
Abstract
In the present study, a high yield of isolated protoplasts from the agronomically important crop Fagopyrum esculentum was obtained by applying a mixture of cellulase, pectolyase, and driselase. We demonstrated that the yield of morphogenic callus-derived protoplasts was 1 × 106 protoplasts per g of fresh tissue. For hypocotyls used as the protoplast source, the number of released cells was twice lower. The protoplasts, embedded in an agarose matrix and cultured in a modified Kao and Michayluk media supplemented with phytosulfokine, re-enter the cell cycle and start to develop, forming microcalli. The plating efficiency was about 20% in the case of hypocotyl- and morphogenic callus-derived protoplasts. For plant regeneration, the medium was supplemented with different combinations of cytokinin. Somatic embryogenesis and organogenesis occur during the cultivation of the protoplast-derived tissues, depending on the applied protoplast source. For the first time, an effective protoplast-to-plant system for F. esculentum has been developed.
Key message
Morphogenic callus- and hypocotyl-derived protoplasts of buckwheat after embedding in agarose beads and culture in phytosulfokine enriched medium regenerated into plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus buckwheat (Fagopyrum) is a promising functional food source that contains various phenolic compounds, especially rutin, quercetin and C-glycosylflavones (orientin, isoorientin, vitexin), which have a positive therapeutic or dietary effect for promoting human health (Zhang et al. 2015). To date, 23 buckwheat species have been identified (Tomasiak et al. 2022). One of the most important species is Fagopyrum esculentum Moench. (common buckwheat), a multipurpose crop with a high nutritional value, mainly high-quality proteins with essential amino acids (Woo et al. 2010). Common buckwheat is also considered a nectariferous and pharmaceutical plant (Kwon et al. 2013). As it has the ability to do well on unproductive soils and does not require extensive fertilisation, common buckwheat is an attractive economic crop and low-cost supplement to cereal grains (Kumar and Saraswat 2018). The biggest problem with common buckwheat is the short life of its single flower (1 day) (Cawoy et al. 2009) and very short growing period (70–90 days). Moreover, the sensitive to ground frost, high temperatures and drought may cause strong flower and embryo abortions. So far, buckwheat F. homotropicum has been cross-pollinated with F. tataricum (Tartary buckwheat) or F. tataricum with F. esculentum in order to transfer genes with a greater resistance to frost and a higher seed yield. Because of the strong barriers that prevent cross-pollination between different species, these studies have been unsuccessful (Shaikh et al. 2001; Woo et al. 2001). Common buckwheat forms dimorphic plants with flowers whose pistils and stamens have different lengths (pin and thrum types), which results in self-incompatibility (Adachi 1990; Cawoy et al. 2009). Therefore, fertilisation occurs between both of these flower types after cross-pollination (Cawoy et al. 2006; Taylor and Obendorf 2001). Among the most important reasons for the low yield of common buckwheat are: (1) self-incompatibility; (2) insufficient fertilisation; (3) embryo abortion; (4) sensitivity to heat and drought stress; and (5) deficiency of the assimilates that occur in ageing plants (Slawinska and Obendorf 2001; Taylor and Obendorf 2001).
