
Abstract
The transport and retardation of radioactive elements in hyper-alkaline conditions of radioactive waste repositories is a challenging field that is still poorly understood. In this study, the transport and attenuation of uranium in a column experiment was modelled by considering kinetic reactions, advection–dispersion and chemical/physical retardation processes. The modelling was first performed for three alluvium samples from Yucca Mountain in circumneutral pH to moderately alkaline conditions. Sorption of uranyl (\({\mathrm{UO}}_{2}^{2+} ({U}_{\mathrm{VI}})\)) was found to strongly depend on the surface complexation model assumed, with no significant removal of \({U}_{\mathrm{VI}}\) by precipitation or ion exchange process. The surface/edge site reaction of Al-hydroxyl group in kaolinite was shown to have a high affinity for uranyl adsorption, while the hydrous ferric oxide edge on hematite adsorbed most of the uranyl ions. The model was then used to interpret uranium transport in a laboratory column filled with Hollington sandstone under hyper-alkaline (pH 13) conditions. The simulation results show that uranium adsorption on the Al-hydroxyl edge of kaolinite exceeds adsorption by the calcium silicate hydrate phase. This result may reflect the lack of surface complexation parameters for calcium silicate hydrate minerals. Hence, further studies are required in the field of surface complexation reactions for calcium silicate hydrate phases.
Graphical Abstract

Article Highlights
-
Sorption of uranyl (\({\mathrm{UO}}_{2}^{2+} ({U}_{\mathrm{VI}})\)) was found to strongly depend on the surface complexation model, with no significant removal of U_VI by precipitation or ion exchange process.
-
The aluminol surface edges in kaolinite were shown to have a higher affinity for uranyl adsorption, while the hydrous ferric oxide edge on hematite adsorbed most of the uranyl ions.
-
Uranium adsorption on the aluminol edge of kaolinite exceeds adsorption by the C-S-H phase. This result may reflect the lack of surface complexation parameters for C-S-H minerals.
Similar content being viewed by others
Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
1 Introduction
Uranium is a radioactive and toxic element which, besides its natural occurrence, can be distributed to the environment through human activities such as oil and gas production, mining processes, and the nuclear industry (Campos et al. 2011; Chandrajith et al. 2010; Merkel and Hasche-Berger 2006, 2008; Ricka et al. 2010). Those industrial processes can enrich the radionuclides activities of uranium and its decay products known as NORM (naturally occurring radioactive material) (Protection 2003). In an oxidising environment the most stable and soluble form of uranium is (Uranyl \({\mathrm{UO}}_{2}^{2+} ({U}_{\mathrm{VI}})\)), which can react with sulphates, carbonates and nitrates to form complex phases with altered speciation and transport characteristics (Zielinski et al. 1997; Meinrath 1998; Tutu et al. 2009; Ribera et al. 1996; Grenthe et al. 1992). In subsurface applications the aqueous concentration and mobility of \({U}_{\mathrm{VI}}\) ions without a complexing ligand are widely recognised to be controlled by sorption at mineral surfaces (Barnett et al. 2002; Prikryl et al. 2001; Turner et al. 1996a; Thomson et al. 1986; Payne and Waite 1991). The interaction between dissolved radionuclides and minerals in the host rock (sorption at solid/solution interface) is critical when evaluating the immobilisation of radionuclides.
Uranyl sorption on mineral surfaces can involve multiple processes (binding sites), such as absorption, adsorption, ion exchange and edge/surface complexation. These processes can significantly influence the transport of pollutants and radionuclides in soils and rocks. For example, adsorption refers to the adherence of ions to the solid surface, while absorption implies uptake of ions into the solid. Conversely, ion exchange involves the substitution of one ion for another over the solid surface (Appelo et al. 2005). In earlier studies, sorption models use the Kd (distribution coefficient) approach, which sums all interactions between the solid/water interaction (Ticknor 1994; Missana et al. 2008; Glynn 2003; Davis et al. 2004; Curtis et al. 2004).
In recent years, a mechanistic approach has been developed based on several absorption/adsorption laboratory experiments conducted on site-specific clay materials to generate a surface complexation model (SCM) that can describe the migration behaviour, stability constants and stoichiometry of different metal/metalloid ions reactions. The SCM can describe and simulate sorption between aqueous uranyl and the surface of clay minerals (sorbent) by fitting the thermodynamic data from other referenced studies under different chemical conditions for various aqueous (uranium) species (Davis 2001; Davis et al. 2001; Bradbury and Baeyens 2005; Zachara and McKinley 1993; Kim 2001; Davis et al. 2002; Koretsky 2000). Through the mass action equation, the surface complexes formed can be then integrated within the reactive transport models (Curtis et al. 2006; Papini et al. 1999).
In high-heat-generating waste-disposal concepts, swelling clays (e.g. bentonite) will be widely used, in part, because of their capability to sorb heavy metals. These clays can sorb heavy metals due to their high osmotic swelling capacity and large specific surface area (Barnett et al. 2000, 2002; McLing 1998). Moreover, clay minerals can simultaneously maintain a fixed, usually negative charge within the particle structure which encourages ion exchange within the interlayer’s spaces, while the variable charge is generated at the clay edges, which may be positive or negative depending on the pH value. Therefore, both cations and/or anions can potentially be sorbed to neutralise the structural charge. Three common surface complexation models are usually used to fit the experimental results: the double/diffuse layer model (DLM), the triple-layer model (TLM), and the constant capacitance model (CCM) (Waite et al. 1994; Turner et al. 1998, 1996a; Hiemstra et al. 1989; Fletcher and Sposito 1989). Due to the low hydraulic conductivity of clay soils, most investigations have been carried out in static systems using batch experiments with powdered material left in contact for periods of time with cement leachate (Claret et al. 2002; Ramı́rez et al. 2002; Fernández et al. 2009; Takahashi et al. 2007). Fewer studies have performed dynamic experiments using diffusion set-ups or advective experiments (Adler et al. 1999).
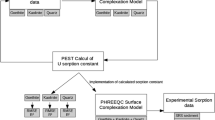
The aim of this work is to establish a reactive transport model with a series of kinetic and equilibrium reactions and incorporate uranyl aqueous speciation and surface charge measurements that control uranium removal from the solution at room temperature and fixed pH value. The modelling results were obtained using the PHREEQC geochemical code. In past numerical modelling attempts, when analysing nuclear waste repositories, most authors assumed the host rock's key properties to be constant. Though, in this research, an adequate approach was used to study the effects of variable porosity, sorption, reactive surface area and pore volume on improving the modelling of rock alteration in the system.
2 Experiment
In this study, a reactive transport model combined with a diffuse layer model (DLM) was used to analyse the adsorption of uranyl ions from aqueous solution in column experiments. The objective was to determine the key parameters that control the sorption of this radionuclide at the solid–solution interface in the host rock of a candidate geological repository. The model was first applied to three alluvium samples from Yucca Mountain that are rich in smectite clay and equilibrated with circumneutral to moderately alkaline solutions. Once the model was validated against the experimental results, it was then used to interpret the transport of uranium in a highly alkaline solution through Hollington Sandstone, a material mainly composed of silicate minerals with minor amounts of kaolinite and hematite. Hollington Sandstone has been used for a mineral analogue for host rocks of UK ILW/LLW disposal.
2.1 Yucca Mountain Alluvium
The Yucca Mountain project in Nevada has been proposed as a geological repository site for the disposal of radioactive wastes. A series of sorption studies (batch, column, and in situ field transport) have been conducted on the alluvium soil to demonstrate its capability to retard the transport of radionuclides in the subsurface.
Three flow-through column experiments were conducted in the early 2000’s with continuous flow and fully saturated conditions using alluvium from the southern area of Yucca Mountain (Scism 2005). The columns were filled with alluvium that had been wet sieved to retain the 2000–75 µm size fraction. The experimental parameters for the three column tests are shown in Table 1. The key mineral phases in the alluvium were identified and quantified using X-ray diffraction (XRD) following the procedure published by Chipera and Bish (2002). Table 2 shows the mineralogy of the alluvium in each of the three columns.
