = 1607 (m). 1H-NMR (Acetone-d 6): δ 8.94 (d, 2H, Ha); 8.75 (t, 1H, Hb); 5.33 (s, 5H, Cp). C20H8FeO9SCl6.2THF calcd.: C, 40.2; H, 2.9; S, 3.8%. Found: C, 40.0; H, 2.8; S, 3.6%.
Crystallographic analysis of CpFe(CO)2SCO-3,5-C6H3(COCl)2, (1)
A single crystal suitable for X-ray structure determination of (1) was obtained by recrystallization from CH2Cl2/hexane mixture. Crystallographic data were as follows: C16H8Cl2FeO5S, formula weight: 439.03, space group P2(1)/n with a = 6.9492(4) Å, b = 20.7699(14) Å, c = 11.9657 Å, β = 102.280o, V = 1687.45(16) Å3, ρcalc = 1.728 g/cm3 and Z = 4. The measurements were collected at 183(2) K on a Kappa CCD diffractometer with a graphite monochromator (λ = 0.71073 Å). There were 11154 independent reflections with 3834 observed reflections (>2σ(I)). The structure was solved by direct methods using SHELXS97 (Sheldrick, 1997) [20] and difmap synthesis using SHELXTL96 (Sheldrick, 1996) [21]. All non-hydrogen atoms were fully refined with anisotropic thermal parameters. The refinement coverage to R1 = 0.0635 and wR2 = 0.1527.
Results and discussion
Synthesis and characterization
The reaction of (μ-S x )[CpFe(CO)2]2 with the trifunctional acid chloride 1,3,5-C6H3(COCl)3 can be controlled to give either the mono-, bi- or tri-iron complexes shown in Scheme 1. The organo-iron chloride, CpFe(CO)2Cl was obtained as a byproduct of these reactions, separated from the products by column chromatography and identified.

Preparation of CpFe(CO)2SCO-3,5-C6H3(COCl)2 (1), [CpFe(CO)2SCO]2-5-C6H3COCl (2) and 1,3,5-[CpFe(CO)2SCO]3 (3)
Complexes (1), (2), and (3) are stable as solids and are also stable in solution. The structures of these complexes were identified by spectroscopic techniques (i.r. and 1H-NMR) as well as by elemental analysis. Their i.r. spectra show 2 bands for the terminal carbonyl groups in the ranges of 2047–2044 and 2003–2000 cm−1, which are comparable to those reported for the corresponding thiocarboxylates CpFe(CO)2SCOR [9, 10]. The bands are shifted to lower wave-numbers as the number of the iron fragment increases, consistent with increasing electron density around the metal centre. The spectra show also a medium band in the range of 1606–1612 cm−1 for the thiocarboxylate-carbonyl group. For (1) and (2) the presence of the acid chloride group is confirmed by the presence a band at 1764–1761 cm−1 [16]. The 1H-NMR spectra of (1)–(3) exhibit a singlet in the range of 5.03–5.09 ppm for the Cp-protons. This chemical shift range is similar to that observed for the thiocarboxylate (4.98–5.13 ppm) [9, 10] and thiocarbonate (4.98–5.12 ppm) analogues [12]. The protons of the phenyl ring appear as two sets for (1) (doublet and triplet) and for (2) (two broad singlets) and as one singlet for (3).
Complex (1) was reacted with amines, thiols, and carboxylic acids to give the substituted complexes CpFe(CO)2SCO-3,5-C6H3(CONR1R2)2 (4), CpFe(CO)2SCO-3,5-C6H3(COSR)2 (5), or CpFe(CO)2SCO-3,5-C6H3(CO2COR)2 (6) respectively as shown in Scheme 2.