Plant biotechnology techniques, specifically tissue and cell cultures, represent a solution to overcome the reproductive barriers for this species. In previous studies related to Tartary and common buckwheat regeneration, the most widely used explants have been cotyledon and hypocotyl segments from seedlings (Adachi et al. 1989; Hou et al. 2015; Kwon et al. 2013; Lachmann 1990). Explants from mature plants, such as petioles, leaves and nodes, have also been used (Slawinska 2009; Woo et al. 2004). The successful regeneration of common buckwheat has been previously reported via organogenesis or somatic embryogenesis using different variants of plant growth regulators (PGRs) (Kwon et al. 2013; Nešković et al. 1987). The application of protoplast cultures guarantees the unicellular origin of the somatic embryos; thus, the recovery of genotypes with novel traits would be favoured. The processes of protoplast isolation from common buckwheat plants have been improving for decades since the first attempt by Holländer-Czytko and Amrhein (1983). Adachi et al. (1989) reported for the first time about plant regeneration from hypocotyl-derived protoplasts of common buckwheat. However, they obtained low plating efficiency (approx. 1%) and abnormal morphology of the regenerated plants. Likewise, Gumerova (2004) achieved plant regeneration from hypocotyl-derived protoplast cultures, but the regenerative capacity of the protoplast-derived callus was low. On the other hand, Lachmann (1990) managed to obtain higher plating efficiency from hypocotyl-derived protoplasts of Tartary buckwheat, but plant regeneration was not induced. As it was mentioned by Woo et al. (1999), it is possible to isolate protoplasts form sperm cells of common buckwheat what can be useful in the case of protoplast fusion. Also hypocotyl-derived protoplasts were applied by Sakamoto et al. (2020) as a valuable tool for analysis of gene function.
As described above, hypocotyls have been commonly used as a source of protoplasts. However, common buckwheat’s morphogenic callus (MC), due to its high regenerative potential, may be a desirable source of protoplasts (Takahata and Jumonji 1985; Yamane 1974). However, using the MC as a source of protoplasts has been little studied (Gumerova 2004). Therefore, in this work, we proposed an efficient protoplast-to-plant regeneration system of common buckwheat via callus formation starting with hypocotyls and the MC as the protoplast source.
Materials and methods
Plant materials for protoplast isolation
For MC induction and the development of etiolated hypocotyls, commercially available seeds of the Panda cultivar (the Malopolska Plant Breeding, Poland) were used. The callus lines (L1 and NL2) were obtained from immature zygotic embryos in the dark at 26 ± 1 °C on a RX medium as previously described (Betekhtin et al. 2019, 2017; Rumyantseva et al. 2005) and maintained with regular subcultures every 2–3 weeks on fresh RX medium composed of Gamborg B5 including vitamins (Gamborg et al. 1968), 2 g L−1 N-Z-amine A, 2 mg L−1 2,4-dichlorophenoxyacetic acid (2,4-D), 0.2 mg L−1 kinetin (KIN), 0.5 mg L−1 3-indoleacetic acid (Sigma-Aldrich, USA), 0.5 mg L−1 1-naphthaleneacetic acid (NAA), 25 g L−1 sucrose and 7 g L−1 phyto agar (Duchefa, Netherland).
For hypocotyl development, the seeds were kept in distilled water overnight, and the seed coat was then removed. The surface sterilisation of the seeds was carried out in a three-step protocol: (1) the seeds were soaked in 70% ethanol for 30 s and then shaken (160 rpm for 30 min) in a 0.2% (v/v) solution of Scorpion 325 SC fungicide (Sygenta, Switzerland) with a drop of Tween 20 (Duchefa); (2) the seeds were immersed in a 20% (w/v) solution of chloramin T (sodium N-chlorotoluene-4-sulphonamide; Chempur, Poland) with 3000 mg L−1 cefotaxime disodium (Duchefa) and one drop of Tween 20 for 30 min; between each step, the seeds were dipped in 70% ethanol for 30–45 s; (3) the seeds were washed three times with sterile distilled water for 5 min each time and soaked overnight in sterile distilled water. The next day, the second and third steps were repeated. After two days of sterilisation, the seeds were air-dried on sterile filter paper and placed in polystyrene Petri dishes (Ø9 cm) with a MS medium supplemented with vitamins (Murashige and Skoog 1962), 200 mg L−1 cefotaxime disodium, 30 g L−1 sucrose and 7 g L−1 plant agar (Duchefa). The Petri dishes were sealed with parafilm and incubated in the dark at 26 ± 1 °C for ten days.