The background solutions (groundwater) used in the experiment were taken from water wells drilled close to each alluvium sample wells. The water chemistry and ion concentration data for each column are shown in Table 3. (A concentrated UO2(NO3)2 solution was diluted in the groundwaters to produce a uranium tracer solution with a concentration of 1 × 10–6 M. The columns were initially allowed to equilibrate with groundwaters and then the uranium tracer solution was injected at an initial flow rate of 10 ml/h, which decreased to 3 ml/h as the experiment continued. The fluid sample at the column outlet was placed in a reciprocating shaker, followed by centrifugation to separate the solids. Simultaneously, control samples that contained only a tracer without alluvium were also shaken and centrifuged to estimate the amount of uranium sorbed to the tube walls. Aqueous uranium concentrations were determined by Liquid Scintillation Counting (LSC) (the concentration in the tracer solution was measured before and after the experiment). Further details of the experimental procedure and protocol are provided in (Scism 2005).
2.2 Hollington Sandstone
In this experiment, uranium in a high-alkaline pH solution will migrate through the sandstone sample and alter the solution composition due to the dissolution of primary silicate minerals in the rock. Later, the solution will be equilibrated by the precipitation of calcium silicate hydrate (C-S-H) phases or C-(Al)–(K)-S-H phases whenever Al and K ions are present in the solution.
Hollington sandstone is part of the Lower Triassic Bromsgrove Sandstone Group. It is mainly composed of silicate minerals with minor quantities of clay and hematite. Its porous nature allows the study of rock–cement–leachate reactive transport reactions in strata with a similar mineralogical composition to that a proposed geological radioactive waste repository. Column experiments on crushed Hollington sandstone permeated with a synthetic young cement leachate at a temperature of 50 °C (Small et al. 2016) are used to predict the transport of aqueous UVI in Hollington sandstone. In this experiment, the aim was to investigate the progression of uranium sorption on the surfaces of the C-S-H phases in an alkaline environment. In fact Langmuir (1997) concluded that hyper-alkalinity conditions can encourage the sorption of uranyl (\({\mathrm{UO}}_{2}^{2+})\) cations due to the availability of neutral and negatively charged surfaces. Furthermore, the C-S-H phases have a high surface area and high retention capacity for radionuclide migration, especially for uranyl (VI) cations due to poor crystallisation (Johnson 2004; Gougar et al. 1996; Ma et al. 2019). Meanwhile Tits et al. (2011) found that C-S-H phases can absorb uranyl cations by forming inner-sphere surfaces (surface complexation) with the silonal edges. Tables 4 and 5 show the mineralogical composition of the sandstone sample used in the experiment and the chemical composition of the synthetic young cement leachate, respectively. Further details of the experimental set-up and procedures are provided in Baqer et al. (2021).
3 Modelling Approach
The geochemical speciation code PHREEQC (version 3.6.1) (Parkhurst and Appelo 2013) was used to simulate the transport and sorption of uranium in the column experiments. PHREEQC is a geochemical software developed by the United States Geological Survey. It includes multiple databases users can use or modify and apply to various aqueous models, including geochemical and industrial applications. Adding to its geochemical modelling tool, it can simulate 1D transport in porous media, making it applicable for the resolution of this research. The software comprises a vast database and geochemical equations describing the equilibria or kinetic interactions of aqueous solutions with solids, gases, and minerals, with the advantage of easily adding or modifying necessary reactions if needed.
In PHREEQC, chemical reactions are integrated based on a built-in or user-defined rate expression, with the possibility of simulating time-dependent geochemical reactions that depend on solute temperature, pH and saturation ratios of minerals. Regarding kinetics reactions, the built-in interpreter contains rate expression (which can also be user-defined) with a Runge–Kutta scheme that solves the differential equations and change the solution speciation within a specific time interval. In addition, the user can define an error tolerance which allows the software to reduce the time interval of the simulation. This scheme effectively achieves equilibrium when multiple kinetic reaction rates are defined, and the rate of each reactant is changed during the reaction.
In this work, the kinetics of dissolution and precipitation, equilibrium reaction and porosity evolution have been incorporated in the coding. A hybrid mixed kinetic-equilibrium approach was used to overcome the shortage and uncertainties of some kinetic reaction parameters (e.g. reactive surface area, specific dissolution/precipitation kinetics) (Chen and Thornton 2018; Van der Lee 1998; Bethke 1996). The LLNL thermochemical database was used as a starting point to compile the details of different chemical reactions (aqueous reactions, mineral dissolution/precipitation, surface complexation, ion exchange) (Delany and Lundeen 1990). This database seemed to be the best option available since it has the thermodynamic information (reactions and equilibrium constants) for variety of minerals and aqueous species, especially carbonate minerals that have major influence on key kinetic reactions and putative reactive pathways that controlled the primary mineral dissolution, mineral evolution, secondary mineral formation and column effluent evolution. Uranium thermodynamic data were obtained from the “Second update on the chemical thermodynamics of uranium, neptunium, plutonium, americium and technetium” published by the OECD Nuclear Energy Agency (Grenthe et al. 2020). Surface complexation and ion exchange reaction were obtained from the literature and then added manually to the database.
3.1 Equilibrium/Kinetic Modelling of Dissolution and Precipitation
The sorption of a soluble radionuclide can be highly affected by dissolution/precipitation processes which occur at mineral surfaces. In some circumstances, the dissolution of some mineral phases can result in the nucleation of clay minerals or hydrous oxides as a coating on the mineral surfaces, which will increase the number and density of sorption sites (Ticknor 1994). Also, precipitation process can sometimes involve radionuclides, removing them from the solution by formation of a secondary phase. The alluvium soil sample contains 11 minerals with different percentages in each column. To ensure consistency in the results and to highlight the sorption effect of each mineral, all the mineral phases were included in the modelling except for "Opal-CT" since it is only found in column 1. The higher percentage minerals (quartz, plagioclase, k-feldspar, clinoptilolite, cristobalite, and smectite) were modelled kinetically while the rest were included in the equilibrium reaction. Equation 1 was used to calculate the rate of mineral dissolution/precipitation reactions (Appelo et al. 2005):
where R is the overall dissolution/precipitation rate (mol L−1 s−1), k is the specific dissolution/precipitation rate (mol m−2 s−1), A is the effective surface area of the mineral (m2 g−1), V is the pore volume (L), M is the moles of solid at a given time, M0 is the initial moles of solid, and \((\mathrm{IAP}/K)\) is equal to the saturation ratio (SR) value of the mineral where IAP is the Ion Activity Product and K is the equilibrium constant. In the above equation, the molar concentration \((M/{M}_{0})\) has n power, which is a function of the initial crystal grain size distribution that affects the solid dissolution rate. A value of 2/3 is assumed in this work (this is a typical value for uniformly dissolving cubes or spheres in the monodisperse population) (Appelo et al. 2005). Table 6 shows the specific kinetic rates and the reactive surface areas for the kinetically modelled minerals. To investigate the effect of precipitation on uranium removal from solution, four uranium mineral phases were added in the equilibrium reactions (In PHREEQC) during the simulation of the Yucca Mountain and Hollington sandstone experiments (CaUO4, Uranophane, Becquerelite, Rutherfordine). These phases were chosen based on the common stable uranium minerals found in the literature (Atkins et al. 1988; Wronkiewicz et al. 1992; Gorman-Lewis et al. 2008; Felipe-Sotelo et al. 2017). Also, Tobermorite-14A, CSH-gel, \({\mathrm{CaUO}}_{4}\), and \({\mathrm{Na}}_{2}{\mathrm{U}}_{2}{\mathrm{O}}_{7}\) were included in the modelling of the Hollington sandstone experiment as a possible precipitating phase in a high-alkaline environment.
3.2 Dynamic Porosity and Reactive Surface Area
In the model, a dynamic porosity was used to emulate changes in the porosity that result from dissolution/precipitation reactions. Equation 2 shows the correlation between mineral mass and surface area during flow of the tracer solution in the column.
where mt is the total mass of the rock sample, \({A}_{0}\) is the initial surface area (\({\mathrm{m}}^{2} {\mathrm{g}}^{-1}\)), and Xi is the mass fraction of each mineral in the rock sample (Beckingham et al. 2016). The surface area is then coupled with the evolved porosity through Eq. (3) (Lichtner 1988):
where \({A}_{r}^{t=0}\) is the reactive surface area of the mineral at the initial porosity \(({\phi }_{t=0})\), and the mineral kinetic rate can be calculated using Eq. (4). The changed porosity can then again be calculated from the volume of the dissolved/precipitated moles of mineral. Further details concerning the mathematical derivation of the dynamic porosity model are provided in Baqer et al. (2021).