Reactions of CpFe(CO)2SCO-3,5-C6H3(COCl)2 (1) with amines, thiols or carboxylic acids
Compounds (4), (5), and (6) were characterized by i.r., 1H-NMR spectroscopy and elemental analysis. The i.r. spectra of each of these complexes exhibit two strong bands in the ranges of 2040–2036 cm−1 and 1996–1985 cm−1 for the terminal carbonyl group coordinated to the iron centre. These ranges are shifted to lower wave-numbers compared to the starting complex (1). The spectra contain a medium band in the range of 1611–1590 cm−1 for the thiocarboxylate-carbonyl ligands. Additionally, the carbonyl group of the amide or thio-ester groups appears as a medium band in the ranges of 1647–1680 cm−1 or 1608–1611 cm−1, respectively. The corresponding carboxylato ligands show two bands in the range of 1697–1696 and at 1728 cm−1. These ranges are found in accordance to the electronegativity of the N(R1)R2, SR or OCOR groups. 1H-NMR spectra of (4), (5), and (6) show singlet peak in the range of 4.90–5.34 ppm for the cyclopentadienyl protons and the expected patterns of the aromatic protons.
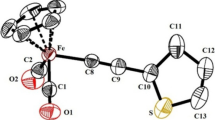
Crystal structure of (1)
The molecular structure of CpFe(CO)2SCO-3,5-C6H3(COCl)2 (1) is shown in Fig. 1. Selected bond distances and the bond angles of (1) are shown in Table 1. The structure of (1) is a typical piano stool with three legs in which the cyclopentadinyl ligand is bonded to iron in an η5-fashion. The Fe–C(Cp) (average = 2.093 Å) and Fe–C(CO) (1.764(7), 1.786(6) Å) bond lengths of (1) are similar to those found in CpFe(CO)2-containing complexes [8–14]. The Fe–S bond distance in (1) of 2.2529(14) Å is comparable to that found in CpFe(CO)2SX complexes [X = SO2CCl3 (2.2803(13) Å), CO2Et (2.2675(10) Å), CO-2-C6H4NO2 (2.266(1) Å), C(S)O-C6H4Cl (2.2765(5) Å)] [9–12]. The C–O bond lengths of the ketonic carbonyl group of the thiocarboxylate ligand (1.216(2) Å) are very comparable to those found in thiocarboxylate complexes [9, 10]. The corresponding bond lengths of the free acid chloride groups are somewhat different from each other (1.391(8), 1.181(7)). This might be due to some disorder of one of these groups. The angles around the iron center (S–Fe–C15, S–Fe–C16), C16–Fe–C15) are around 90º, consistent with a pseudo-octahedral structure. The sulfur atom in this complex has a sp3 hybridization as predicted from the Fe–S–C1 angle. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC No. 711242.

ORTEP drawing of CpFe(CO)2SCO-3,5-C6H3(COCl)2 (1) at 30% probability
References
Patai S, Rappoport Z (eds) (1986/1987) The chemistry of organic selenium and tellurium compounds, vols 1 and 2. Wiley, New York
Murray SG, Hartley FR (1981) Chem Rev 81:365
Gysling HJ (1982) Coord Chem Rev 42:133
Ng MT, Vittal JJ (2006) Inorg Chem 45:10247
Jacob JH, Khalil AM, Muslat AO (2004) J Carcinog 3:1477
Muslat AO, Jibril I, Abussaud M, Abd-Alhadi E, Hamadeh Z (2002) J Appl Organomet Chem 16:44
Delgado JE, Donnadieu B, Hernandez E, Zamora PF (2004) J Organomet Chem 689:552
El-Hinnawi MA, Aruffo AA, Santarsiero BD, McAlister DR, Shomaker V (1983) Inorg Chem 22:1585
El-Hinnawi MA, Ajlouni A (1987) J Organomet Chem 332:321
El-Hinnawi MA, Ajlouni A, Abu-Nasser JS, Powell AK, Wahrenkamp H (1989) J Organomet Chem 359:79
El-khateeb M, Obidate T (2001) Polyhedron 20:2393
El-khateeb M, Asali KJ, Lataifeh A (2003) Polyhedron 22:3105
El-khateeb M, Asali KJ, Lataifeh A (2006) Polyhedron 25:1605
El-khateeb M, Roller A (2007) Polyhedron 26:3920
El-khateeb M, Goerls H, Weigand W (2006) Inorg Chim Acta 360:705
Jibril I, Ali AK (1997) Indian J Chem 36A:987
Jibril I, Ali AK, Omar JT (1997) Polyhedron 16:3327
Jibril I, Abd-Alhadi EH (2000) Indian J Chem 39A:1055
Jibril I, Abd-Alhadi EH, Hamadeh Z (2000) Trans Met Chem 25:407
Sheldrick GM (1997) SHELXS-97, program for the solution of crystal structures. University of Göttingen, Germany
Sheldrick GM (1996) SHELXL-96, program for the refinment of crystal structures. University of Göttingen, Germany
Acknowledgments
The financial support (grant no. 42/2007) from the Deanship of Scientific Research, Jordan University of Science and Technology is gratefully acknowledged. ME thanks Alexander von Humboldt foundation for equipment donation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
El-khateeb, M., Asali, K.J., Jibril, I. et al. Multifunctional iron thiocarboxylate complexes: synthesis, reactivity and structure of CpFe(CO)2SCO-3,5-C6H4(COCl)2 . Transition Met Chem 34, 419–424 (2009). https://doi.org/10.1007/s11243-009-9211-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-009-9211-2