Protoplast isolation and culture
The protoplasts were isolated from two types of source materials including around 12-day-old MC and 10-day-old hypocotyls. First, a pre-plasmolysis step was performed. One gram of 8-day-old MC L1 line or 2 g of 12-day-old NL2 line were incubated with PSII/F solution (Table 1) in a glass Petri dish (Ø9 cm). For the hypocotyls, 1 g of plant material was cut into 1 cm long pieces, and these were cut longitudinally in PSII solution (Table 1). In both cases, the pre-plasmolysis step took place in the dark at room temperature (RT) for 1 h. After this, the solution was removed, and enzymatic maceration was carried out by adding the enzyme solution E1, E2 or E3 (Table 1) for the L1, NL2 and hypocotyls, respectively. This step was performed overnight for 16 h with gentle shaking (50 rpm at RT) in the dark. The quality of the released protoplasts was checked using an inverted microscope (Axiovert S100; Carl Zeiss, Germany). The suspension was filtered using nylon filters (mesh size 100 μm; Millipore, USA) and then centrifuged for 5 min (1000 rpm at RT). The pellet was re-suspended in a Suc/MES solution (Table 1) for the callus or a Suc-2/MES solution (Table 1) for the hypocotyls, then W5 solution (Table 1) was carefully overlaid and it was centrifuged for 10 min (1200 rpm at RT). The ring of viable protoplasts, formed at the interphase of these two solutions, was collected in a new centrifuge tube. The collected protoplasts were dissolved in W5 solution and centrifuged for 5 min (1000 rpm at RT). Next, the supernatant was removed, and the culture medium added. The culture medium was based on CPP medium according to Dirks et al. (1996) and supplemented with 1.0 mg L−1 6-benzylaminopurine (BAP) and 2.0 mg L−1 NAA, and then called the basal medium (BM; Table 2). The density of the protoplasts was determined using a Fuchs-Rosenthal haemocytometer (Heinz Herenz, Germany) and then adjusted with the BM to 8 × 105 or 5 × 105 cells per ml for the MC lines and hypocotyls, respectively. The protoplasts were embedded in a filter-sterilised solution of 1.2% (w/v) low melting point agarose (LMPA; Duchefa), according to Grzebelus et al. (2012b). The mixture of protoplasts and agarose was dropped at a rate of four beads per Petri dish (Ø6 cm). After solidification of the agarose beads, the BM was added to each dish and the medium was additionally supplemented with 100 nM phytosulfokine (PSK), 0.25–0.75 mg L−1 chloropyridin phenylurea (CPPU; Sigma-Aldrich), 8 mg L−1 putrescine (PUT; Sigma-Aldrich) or 0.025–0.05% polyvinylpyrrolidone (PVP, MW 40,000) in different combinations. To prevent bacterial contamination, the culture media of hypocotyl-derived protoplasts were supplemented with 200 mg L−1 cefotaxime disodium. The protoplast cultures were incubated at 26 ± 1 °C in the dark for 60 days. The medium, with all supplements, was renewed on the 10th day of culture.
Plant regeneration
After two months of protoplast culture, agarose beads overgrown with the protoplast-derived callus was transferred to a callus multiplication medium (CM; Table 2). The cultures were incubated at 26 ± 1 °C in the dark and subcultured every three weeks on the same CM. Next, the callus was transferred to the regeneration medium (RM; Table 2) and maintained in a climate room at 28 ± 1 °C with a 16/8 h (light/dark) photoperiod (a light intensity of 55 µmol m−2 s−1; fluorescent lamps Sylvania Gro-lux T8, USA). The RM_MS3 regeneration medium was used for the protoplast-derived callus originating from the NL2 line, while the regeneration of the protoplast-derived callus originating from the L1 line and hypocotyls was carried out on the RM_MS4 medium (Table 2). Initially, the NL2 line was also tested on RM_MS4 medium; however, there was no evidence of regeneration after several months of subculture. The NL2 callus-derived somatic embryos were separated, transferred to the RM_MS3 medium and subcultured every two weeks in the same medium until shoots were obtained. The callus originating from the L1 line and hypocotyls were subcultured every three weeks on RM_MS4 medium. For rooting, shoots were transferred to sterile vessels (150 mm L × 90 mm W) with rooting medium (Table 2) and maintained in a climate room at 25 ± 1 °C with a 16/8 h (light/dark) photoperiod (a light intensity of 55 µmol m−2 s−1; fluorescent lamps Sylvania Gro-lux T8, USA). When the roots had grown large enough, the plants were transferred to a moss-coconut fiber substrate (Ceres International Ltd., Pyzdry, Poland) and placed in greenhouse conditions at 25 ± 1 °C, 16/8 h (light/dark) photoperiod (light intensity 90 µmol m−2 s−1).