3.3 Transport Process
The transport of the uranium tracer solution through the column was modelled using the one-dimensional (1D) mixed cells concept with flux type boundary conditions and a mass entering the column per unit time. This is applicable to a laboratory column with a diameter much smaller than the column length (Table 1) because of the near-zero concentration gradient at the column end (Appelo et al. 2005). The column was divided into ten equal length cells along the flow path, with additional cells at each end for inflow and outflow. The simulation was performed for a series of time steps in which each step is equal to the time taken for the pore solution in one cell to move into the next cell. The simulation was performed for 700 h, and the advection–dispersion process through the column was modelled using the equation of advection–reaction–dispersion described below (Eq. 5) (Nardi et al. 2014):
where \({C}_{i}\) is the total concentration of component i in water (mol Kg w−1), \(t\) is time (s), \(x\) is the distance (m), \({D}_{L}\) is hydrodynamic dispersion coefficient (m2 s−1), \({\nu }_{im}\) is the stoichiometric coefficient of component i in kinetic reactant m (dimensionless), \({R}_{m}\) is the overall dissolution/precipitation rate of kinetic reactant m (mol Kg w−1 s−1), and n is the number of kinetic reactants. For each time step, advection was modelled first, followed by dispersion, then finally the chemical reactions were modelled, so the solution composition in each cell could be updated. The reader is referred to Baqer et al. (2021) for more details concerning the transport model.
In the Yucca Mountain experiment, different flow rates for the pulse solution were used for each column. Based on the time step in each column, the velocity will vary, and therefore each column will have a different dispersivity value. Table 7 shows the modelling input parameters for the transport process simulation in the three columns.
3.4 Uranium Speciation
The aqueous speciation of the uranyl (\({\mathrm{UO}}_{2}^{2+}\)) ion is strongly influenced by pH, as it determines the stability of different ion complexes and the distribution of the surface sites, which will consequently shape the sorption mechanism (Chisholm-Brause et al. 1994). For example, where CO2 is present, neutral uranyl–carbonate species start to form at pH values > 6.5, and negatively charged uranyl–carbonate species dominate in alkaline conditions (Nair et al. 2014). Formation of negatively charged complexes will affect adsorption to partially ionised surface/edges sites (Guimarães et al. 2016; Ticknor 1994). Table 8 lists equilibrium constants for uranium aqueous speciation reactions at 25 °C and pCO2 = 10–3.5 atm (Grenthe et al. 2020).
3.5 Sorption
Smectite contains several types of sorption site with high affinity for aqueous uranium complexes (Barnett et al. 2000; Davis et al. 2004), and kaolinite contains aluminol surface/edges that also have high affinity for uranium species (Borovec 1981; Kohler et al. 1992). Furthermore, the presence of iron ions in hematite can further enhance uranium sorption, hence it has also been included in the surface complexation model (Davis et al. 2004; Dong and Wan 2014). Most published literature model sorption with cation exchange and surface complexation mechanisms (Turner et al. 1996a; Missana et al. 2008; Bachmaf and Merkel 2011; Korichi and Bensmaili 2009; Nair et al. 2014). The advance in this study is that it models both ion exchange and surface complexation as multi-site complexation phenomena at the clay–water interface (Fig. 1).

Multi-site complexation model of uranium on smectite clay
3.5.1 Ion Exchange
Uranyl sorption at smectite interlayer cation exchange sites is included in the model. It assumed that these sites are initially occupied by either Ca2+ or Na+ cations [Na+ is readily displaced by \({\mathrm{UO}}_{2}^{2+}\), whereas Ca2+ has a similar affinity to interlayer cation exchange sites to uranyl (Tsunashima et al. 1981; McKinley et al. 1995; Hiemstra et al. 1989)]. The stoichiometry and equilibrium constants assumed for these cation exchange reactions are shown in Table 9. The site density (X−) is assumed to equal the cation exchange capacity (CEC), which for smectite equals 810 meq/kg (Appelo et al. 2005).
3.5.2 Surface Complexation
A surface complexation model (SCM) Guimarães et al. (2016), Dong and Wan (2014), Dangelmayr et al. (2017), Korichi and Bensmaili (2009), Nair et al. (2014) is used for uranium sorption onto various surface and edge sites on clay minerals to overcome the limitation of reactive solute transport using the average Kd value (laboratory calculated) approach (which depends on, and is limited to, a specific water composition) (Glynn 2003; Reardon 1981; Zhu 2003; Bethke and Brady 2000; Davis et al. 1978; Kent et al. 1988). The model assumes that the mineral surface includes a group of functional hydroxyl surfaces denoted as \((\equiv \mathrm{SOH})\), and that the sorption of uranium is highly dependent on the behaviour of these functional groups at surface and edge sites on clay minerals. Such sites are amphoteric, so protonation and deprotonation equilibrium reactions of these sites are included in the model (Missana et al. 2008)
where K1 and K2 are the equilibrium constants (Table 10).
In this study a double/diffuse layer SCM model was used, and the moles of each aqueous species are computed based on a constant thickness for the diffuse layer. Before applying the DLM for the adsorption of uranyl ions onto clay surfaces, several variables must be defined: (1) the chemical reaction at the clay–water interface, (2) the surface complexation equilibrium constant (Log k), and (3) surface site density and amount of available binding sites.
Usually, for smectite, the binding of uranyl aqueous complexes takes place on the aluminol and silanol edges, and on the aluminol edge site for kaolinite (Bachmaf and Merkel 2011; McKinley et al. 1995; Turner et al. 1996b; Zachara and McKinley 1993). The amount of edge/surfaces associated with surface complexation is usually 10–20% of the sorption sites (Anderson and Sposito 1991; Pabalan and Turner 1996; McKinley et al. 1995; Hiemstra et al. 1989). Table 10 shows the surface edge site reactions on aluminol/silanol hydroxyl groups included in the PHREEQC modelling to best fit the experimental uranyl sorption results. Dzombach and Morel (1990) recommend using 10% of the total specific surface area measured by the N2-BET method to account for the area of the crystallite surface edge sites, or site density value of 2.3 sites/nm2 for all minerals. Therefore, the total number of moles of binding site surfaces was calculated from the weight percentage and surface area of each mineral in the bulk sample of alluvium in the column. Note that the surface complexation reaction of carbonate complexes has been ignored in this study since they have high solubility.
4 Result and Discussion
4.1 Yucca Mountain Alluvium
The final composition of the effluent solution was calculated after a series of geochemical reactions combined with fluid flow and uranium sorption. The results were obtained by implementing multiple binding site ion complexation containing fixed and edge aluminol/silonal sites. Table 11 shows the predicted composition of the site groundwater after equilibration with mineral phases in Yucca Mountain alluvium and atmospheric carbon dioxide.
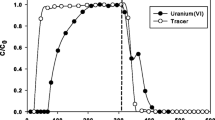
In all cases the initial breakthrough of uranium almost occurs at the same time as the HTO-tracer (Fig. 2). The modelling results describe the adsorption of uranyl well: the uranium concentration starts to increase in the existing fluid until it reaches a maximum concentration (C/C0) of 0.0078 mol L−1, 0.023 mol L−1, and 0.08 mol L−1 for columns 1, 2 and 3, respectively (Fig. 3). However, the highest value of the uranium concentration was still much lower than the pulse solution concentration (0.24 mol L−1), implying that uranium retardation happens simultaneously with the fluid flow.

Uranium and HTO-tracer breakthrough curves for Yucca Mountain alluvium columns 1, 2 and 3

Experimental and modelling results for uranium breakthrough curves in Yucca Mountain alluvium columns 1, 2 and 3
The simulated saturation indices of uranium phases (CaUO4, uranophane, becquerelite, rutherfordine, Fig. 4) indicate that they are undersaturated and the precipitation of solid uranium phases is unlikely to occur in the experiment. Therefore, precipitation can be excluded as a possible uranium removal mechanism in this experiment. This is usually the case since the nucleation kinetics of solid phases is considered to be very slow to affect uranium removal significantly (Dangelmayr et al. 2017). Consequently, the decrease in uranium concentration is due to the sorption of uranyl ions by ion exchange and surface complexation reactions only.