Histological analysis
The fixation was carried out following the methodology proposed by Betekhtin et al. (2019). The calli derived from protoplasts were fixed in a mixture of 4% paraformaldehyde and 1% glutaraldehyde in phosphate-buffered saline (PBS) overnight at 4 ± 1 °C. The samples were washed with PBS, followed by a dehydration process in increasing ethanol concentrations. Next, the samples were embedded in LR White resin (London Resin, St. Louis, USA) and left to polymerise for 24–48 h at 58 ± 1 °C. The samples were then cut into 1.5 μm thick sections using an EM UC6 ultramicrotome (Leica Biosystems, Wetzlar, Germany) and placed on glass slides coated with poly-L-lysine. The slides were stained with 0.05% Toluidine Blue O (Sigma-Aldrich) for 5 min and washed twice with distilled water. The stained sections were examined under an Olympus BX43F microscope equipped with the Olympus XC50 digital camera.
Data collection and statistical analysis
The protoplast yield was presented as the protoplast number per gram of fresh weight (FW) in 1 ml of suspension. The viability of the protoplasts was assessed, immediately after embedding the cells in an agarose matrix, by staining with fluorescein diacetate (FDA; Sigma-Aldrich), according to Grzebelus et al. (2012a). The viability was expressed as a percentage of protoplasts with apple-green fluorescence out of the total observed cells. Plating efficiency was determined in 10-day-old cultures and expressed as a percentage of cell aggregates per total number of observed undivided cells and cell colonies. Microscopic observations were performed under an inverted Axiovert S100 microscope with a filter set appropriate for FDA visualisation (λEx = 485 nm, λEm = 515 nm). Image acquisition was performed under an inverted Lecia DMi8 microscope (Leica Microsystems, Germany) equipped with a Leica DFC 7000 T camera conjugated with LAS X Extended Depth of Field and Deconvolution Modules.
At least three independent protoplast isolation experiments with a single treatment represented by three to four Petri dishes were carried out as repetitions. Microscopic observations were carried out on 100 cells per Petri dish. The mean values and standard errors were calculated. The overall effect of treatments was determined using analysis of variance (ANOVA) in Statistica ver. 13 (TIBCO Software Inc., USA) at P ≤ 0.05. Tukey’s honestly significant difference test was used to determine significant differences between the means.
Results
Plant materials
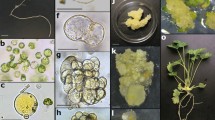
For protoplast isolation, two lines of the MC (L1, NL2; Fig. 1a, b) and etiolated hypocotyls (Fig. 2a) were used. The calli lines differed in age (L1, two-years-old; NL2, one-year-old). A dense globular milky-white structure characterises this type of calli due to the accumulation of starch grains (in the cytoplasm of storage cells). The surface of the calli is covered by an epidermal-like layer under which the meristematic cells and parenchymatous cells are located.