Saturation indices for uranium secondary phases in Yucca Mountain alluvium columns 1, 2 and 3
Figure 5 shows the number of moles of uranium adsorbed by cation exchange processes. As the experiment proceeds, the pulse solution is exposed more to the smectite mineral; thus, more uranyl is exchanged on the smectite binding sites. However, the amount of adsorbed uranyl is minimal throughout the experiment, which agrees well with other studies that highlight the dominance of ion exchange in the region of low pH only (McKinley et al. 1995; Zachara and McKinley 1993; Davis and Kent 2018). Therefore, uranium binding to a fixed site by ion exchange process is most likely not the dominant sorption mechanism in this model as well.

Number of adsorbed moles by cation exchange in Yucca Mountain alluvium columns 1, 2 and 3
Figure 6 illustrates the results of the four surface edges sites (\({\mathrm{AlOH}}_{\mathrm{Smectite}}\), \(\mathrm{SiOH}\), \({\mathrm{AlOH}}_{\mathrm{Koalinite}},\) \(\mathrm{FeOH}\)), which confirm the modelling approach for uranium sorption of binding to a variable charge site by surface complexation. As expected, uranyl ions form an inner surface complex on smectite, kaolinite and hydrous ferric oxide. When assessing the adsorption results of each surface site, column 3 shows the highest adsorption value in all four sites, followed by column 2 and finally column 1. This agrees well with the highest smectite and hematite weight per cent in column 3 and the lowest weight percentage in column 1. Moreover, from Table 11, column 3 has the highest ionic strength while column 1 has the lowest, which agrees with the results of other literature that shows a more elevated surface complexation potential with higher ionic strength (Chisholm-Brause et al. 2001; Hennig et al. 2002; Sylwester et al. 2000; Bauer et al. 2001; Korichi and Bensmaili 2009; Schindler et al. 2015; Turner and Sassman 1996).

Number of adsorbed moles for surface complexation model in Yucca Mountain alluvium columns 1, 2 and 3
The number of adsorbed moles on the aluminol edge \((\equiv \mathrm{AlO}-{\mathrm{UO}}_{2}^{+})\) is much higher (6 orders of magnitude) than the number of adsorbed moles on the silonal sites (\(\equiv \mathrm{SiO}-{\mathrm{UO}}_{2}^{+})\), which agrees well with higher uranyl affinity towards aluminol edges. This may be justified by the lower tendency of the (\(\equiv \mathrm{SiO}-{\mathrm{UO}}_{2}^{+})\) group to donate its oxygen and form inner-sphere surface species (Morris et al. 1994; Kowal-Fouchard et al. 2004). Even with the high weight per cent of smectite clay in the alluvium samples, it has the lowest sorption capacity for uranium. The aluminol surface edge sites in kaolinite have shown a higher affinity for uranyl adsorption, which is also reported in other studies (Borovec 1981; Kohler et al. 1992; Payne et al. 2004) for a pH range between 5–9. This can be related to the higher number of the exposed surface sites at the Al-octahedral sheet (Kaolinite has a 1:1 clay structure), which results in greater uptake of uranyl ions.
In all three columns and among the four surface complexes, the \(\equiv \mathrm{FeOH}\) site has the highest number of adsorbed uranyl moles. The same behaviour has also been reported in other studies (Dzombak and Morel 1991; Waite et al. 1994; Sherman et al. 2008; Hsi and Langmuir 1985), in which for the region of neutral and alkaline pH, a very small mass weight percentage \((\approx 1\%)\) of iron hydroxides (goethite, hematite, ferrihydrite) is a major sink for uranium. More importantly, (Liger et al. 1999) found that in the presence of hematite, uranium reduction through sorption can occur within hours at neutral pH; the reaction kinetics between the adsorbed uranium and ferrous iron is enhanced by hematite according to a first-order pseudo-kinetic law.
The distribution of uranium species in PHREEQC also shows the presence and dominance of \({\mathrm{Ca}}_{2}{\mathrm{UO}}_{2}{({\mathrm{CO}}_{3})}^{3}\), \({\mathrm{CaUO}}_{2}{({\mathrm{CO}}_{3})}_{3}^{-2}\) complexes. This is consistent with the view of several authors that neutral and negatively charged uranium–carbonate ions are unlikely to bond with the negatively ferric-hydroxide surfaces (Morrison et al. 1995; Geipel et al. 1998; Ho and Miller 1986). Moreover Fox et al. (2006), Dong and Brooks (2008), Stewart et al. (2010), Nair and Merkel (2011a), Nair and Merkel (2011b) found that the availability of Ca and Mg ions in the solution can also shift the aqueous speciation of uranium towards more stable (neutral) or negatively charged Ca–Mg-ternary complexes. The simulation results also indicate a general dissolution of smectite, clinoptilolite, and cristobalite in all three columns (Fig. 7), which will result in an increase in the pore volume of each column and hence the porosity (Fig. 8).

Saturation indices for smectite, clinoptilolite, and cristobalite in Yucca Mountain alluvium columns 1, 2 and 3

Predicted porosity evolution in the Yucca Mountain alluvium column experiments
4.2 Hollington Sandstone
The experiment was conducted in a CO2 free environment which is the usual case in a cementitious geological repository due to the interaction with cement-Ca and precipitation of calcite (CaCO3) (Disposal 2010; Vines and Lever 2013; Auroy et al. 2013). Therefore, in an oxidising environment, \({U}_{\mathrm{VI}}\) ions would be expected to be in the form of uranyl hydroxide complexes with limited solubility compared with the carbonate species (Tits et al. 2011; Bourdon et al. 2003).
The model simulations show that there is no significant precipitation of C-S-H or uranium composed phases (Fig. 9). Tobermorite-14A, CSH-gel, and \({\mathrm{CaUO}}_{4}\) are all in multiple cycles of precipitation and dissolution that cancel out the effect of both processes. Meanwhile, \({\mathrm{Na}}_{2}{\mathrm{U}}_{2}{\mathrm{O}}_{7}\) approaches but never reaches equilibrium. Therefore, no significant retardation is expected by the edge surfaces of the C-S-H phases or precipitation of uranium. This result is also represented in Fig. 10, which shows that in the case of no sorption, the uranium concentration at the column end reaches its initial injected concentration (0.24 mol L−1) and achieves a steady-state value. Conversely, in the full sorption model uranium breakthrough occurs simultaneously as in the previous case but with significant retardation of uranium.

Modelled saturation indices for Tobermorite-14A, CSH-gel, CaUO4, and Na2U2O7 in the Hollington sandstone experiment

Simulated uranium breakthrough curves in the Hollington sandstone experiment. The solid line represents the model with sorption kinetics, and the dotted line represents the model without sorption kinetics
As there was no significant precipitation of secondary C-S-H phases, an assumption was made to treat the smectite clay in the Hollington sandstone as a secondary C-S-H mineral. This assumption has also been made due to the lack of surface complexation parameters for C-S-H minerals. The modelled results for this assumption are shown in Fig. 11. Again, the results show that aluminol surface complexation on kaolinite dominates uranyl adsorption. So, even in high-alkaline conditions, it still overcomes the adsorption of both aluminol and silonal edges on the C-S-H phase. But it is worth mentioning that (Harfouche et al. 2006; Evans 2008; Pointeau et al. 2004) have experimentally found that uranyl \(({\mathrm{UO}}_{2}^{2+})\) can substitute for Ca2+ in the interlayer of C-S-H phases. Unfortunately, this reaction could not be modelled in this study due to a lack of the relevant surface complexation and ion exchange parameters.

Number of adsorbed moles on kaolinite and C-S-H phase by surface complexation model in Hollington sandstone experiment
In the study of Korichi and Bensmaili (2009), the authors also found that increasing the uranium concentration in the pulse solution will decrease the adsorption percentage, and in the case of low initial concentration, uranyl ions will have high mobility at the beginning before adsorption occurs. This behaviour was perfectly captured in the modelling results of this study, where a small peak in uranium concentration occurs at the beginning of the experiment before it drops down once the adsorption reaction dominates (Figs. 10, 11).
The applied uranium transport and sorption methods in this study provide insight into the key parameters that have the most significant impact on minimising uranyl mobility in a geological repository. It also indicates the required data needed to construct a consistent and reliable model for the reactive transport and sorption process of radionuclide migration in the geochemical application. Finally, the result of this study shows that the prediction of uranium migration is highly site-specific as it depends on the mineralogy and the geochemistry of the geosphere.
5 Conclusion
Modelling uranium retardation is still a challenging task with all the variables included in the process, such as characterisation of the sorbent material (mineral type and composition, density of sorption sites, surface area), solid–solution ratio, solution composition, carbon dioxide presence, and pH value. Usually, in the region of low pH and ionic strength, uranium is retarded by forming an outer sphere complex with the fixed charge surface (ion exchange process). In contrast, at neutral pH and high ionic strength, the formation of inner-sphere complexation due to amorphic surface/edge sites occurs (surface complexation) controls the sorption process. Precipitation of uranium phases is most likely to be neglected in modelling experimental uranium sorption since the nucleation kinetics of solid phases is considered very slow to affect uranium reduction significantly. In this study, reactive transport with a double-layer surface complexation model has been implemented to model uranium sorption in a column experiment with a dynamic transport process. The model was first applied to an alluvium soil sample from Yucca Mountain in a neutral pH environment. The result shows that the aluminol surface edge sites in kaolinite have a higher affinity for uranyl adsorption than both aluminol and silonal edges in smectite clay.
Meanwhile, hydrous ferric oxide edge \((\equiv {\text{FeOH)}}\) on hematite adsorbed most of the uranyl ions. Subsequently, the model was applied to Hollington sandstone in a high-pH environment. The modelling simulation shows that there is no significant precipitation of C-S-H or uranium composed phases. Tobermorite-14A, CSH-gel, and \({\text{CaUO}}_{{\text{4}}}\) are all in multiple cycles of precipitation and dissolution that cancel out the effect of both processes. Meanwhile, \({\text{Na}}_{2} {\text{U}}_{2} {\text{O}}_{7}\) is almost close to equilibrium but never reached. Like Yucca Mountain, the aluminol surface edge sites in kaolinite overcome the adsorption of both aluminol and silonal edges on the C-S-H phase. It is worth mentioning that the surface complexation reaction on C-S-H phases was not modelled adequately due to the lack of modelling parameters and further studies are needed in this field.
5.1 Limitations of the mathematical model
Whether an experiment can be modelled correctly by numerical methods depends on (1) the experimental data obtained, (2) the modelling tools and methods, (3) the theoretical analysis, and (4) the relationship between the experiment and modelling (some parameters might be important for modelling, however, is not necessary for the experiment). One of the challenges of modelling this kind of experiment was the unknown parameters caused by the different scientific focus for the experiment. For example, one of the major uncertainties in modelling evolving porous media arises from the uncertainty in identifying the value of the reactive surface area, which has a first-order effect on the timing and the level of dissolution/precipitation reactions. Commonly, the rate of dissolution in the experiment is 1–3 orders of magnitude higher than in the natural system. This can be explained by the difference in reactive and total surface area between natural and experimental systems (White and Peterson 1990; White and Brantley 2003; Velbel 1993).
Another area which requires improvement is the development of strategies for coupling fluid flow and reactive transport. Global implicit, sequential-iterative or sequential non-iterative schemes are often used to solve most reactive transport problems (Steefel et al. 2015). Irrespective of the numerical algorithm used, permeabilities and reactivities are updated with a time lag. Although this is not often done for porosity and diffusion coefficients, it does suggest that there is the possibility of errors in small mass balance at each time step. In addition, the simulation of an evolving porous media may be complicated when attempting to update the relevant parameters of the media as the evolution proceeds because of the interactions between flow and transport. In many published models, both multi-component and groundwater flow problems are solved sequentially. This does not apply, however, to evolving porous media because the evolution of porosity affects the flow equation through the storage term and modifies the permeability. Decoupling flow and reactive transport should help avoid numerically induced minor mass balance errors. Interestingly, there have been recent attempts to recouple both multiphase flow and reactive transport through the variation of porosity (Seigneur et al. 2018).
Processes occurring at the pore and macroscopic scales cannot be adequately described by porosity evolution alone. Unfortunately, the rigorous mathematical frameworks required for such an integrated approach are not yet available. Instead of these formulations, the geometrical organisation of the pore scale may be adequate. For instance, fluid–skeleton interactions could be described based on the surface area as pathways with high tortuosity. This is known to have lower transport properties caused by increased surface area.
Finally, In PHREEQC, the 1D advection–dispersion transport is calculated by an explicit finite difference algorithm. Sometimes, when the grid is coarse, the algorithm will compute numerical dispersion, which can be large or small in magnitude depending on the modelled reactions. Likewise, for the sorption reaction, the software has some uncertainties in defining the composition of the sorbed species, the number of sites, and the surface area. Therefore, most models of surface complexation applications depend on experimental data for specific materials from the studied site. PHREEQC also has a limited capability to integrate a system with complex kinetic reactions due to its explicit time simulation method (MacQuarrie and Mayer 2005).
Availability of Data and Materials
Not applicable.
References
Adler, M., Mäder, U., Waber, H.N.: High-pH alteration of argillaceous rocks: an experimental study. Schweiz. Mineral. Petrogr. Mitt 79, 445–454 (1999)
Anderson, S.J., Sposito, G.: Cesium-adsorption method for measuring accessible structural surface charge. Soil Sci. Soc. Am. J. 55, 1569–1576 (1991)
Appelo, C., Postma, D.J.B.: Geochemistry, groundwater and pollution. CRC, Roterdam (2005)
Atkins, M., Beckley, A.N., Glasser, F.P.: Influence of cement on the near field environment and its specific interactions with uranium and iodine. Radiochim. Acta 44–45, 255–262 (1988)
Auroy, M., Poyet, S., Le Bescop, P., Torrenti, J.: Impact of carbonation on the durability of cementitious materials: water transport properties characterization. In: EPJ Web of Conferences, 2013, p. 01008. . EDP Sciences (2013)
Bachmaf, S., Merkel, B.J.: Sorption of uranium(VI) at the clay mineral–water interface. Environ. Earth Sci. 63, 925–934 (2010)
Bachmaf, S., Merkel, B.J.: Sorption of uranium (VI) at the clay mineral–water interface. Environ. Earth Sci. 63, 925–934 (2011)
Baqer, Y., Bateman, K., Tan, V., Stewart, D.I., Chen, X., Thornton, S.F.: The influence of hyper-alkaline leachate on a generic host rock composition for a nuclear waste repository: experimental assessment and modelling of novel variable porosity and surface area. Transp. Porous Media 140, 559–580 (2021)
Barnett, M., Jardine, P., Brooks, S., Selim, H.: Adsorption and transport of uranium (VI) in subsurface media. Soil Sci. Soc. Am. J. 64, 908–917 (2000)
Barnett, M.O., Jardine, P.M., Brooks, S.C.: U (VI) adsorption to heterogeneous subsurface media: application of a surface complexation model. Environ. Sci. Technol. 36, 937–942 (2002)