Plant regeneration from morphogenic callus (MC)-derived protoplasts in Fagopyrum esculentum. Lines of MC used for protoplast isolation: a L1 and b NL2; c freshly MC-derived protoplasts; multicellular aggregate in d 6-, e 20- and f 30-day-old protoplast cultures; subsequent stages of plant regeneration from NL2 line-derived protoplast cultures on the regeneration medium: g formation of somatic embryos (examples shown by arrows) in 6-day-old cultures and h somatic embryos converted into plants (examples shown by arrows) in 15-day-old cultures; i three-month-old protoplast-derived-flowering plant acclimatised to ex vitro conditions. Scale bars: 0.5 mm (a–b), 100 µm (c), 50 µm (d–f), 0.5 mm (g), 1 mm (h), 1.5 cm (i)

Plant regeneration from hypocotyl-derived protoplasts of Fagopyrum esculentum. a 10-day-old etiolated hypocotyls used for protoplast isolation; b freshly hypocotyl-derived protoplasts; c 2-cell aggregate after the first mitotic division of the protoplast-derived cell; multicellular aggregate in d 10- and e 20-day-old protoplast cultures; f protoplast-derived microcalli with the somatic embryo (black arrow) in two-month-old protoplast cultures; g shoot formation (red arrow) from the protoplast-derived callus after two weeks on the regeneration medium; h flowering protoplast-derived plant acclimatized to ex vitro conditions. Scale bars: 2 cm (a), 100 µm (b), 50 µm (c–e), 1 mm (f–g), 1 cm (h)
Protoplast isolation efficiency and viability
After overnight incubation in the enzyme solution, spherical protoplasts from both MC lines and hypocotyls were successfully released (Fig. 1c, 2b). The mean yield of MC-derived protoplasts varied from 0.83 to 1.54 × 106 (Table 3). The most efficient protoplast isolation was achieved for the NL2 line in the presence of E2 enzyme solution (1.54 × 106). The use of E1 enzyme solution reduced the number of released protoplasts by approximately half. The callus age (between 8 and 12 days) had no effect on the protoplast isolation efficiency.
Although different enzyme solutions differing in enzyme activity and composition were used (Supplementary Table 1) in the preliminary experiments on protoplast isolation from hypocotyl tissue, the number of released cells was very low. Only after applying driselase to the enzyme solution was an adequate number of protoplasts was recorded. Two concentrations of driselase for tissue digestion were tested, however, different numbers of released cells were not observed (Table 4). The average yield of hypocotyl-derived protoplasts was 0.44 × 106 per g FW. Nevertheless, the efficiency of protoplast isolation from hypocotyls was more than twice-fold lower than from MC sources (P = 0.013).
The quality of released MC-derived protoplasts assessed by FDA staining just after embedding in agarose varied from 73 to 81% (Table 3). Higher protoplast viability was recorded for the L1 line (81%) compared to NL2 line (74%). However, significant differences in protoplast viability were not observed after applying different enzyme mixtures and the callus lines used. The viability of hypocotyl-derived protoplasts reached an average of 75% (Table 4). The higher protoplast viability (80%) was noted after using 0.1% driselase in the enzyme solution. The higher concentration of driselase (0.15%) resulted in lower viability of hypocotyl-derived protoplasts (69%). Nevertheless, the observed differences were not statistically significant.
Development of protoplast cultures
Protoplasts from all source materials revealed the ability to undergo cell division after PSK was applied to the BM. The first division occurred between the third and seventh day of the culture (Fig. 2c), then the next ones took place resulting in the formation of multicellular aggregates (Fig. 1d, 2d–e) in around 10-day-old protoplast cultures. The plating efficiency of the MC L1 line depended on the culture medium variant used and was from 23 to 35% (Fig. 3a). However, no significant differences were observed. PVP was also applied to the culture media to absorb toxic metabolites and phenolic compounds and to support the development of the cells, but a clear positive effect of PVP on the development of protoplast cultures was not recorded.

Effect of different medium variants on plating efficiency in Fagopyrum esculentum 10-day-old protoplast cultures. Plant growth regulator composition in BM = BAP 1.0 mg L−1 + NAA 2.0 mg L−1; BAP = 6-Benzylaminopurine; NAA = naphthylacetic acid; 100 PSK = 100 nM phytosulfokine; CPPU 0.25, CPPU 0.5, CPPU 0.75 = 0.25, 0.5 or 0.75 mg L−1 N-(2-chloro-4-pyridyl)-N’-phenylurea, respectively; PUT 8.0 = 8 mg L−1 putrescine; 0.025, 0.05 PVP = 0.025% or 0.05% polyvinylpyrrolidone, respectively. Bars represent the means from three independent experiments ± standard error. Means marked with the same letters were not significantly different at P ≤ 0.05
In preliminary experiments with the NL2 line, the E1 enzyme solution was used to release the cells, which resulted in low efficiency (about 9%) of cell aggregate formation in culture medium variants I, II, IV, V, and VI as shown in Fig. 3. Based on these results, the E2 enzyme solution was applied in the following experiments. The plating efficiency in protoplast cultures originating from callus digestion in E2 solution was from 21 to 25% (Fig. 3b), but the differences were statistically not significant. For the hypocotyl-derived protoplast cultures, the efficiency of cell aggregate formation was around 21% (Fig. 3c) in all culture medium variants used. A lower level of plating efficiency characterised the NL2 line and hypocotyl protoplast cultures compared to the L1 line (P ≤ 0.01).
The continued growth of aggregates (Fig. 1e–f, 2e) resulted in the formation of microcalli for all protoplast donor sources used. After 30 days of culture, microcalli originating from the NL2-derived protoplast cultures had overgrown the agarose beads and pro-embryogenic masses were observed. For the L1- and hypocotyl-derived protoplast cultures, agarose beads were overgrown by microcalli in 60-day-old cultures; moreover, somatic embryos were occasionally observed in the hypocotyl protoplast cultures (Fig. 2f).
Histological observations of callus-derived protoplast cultures
Histological observations revealed that the microcalli originating from the protoplasts of the L1 line were composed of parenchymatous cells with a big vacuole and an irregularly shaped nucleus with one or two nucleoli (Fig. 4a, inset 1). It should be noted that few phenol-containing cells were detected on the surface of the callus (Fig. 4a, inset 2). Microcalli originating from protoplasts of the NL2 line were mainly composed of meristematic cells characterised by dense cytoplasm, small vacuoles and a round-shaped nucleus with one or two nucleoli (Fig. 4b, inset 1). Moreover, few phenolic-containing cells were observed on the surface of the microcalli (Fig. 4b, inset 2). Microcalli obtained from hypocotyl-derived protoplasts also contained phenolic-containing cells on the surface of the calli (Fig. 4c, inset 1). These microcalli were composed chiefly of parenchymatous cells with an irregularly shaped nucleus located near the cell wall (Fig. 4c, inset 2) and small regions of meristematic-like cells with two or three nucleoli and small vacuoles (Fig. 4c, inset 3).