Bauer, A., Schäfer, T., Dohrmann, R., Hoffmann, H., Kim, J.: Smectite stability in acid salt solutions and the fate of Eu, Th and U in solution. Clay Miner. 36, 93–103 (2001)
Beckingham, L.E., Mitnick, E.H., Steefel, C.I., Zhang, S., Voltolini, M., Swift, A.M., Yang, L., Cole, D.R., Sheets, J.M., Ajo-Franklin, J.B.: Evaluation of mineral reactive surface area estimates for prediction of reactivity of a multi-mineral sediment. Geochim. Cosmochim. Acta 188, 310–329 (2016)
Bethke, C.M., Brady, P.V.: How the Kd approach undermines ground water cleanup. Groundwater 38, 435–443 (2000)
Bethke, C.: Geochemical Reaction Modeling: Concepts and Applications. Oxford University Press on Demand (1996)
Borovec, Z.: The adsorption of uranyl species by fine clay. Chem. Geol. 32, 45–58 (1981)
Bourdon, B., Turner, S., Henderson, G.M., Lundstrom, C.C.: Introduction to U-series geochemistry. Rev. Mineral. Geochem. 52, 1–21 (2003)
Bradbury, M.H., Baeyens, B.: Modelling the sorption of Mn (II), Co (II), Ni (II), Zn (II), Cd (II), Eu (III), Am (III), Sn (IV), Th (IV), Np (V) and U (VI) on montmorillonite: Linear free energy relationships and estimates of surface binding constants for some selected heavy metals and actinides. Geochim. Cosmochim. Acta 69, 875–892 (2005)
Campos, M.B., de Azevedo, H., Nascimento, M.R.L., Roque, C.V., Rodgher, S.: Environmental assessment of water from a uranium mine (Caldas, Minas Gerais State, Brazil) in a decommissioning operation. Environ. Earth Sci. 62, 857–863 (2011)
Carroll, S.A., Walther, J.V.: Kaolinite dissolution at 25 degrees, 60 degrees, and 80 degrees C. Am. J. Sci. 290, 797–810 (1990)
Chandrajith, R., Seneviratna, S., Wickramaarachchi, K., Attanayake, T., Aturaliya, T., Dissanayake, C.: Natural radionuclides and trace elements in rice field soils in relation to fertilizer application: study of a chronic kidney disease area in Sri Lanka. Environ. Earth Sci. 60, 193–201 (2010)
Chen, X., Thornton, S.: Multi-mineral reactions controlling secondary phase evolution in a hyper-alkaline plume. Environ. Geotech. 6, 521–527 (2018)
Chen, X., Thornton, S.F., Small, J.: Influence of hyper-alkaline pH leachate on mineral and porosity evolution in the chemically disturbed zone developed in the near-field host rock for a nuclear waste repository. Transp. Porous Media 107, 489–505 (2015)
Chipera, S.J., Bish, D.L.: FULLPAT: a full-pattern quantitative analysis program for X-ray powder diffraction using measured and calculated patterns. J. Appl. Crystallogr. 35, 744–749 (2002)
Chisholm-Brause, C., Conradson, S.D., Buscher, C., Eller, P.G., Morris, D.E.: Speciation of uranyl sorbed at multiple binding sites on montmorillonite. Geochim. Cosmochim. Acta 58, 3625–3631 (1994)
Chisholm-Brause, C.J., Berg, J.M., Matzner, R.A., Morris, D.E.: Uranium (VI) sorption complexes on montmorillonite as a function of solution chemistry. J. Colloid Interface Sci. 233, 38–49 (2001)
Claret, F., Bauer, A., Schäfer, T., Griffault, L., Lanson, B.: Experimental investigation of the interaction of clays with high-pH solutions: a case study from the Callovo–Oxfordian formation, Meuse-Haute Marne underground laboratory (France). Clays Clay Miner. 50, 633–646 (2002)
Curtis, G.P., Fox, P., Kohler, M., Davis, J.A.: Comparison of in situ uranium KD values with a laboratory determined surface complexation model. Appl. Geochem. 19, 1643–1653 (2004)
Curtis, G. P., Davis, J.A., Naftz, D.L.: Simulation of reactive transport of uranium (VI) in groundwater with variable chemical conditions. Water Resour. Res. 42, 1 (2006)
Dangelmayr, M.A., Reimus, P.W., Wasserman, N.L., Punsal, J.J., Johnson, R.H., Clay, J.T., Stone, J.J.: Laboratory column experiments and transport modeling to evaluate retardation of uranium in an aquifer downgradient of a uranium in-situ recovery site. Appl. Geochem. 80, 1–13 (2017)
Davis, J.: Surface Complexation Modeling of Uranium (VI) Adsorption on Natural Mineral Assemblage NUREG/CR-6708, US Nuclear Regulatory Commission, Rockville, MD (2001)
Davis, J.A., Kent, D.B.: Surface complexation modeling in aqueous geochemistry. In: Mineral-Water Interface Geochemistry, pp. 177–260 (2018)
Davis, J.A., James, R.O., Leckie, J.O.: Surface ionization and complexation at the oxide/water interface: I. Computation of electrical double layer properties in simple electrolytes. J. Colloid Interface Sci. 63, 480–499 (1978)
Davis, J.A., Payne, T.E., Waite, T.D.: Simulating adsorption by a weathered schist with surface complexation models. Geochem. Soil Radionucl. Spec. Publ. 59, 61–86 (2001)
Davis, J.A., Payne, T.E., Waite, T.D.: Simulating the pH and pCO2 dependence of uranium (VI) adsorption by a weathered schist with surface complexation models. Geochem. Soil Radionucl. 59, 61–86 (2002)
Davis, J.A., Meece, D.E., Kohler, M., Curtis, G.P.: Approaches to surface complexation modeling of Uranium(VI) adsorption on aquifer sediments. Geochim. Cosmochim. Acta 68, 3621–3641 (2004)
de Windt, L., Marsal, F., Tinseau, E., Pellegrini, D.: Reactive transport modeling of geochemical interactions at a concrete/argillite interface, Tournemire site (France). Phys. Chem. Earth Parts a/b/c 33, S295–S305 (2008)
Delany, J., Undeen, S.: The LLNL thermochemical database. In: Lawrence Livermore National Laboratory Report UCRL-21658, vol. 150 (1990)
Disposal, G.: Radionuclide behaviour status report, NDA (Radioactive Waste Management Directorate) Report no. NDA/RWMD/034, Didcot, Oxon, OX11 0RH, UK (2010)
Dong, W., Brooks, S.C.: Formation of aqueous MgUO2(CO3)32− complex and uranium anion exchange mechanism onto an exchange resin. Environ. Sci. Technol. 42, 1979–1983 (2008)
Dong, W., Wan, J.: Additive surface complexation modeling of uranium (VI) adsorption onto quartz-sand dominated sediments. Environ. Sci. Technol. 48, 6569–6577 (2014)
Dzombach, D., Morel, F.: Surface Complexing Modeling Hydrous Ferric Oxide. Wiley-Interscience, New York (1990)
Dzombak, D.A., Morel, F.M.: Surface Complexation Modeling: Hydrous Ferric Oxide. Wiley, New York (1991)
Evans, N.D.: Binding mechanisms of radionuclides to cement. Cem. Concr. Res. 38, 543–553 (2008)
Felipe-Sotelo, M., Hinchliff, J., Field, L., Milodowski, A., Preedy, O., Read, D.: Retardation of uranium and thorium by a cementitious backfill developed for radioactive waste disposal. Chemosphere 179, 127–138 (2017)
Fernández, R., Mäder, U.K., Rodríguez, M., Vigil de la Villa, R., Cuevas, J.: Alteration of compacted bentonite by diffusion of highly alkaline solutions. Eur. J. Mineral. 21, 725–735 (2009)
Fernández, R., Cuevas, J., Mäder, U.K.: Modeling experimental results of diffusion of alkaline solutions through a compacted bentonite barrier. Cem. Concr. Res. 40, 1255–1264 (2010)
Fletcher, P., Sposito, G.: The chemical modelling of clay/electrolyte interactions for montmorillonite. Clay Miner. 24, 375–391 (1989)
Fox, P.M., Davis, J.A., Zachara, J.M.: The effect of calcium on aqueous uranium (VI) speciation and adsorption to ferrihydrite and quartz. Geochim. Cosmochim. Acta 70, 1379–1387 (2006)
Geipel, G., Bernhard, G., Brendler, V., Nitsche, H.: Complex formation between UO2+2 and CO2−3: studied by laser-induced photoacoustic spectroscopy (LIPAS). Radiochim. Acta 82, 59–62 (1998)
Glynn, P.D.: Modeling Np and Pu transport with a surface complexation model and spatially variant sorption capacities: implications for reactive transport modeling and performance assessments of nuclear waste disposal sites. Comput. Geosci. 29, 331–349 (2003)
Golubev, S.V., Bauer, A., Pokrovsky, O.S.: Effect of pH and organic ligands on the kinetics of smectite dissolution at 25 C. Geochim. Cosmochim. Acta 70, 4436–4451 (2006)
Gorman-Lewis, D., Burns, P.C., Fein, J.B.: Review of uranyl mineral solubility measurements. J. Chem. Thermodyn. 40, 335–352 (2008)
Gougar, M., Scheetz, B., Roy, D.: Ettringite and C-S-H Portland cement phases for waste ion immobilization: a review. Waste Manag. 16, 295–303 (1996)