Histological sections of protoplast-derived callus originating from morphogenic callus lines L1 (a), NL2 (b) and hypocotyls (c). a Calli originating from L1-derived protoplasts was composed chiefly of parenchymatous cells (black asterisk) with a big vacuole and an irregularly shaped nucleus with one or two nucleoli (red asterisk) with a nucleus with two or three nucleoli (inset 1); it also has a layer of phenolic-containing cells on the surface (insert 2). b Calli derived from protoplast cultures of the NL2 line presented abundant meristematic cells (red asterisk) with a nucleus with two nucleoli (inset 1) and some phenolic-containing cells (inset 2). c Calli derived from the protoplasts of hypocotyls were characterised by the abundance of parenchymatous cells (black asterisk) with a large vacuole, a peripherally positioned nucleus (inset 2) and small regions of meristematic-like cells (red asterisk) with a nucleus with two or three nucleoli (inset 3). Black arrows indicate nuclei with two or three nucleoli; double black arrows indicate peripheral nuclei in parenchymatous cells. Scale bars: 10 μm (insets), 100 μm (a, b, c)
Plant regeneration
Microcalli originating from all protoplast sources proliferated successfully on the CM medium. The L1 line was cultured on CM medium leading to the formation of dense globular calli that were transferred to the RM. After three months, yellow and brown calli were noted, and, sporadically, green or transparent-green shoot-like structures. After four months of regeneration, shoots started appearing, and plants developed. For the NL2 line, abundant growth of somatic embryos was observed in the first week of culture on the RM (Fig. 1g). After 15 days, green structures and some shoots (Fig. 1h) developed. To develop strong root system plants from all callus protoplast sources were kept in a rooting medium for 4 to 5 weeks and then successfully transferred to soil (Fig. 1i). The tissue derived from hypocotyl protoplast cultures doubled in mass after one month of cultivation on the CM medium. Plant regeneration occurred via somatic embryogenesis (Fig. 2f) or organogenesis (Fig. 2g). Scarce plant regeneration was noted after three months of regeneration (around nine plants from all the experiments undertaken) (Fig. 2h).
Discussion
Single cells like protoplasts may be applied in many fields, such as genetic manipulation, genome editing, the characterisation of plant genes, and somatic hybridisation (Grosser et al. 2010). Significantly, the last method may help overcome incompatibility and hybridisation barriers and develop plant hybrids (Eeckhaut et al. 2013). As mentioned in the introduction, only a few attempts have been reported on the isolation and culture of protoplasts from common buckwheat (Adachi et al. 1989; Gumerova 2004; Rumyanzeva and Lozovaya 1988).
The establishment of a protoplast-to-plant system is affected by many factors, such as the protoplast isolation procedure, yield and quality of protoplasts, culture system, and medium composition (Rahmani et al. 2016). Several factors determine the satisfactory release of protoplasts from the source tissue. Protoplast source tissue, pre-treatment of the tissue before enzymatic maceration, composition of the enzyme solution, tissue digestion conditions, and the protoplast isolation method significantly affected the yield and viability of protoplasts. In the presented investigation, two lines of MC and hypocotyls of common buckwheat were used as the source material for protoplast isolation. The mean yield was around 1 × 106 or 0.44 × 106 protoplasts per g of FW released from the MC and hypocotyls, respectively. Adachi et al. (1989) mentioned releasing 5 × 106 protoplasts per 30 hypocotyls. Also, other materials such as the callus (Gumerova 2004; Rumyanzeva and Lozovaya 1988), mesophyll tissue (Lachmann 1994) and sperm cells (Woo et al. 1999) of common buckwheat were used for protoplast isolation. It has been noted that the authors did not mention the protoplast yield. We presumed that the differences in protoplast yield in our study might result from the different source tissue applied, the growth conditions, and the consequent differences in cell wall composition. Maceration of plant tissue and digestion of cell walls is connected with pectinase and cellulase activity (Noguchi et al. 1978) which characterise, for example, driselase (Kawai et al. 1979). Moreover, driselase had a better enzymolysis effect on the cell wall containing xylan, laminarin and cellulose (Ning et al. 2022). In our research, driselase was applied to improve cell wall digestion and increase tissue maceration. For the L1 line, the application of driselase was not necessary because of the lack of undigested tissue. However, for the NL2 line the beneficial effect of driselase application on the digestion activity and amount of released protoplasts was noted. Additionally, from the amount of enzyme mixture used to release protoplasts from hypocotyl tissue, driselase application resulted in a satisfactory number of obtained protoplasts. Similarly, in our previous work, protoplast isolation from the hypocotyls of Tartary buckwheat was possible after driselase treatment (personal communication). According to Kawai et al. (1979), driselase partially injures the cell wall of Irpex lacteus cotyledons and allows other enzymes to digest the source material. The application of driselase increases the protoplast yield of Brassica oleracea (Robertson and Earle 1986), Spathiphyllum wallisii, Anthurium scherzerianum (Duquenne et al. 2007) and Kalanchoe blossfeldiana (Castelblanque et al. 2009).