Grenthe, I., Fuger, J., Konings, R.J., Lemire, R.J., Muller, A.B., Nguyen-trung, C., Wanner, H.: Chemical Thermodynamics of Uranium, North-Holland Amsterdam (1992)
Grenthe, I., Gaona, X., Rao, L., Plyasunov, A., Runde, W., Grambow, B., Konings, R., Smith, A., Moore, E., Ragoussi, M.-E.: Second update on the chemical thermodynamics of uranium, neptunium, plutonium, americium and technetium. In: Chemical Thermodynamics volume 14. Organisation for Economic Co-Operation and Development (2020)
Guimarães, V., Rodríguez-Castellón, E., Algarra, M., Rocha, F., Bobos, I.: Influence of pH, layer charge location and crystal thickness distribution on U(VI) sorption onto heterogeneous dioctahedral smectite. J. Hazard. Mater. 317, 246–258 (2016)
Harfouche, M., Wieland, E., Dähn, R., Fujita, T., Tits, J., Kunz, D., Tsukamoto, M.: EXAFS study of U (VI) uptake by calcium silicate hydrates. J. Colloid Interface Sci. 303, 195–204 (2006)
Hennig, C., Reich, T., Dähn, R., Scheidegger, A.: Structure of uranium sorption complexes at montmorillonite edge sites. Radiochim. Acta 90, 653–657 (2002)
Hiemstra, T., van Riemsdijk, W., Bolt, G.: Multisite proton adsorption modeling at the solid/solution interface of (hydr) oxides: A new approach: I. Model description and evaluation of intrinsic reaction constants. J. Colloid Interface Sci. 133, 91–104 (1989)
Ho, C., Miller, N.: Adsorption of uranyl species from bicarbonate solution onto hematite particles. J. Colloid Interface Sci. 110, 165–171 (1986)
Hsi, C.-K.D., Langmuir, D.: Adsorption of uranyl onto ferric oxyhydroxides: application of the surface complexation site-binding model. Geochim. Cosmochim. Acta 49, 1931–1941 (1985)
Johnson, C.A.: Cement stabilization of heavy-metal-containing wastes. Geol. Soc. Lond. Spec. Publ. 236, 595–606 (2004)
Kent, D. B., Tripathi, V. S., Ball, N. B., Leckie, J., Siegel, M.: Surface-Complexation Modeling of Radionuclide Adsorption in Subsurface Environments. Stanford University (1988)
Kim, S.: Sorption mechanism of U (VI) on a reference montmorillonite: binding to the internal and external surfaces. J. Radioanal. Nucl. Chem. 250, 55–62 (2001)
Klajmon, M., Havlová, V., Červinka, R., Mendoza, A., Franců, J., Berenblyum, R., Arild, Ø.: REPP-CO2: equilibrium modelling of CO2-rock-brine systems. Energy Procedia 114, 3364–3373 (2017)
Knauss, K.G., Wolery, T.J.: The dissolution kinetics of quartz as a function of pH and time at 70 C. Geochim. Cosmochim. Acta 52, 43–53 (1988)
Kohler, M., Wieland, E., Leckie, J. Metal-ligand-surface interaction during sorption of uranyl and neptunyl on oxides and silicates. In: International Symposium on Water-Rock Interaction, pp. 51–54 (1992)
Koretsky, C.: The significance of surface complexation reactions in hydrologic systems: a geochemist’s perspective. J. Hydrol. 230, 127–171 (2000)
Korichi, S., Bensmaili, A.: Sorption of uranium (VI) on homoionic sodium smectite experimental study and surface complexation modeling. J. Hazard. Mater. 169, 780–793 (2009)
Kowal-Fouchard, A., Drot, R., Simoni, E., Ehrhardt, J.: Use of spectroscopic techniques for uranium (VI)/montmorillonite interaction modeling. Environ. Sci. Technol. 38, 1399–1407 (2004)
Langmuir, D.: Aqueous Environmental, vol. 600. Geochemistry Prentice Hall, Upper Saddle River (1997)
Lichtner, P.C.: The quasi-stationary state approximation to coupled mass transport and fluid–rock interaction in a porous medium. Geochim. Cosmochim. Acta 52, 143–165 (1988)
Liger, E., Charlet, L., van Cappellen, P.: Surface catalysis of uranium (VI) reduction by iron (II). Geochim. Cosmochim. Acta 63, 2939–2955 (1999)
Ma, B., Charlet, L., Fernandez-Martinez, A., Kang, M., Madé, B.: A review of the retention mechanisms of redox-sensitive radionuclides in multi-barrier systems. Appl. Geochem. 100, 414–431 (2019)
Macquarrie, K.T., Mayer, K.U.: Reactive transport modeling in fractured rock: a state-of-the-science review. Earth Sci. Rev. 72, 189–227 (2005)
Marques Fernandes, M., Baeyens, B., Dähn, R., Scheinost, A.C., Bradbury, M.H.: U(VI) sorption on montmorillonite in the absence and presence of carbonate: a macroscopic and microscopic study. Geochim. Cosmochim. Acta 93, 262–277 (2012)
Mckinley, J.P., Zachara, J.M., Smith, S.C., Turner, G.D.: The influence of uranyl hydrolysis and multiple site-binding reactions on adsorption of U (VI) to montmorillonite. Clays Clay Miner 43(5), 586–598 (1995)
Mckinley, J., Zachara, J., Smith, S.: An experimental and modeling investigation of UO22+ interaction with smectite clays. In: 205th ACS National Meeting (1993)
Mcling, T.: Uranium sorption onto natural sands as a function of sediment characteristics and solution pH. In: Adsorption of Metals by Geomedia: Variables, Mechanisms, and Model Applications, vol. 181 (1998).
Meinrath, G.: Speciation of uranium. In: Uranium Mining and Hydrogeology II. Proceedings (1998)
Merkel, B.J., Hasche-Berger, A.: Uranium in the environment. In: Mining Impacts and (2006).
Merkel, B.J., Hasche-Berger, A.: Uranium, Mining and Hydrogeology. Springer, New York (2008)
Missana, T., Alonso, U., Garcia-Gutierrez, M., Albarran, N., Lopez, T.:. Experimental study and modeling of uranium (VI) sorption onto a Spanish smectite. In: MRS Online Proceedings Library (OPL), vol. 1124 (2008)
Morris, D.E., Chisholm-Brause, C.J., Barr, M.E., Conradson, S.D., Eller, P.G.: Optical spectroscopic studies of the sorption of UO2+ 2 species on a reference smectite. Geochim. Cosmochim. Acta 58, 3613–3623 (1994)
Morrison, S.J., Spangler, R.R., Tripathi, V.S.: Adsorption of uranium (VI) on amorphous ferric oxyhydroxide at high concentrations of dissolved carbon (IV) and sulfur (VI). J. Contam. Hydrol. 17, 333–346 (1995)
Nair, S., Merkel, B.J.: Impact of alkaline earth metals on aqueous speciation of uranium (VI) and sorption on quartz. Aquat. Geochem. 17, 209–219 (2011b)
Nair, S., Karimzadeh, L., Merkel, B.J.: Surface complexation modeling of Uranium (VI) sorption on quartz in the presence and absence of alkaline earth metals. Environ. Earth Sci. 71, 1737–1745 (2014)
Nair, S., Merkel, B.J.: Effect of Mg–Ca–Sr on the sorption behavior of uranium (VI) on silica. In: The New Uranium Mining Boom. Springer (2011a)
Nardi, A., Idiart, A., Trinchero, P., de Vries, L.M., Molinero, J.: Interface COMSOL-PHREEQC (iCP), an efficient numerical framework for the solution of coupled multiphysics and geochemistry. Comput. Geosci. 69, 10–21 (2014)
Pabalan, R.T., Turner, D.R.: Uranium(6+) sorption on montmorillonite: experimental and surface complexation modeling study. Aquat Geochem 2, 203–226 (1996)
Papini, M.P., Kahie, Y.D., Troia, B., Majone, M.: Adsorption of lead at variable pH onto a natural porous medium: Modeling of batch and column experiments. Environ. Sci. Technol. 33, 4457–4464 (1999)
Parkhurst, D.L., Appelo, C.: Description of input and examples for PHREEQC version 3: a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. US Geological Survey (2013)
Payne, Τ, Waite, T.: Surface complexation modelling of uranium sorption data obtained by isotope exchange techniques. Radiochim. Acta 52, 487–494 (1991)
Payne, T., Davis, J., Lumpkin, G., Chisari, R., Waite, T.: Surface complexation model of uranyl sorption on Georgia kaolinite. Appl. Clay Sci. 26, 151–162 (2004)
Pointeau, I., Landesman, C., Giffaut, E., Reiller, P.: Reproducibility of the uptake of U (VI) onto degraded cement pastes and calcium silicate hydrate phases. Radiochim. Acta 92, 645–650 (2004)
Prikryl, J.D., Jain, A., Turner, D.R., Pabalan, R.T.: UraniumVI sorption behavior on silicate mineral mixtures. J. Contam. Hydrol. 47, 241–253 (2001)
Protection, R. The management of radioactive waste in the oil and gas industry. In: Safety Reports Series, vol. 34 (2003).
Ramírez, S., Cuevas, J., Vigil, R., Leguey, S.: Hydrothermal alteration of “La Serrata” bentonite (Almeria, Spain) by alkaline solutions. Appl. Clay Sci. 21, 257–269 (2002)
Reardon, E.: Kd’s—can they be used to describe reversible ion sorption reactions in contaminant migration? Groundwater 19, 279–286 (1981)
Ribera, D., Labrot, F., Tisnerat, G., Narbonne, J.-F. Uranium in the environment: occurrence, transfer, and biological effects. In: Reviews of Environmental Contamination and Toxicology. Springer (1996)
Ricka, A., Kuchovsky, T., Sracek, O., Zeman, J.: Determination of potential mine water discharge zones in crystalline rocks at Rozna, Czech Republic. Environ. Earth Sci. 60, 1201–1213 (2010)
Schindler, M., Legrand, C.A., Hochella, M.F., Jr.: Alteration, adsorption and nucleation processes on clay–water interfaces: mechanisms for the retention of uranium by altered clay surfaces on the nanometer scale. Geochim. Cosmochim. Acta 153, 15–36 (2015)
Scism, C.D.: The Sorption/Desorption Behavior of Uranium in Transport Studies Using Yucca Mountain Alluvium. Los Alamos National Lab.(LANL), Los Alamos, NM (United States) (2005)
Seigneur, N., Lagneau, V., Corvisier, J., Dauzères, A.: Recoupling flow and chemistry in variably saturated reactive transport modelling-An algorithm to accurately couple the feedback of chemistry on water consumption, variable porosity and flow. Adv. Water Resour. 122, 355–366 (2018)
Sherman, D.M., Peacock, C.L., Hubbard, C.G.: Surface complexation of U (VI) on goethite (α-FeOOH). Geochim. Cosmochim. Acta 72, 298–310 (2008)
Small, J., Bryan, N., Lloyd, J., Milodowski, A., Shaw, S., Morris, K.: Summary of the BIGRAD project and its implications for a geological disposal facility. In: National Nuclear Laboratory, Report NNL (16), p. 1381 (2016)
Steefel, C., Appelo, C., Arora, B., Jacques, D., Kalbacher, T., Kolditz, O., Lagneau, V., Lichtner, P., Mayer, K.U., Meeussen, J.: Reactive transport codes for subsurface environmental simulation. Comput. Geosci. 19, 445–478 (2015)
Stewart, B.D., Mayes, M.A., Fendorf, S.: Impact of uranyl−calcium−carbonato complexes on uranium (VI) adsorption to synthetic and natural sediments. Environ. Sci. Technol. 44, 928–934 (2010)
Sylwester, E., Hudson, E., Allen, P.: The structure of uranium (VI) sorption complexes on silica, alumina, and montmorillonite. Geochim. Cosmochim. Acta 64, 2431–2438 (2000)
Takahashi, T., Ohkubo, T., Suzuki, K., Ikeda, Y.: High resolution solid-state NMR studies on dissolution and alteration of Na-montmorillonite under highly alkaline conditions. Microporous Mesoporous Mater. 106, 284–297 (2007)
Thomson, B.M., Longmire, P.A., Brookins, D.G.: Geochemical constraints on underground disposal of uranium mill tailings. Appl. Geochem. 1, 335–343 (1986)
Ticknor, Κ: Uranium sorption on geological materials. Radiochim. Acta 64, 229–236 (1994)
Tits, J., Geipel, G., Macé, N., Eilzer, M., Wieland, E.: Determination of uranium (VI) sorbed species in calcium silicate hydrate phases: a laser-induced luminescence spectroscopy and batch sorption study. J. Colloid Interface Sci. 359, 248–256 (2011)
Tsunashima, A., Brindley, G., Bastovanov, M.: Adsorption of uranium from solutions by montmorillonite; compositions and properties of uranyl montmorillonites. Clays Clay Miner. 29, 10–16 (1981)
Turner, D.R., Sassman, S.A.: Approaches to sorption modeling for high-level waste performance assessment. J. Contam. Hydrol. 21, 311–332 (1996)
Turner, G., Zachara, J., McKinley, J., Smith, S.: Surface-charge properties and UO22+ adsorption of a subsurface smectite. Geochim. Cosmochim. Acta 60, 3399–3414 (1996a)
Turner, G.D., Zachara, J.M., Mckinley, J.P., Smith, S.C.: Surface-charge properties and UO22+ adsorption of a subsurface smectite. Geochim. Cosmochim. Acta 60, 3399–3414 (1996b)
Turner, D.R., Pabalan, R.T., Bertetti, F.P.: Neptunium (V) sorption on montmorillonite: An experimental and surface complexation modeling study. Clays Clay Miner. 46, 256–269 (1998)
Tutu, H., Cukrowska, E.M., McCarthy, T.S., Hart, R., Chimuka, L.: Radioactive disequilibrium and geochemical modelling as evidence of uranium leaching from gold tailings dumps in the Witwatersrand Basin. Int. J. Environ. Anal. Chem. 89, 687–703 (2009)
van der Lee, J.: Thermodynamic and Mathematical Concepts of CHESS (1998)
Velbel, M.A.: Constancy of silicate-mineral weathering-rate ratios between natural and experimental weathering: implications for hydrologic control of differences in absolute rates. Chem. Geol. 105, 89–99 (1993)
Vines, S., Lever, D. An integrated approach to geological disposal of UK wastes containing carbon-14. In: International Conference on Radioactive Waste Management and Environmental Remediation, 2013. American Society of Mechanical Engineers, V001T01A060 (2013)
Waite, T., Davis, J., Payne, T., Waychunas, G., Xu, N.: Uranium (VI) adsorption to ferrihydrite: application of a surface complexation model. Geochim. Cosmochim. Acta 58, 5465–5478 (1994)
Watson, C., Hane, K., Savage, D., Benbow, S., Cuevas, J., Fernandez, R.: Reaction and diffusion of cementitious water in bentonite: results of ‘blind’modelling. Appl. Clay Sci. 45, 54–69 (2009)
White, A.F., Brantley, S.L.: The effect of time on the weathering of silicate minerals: why do weathering rates differ in the laboratory and field? Chem. Geol. 202, 479–506 (2003)
White, A.F., Peterson, M.L.: Role of Reactive-Surface-Area Characterization in Geochemical Kinetic Models. ACS Publications, Washington (1990)
Wronkiewicz, D.J., Bates, J.K., Gerding, T.J., Veleckis, E., Tani, B.S.: Uranium release and secondary phase formation during unsaturated testing of UO2 at 90 C. J. Nucl. Mater. 190, 107–127 (1992)
Zachara, J.M., McKinley, J.P.: Influence of hydrolysis on the sorption of metal cations by smectites: importance of edge coordination reactions. Aquat. Sci. 55, 250–261 (1993)
Zhu, C.: A case against Kd-based transport models: natural attenuation at a mill tailings site. Comput. Geosci. 29, 351–359 (2003)
Zielinski, R., Asher-Bolinder, S., Meier, A., Johnson, C., Szabo, B.: Natural or fertilizer-derived uranium in irrigation drainage: a case study in southeastern Colorado, USA. Appl. Geochem. 12, 9–21 (1997)
Acknowledgements
The first, fourth, and fifth authors acknowledge Kuwait Petroleum Company (KPC) for sponsoring this work. The authors would like to acknowledge the edits and comments on early draft of this paper from Dr Matt Kirby (Nuclear Waste Services., Harwell, UK).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
YB is leading writing the paper, including setting up the geochemical code for the studied system, performed computer modelling and simulation, interpreted the behaviour of the modelling resulting compared with the experimental data, and prepared the manuscript. TS assisted in the interpretation of the modelling results and contributed to the manuscript editing. SDI assisted in the interpretation of the modelling results and contributed to the manuscript editing. NS refined the manuscript and provided deeper insight into the results. CX supported in understanding and building the geochemical model.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baqer, Y., Thornton, S., Stewart, D.I. et al. Analysis of Uranium Sorption in a Laboratory Column Experiment Using a Reactive Transport and Surface Complexation Model. Transp Porous Med 149, 423–452 (2023). https://doi.org/10.1007/s11242-023-01956-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11242-023-01956-y