The establishment of an appropriate protoplast regeneration protocol is based on optimisation of the different culture conditions, such as the protoplast plating density, the type of cell culture system (e.g., embedding the cells in different gel matrixes) and the composition of the culture media (Davey et al. 2005b; Fehér and Dudits 1994). The overall protoplast density is crucial for cell wall regeneration and daughter cell formation (Davey et al. 2005b). According to Davey et al. (2005b), the typical range of protoplast density in the culture varied from 5 × 104 to 1 × 106 protoplasts per ml. The cell density applied for common buckwheat was from 104 to 105 or 5 × 104 cells per ml (Adachi et al. 1989; Rumyanzeva and Lozovaya 1988). In our study, a higher culture density (2.5 × 105 and 4 × 105 per ml) was applied than was optimal for protoplast development. In Petunia hybrida protoplast cultures (Kang et al. 2020), a plating density of 10 × 104 protoplasts per ml, in contrast to 5 × 104, resulted in a higher frequency of division and the number of calli formed. Similar sightings were recorded by Adedeji et al. (2020) in Chrysanthemum cv. ‘White ND’ protoplast cultures. Furthermore, cultured protoplasts released growth factors that can stimulate the mitotic divisions of neighbouring cells (Davey et al. 2005b). Nevertheless, too high a cell density may result in the accumulation of phenolic compounds in the culture media leading to development of the culture stopping (Adedeji et al. 2020; Kang et al. 2020).
The protoplast embedding technique is the second factor that may significantly affect protoplast culture development. Embedding protoplasts in a semi-solid medium, such as agarose, enables the avoidance of cell agglutination that causes the accumulation of toxic substances, such as polyphenols, that may inhibit cell growth (Davey et al. 2005a; Deryckere et al. 2012). The gel matrix may affect membrane stabilisation by inhibiting lipid peroxidation and reducing metabolites and the diffusion of molecules essential for cell wall synthesis and protoplast division (Eeckhaut et al. 2013; Fehér and Dudits 1994). Furthermore, Deryckere et al. (2012) mentioned that the exchange of nutrients and gases may be more accessible due to the decreased concentration of the LMPA. The culture of Tartary buckwheat protoplasts in agarose beads in comparison to alginate layers had a positive impact on their development (personal communication). Following these results, an agarose embedding matrix was applied in the present study. LMPA beads were noted as a standard method for developing a protoplast-to-plant system in Cichorium. The authors noted that a solid or liquid medium was not optimal for protoplast cultures of the Cichorium genotypes used as the protoplasts burst and died. For the first time, this technique enables plant regeneration from protoplasts of Cichorium endivia genotypes (Deryckere et al. 2012). Also, Ulmus americana protoplasts did not survive in liquid or alginate bead culture systems compared to LMPA beads (Jones et al. 2015).
The optimal protoplast culture media may depend on the species, genotype, and source tissue used (Davey et al. 2005b). The appropriate nutrients, supplements, and PGRs are essential in protoplast cultures. Auxins and cytokinins are necessary for protoplast development (Davey et al. 2005b; Reed and Bargmann 2021). Most media are based on MS or B5 (Gamborg et al. 1968) compounds (Davey et al. 2005b); however, the type of PGRs and ratio may vary. In our study, the medium for protoplast cultures based on a Kao and Michaluk composition was applied and supplemented with NAA and BAP. For the development of protoplasts originating from the callus of common buckwheat, Gamborg’s B5 mineral salts were added (Rumyanzeva and Lozovaya 1988). The authors reported the first mitotic divisions of protoplast-derived cells on days 6–7 of culture but further development of the culture was not observed. Adachi et al. (1989) applied MS salts to hypocotyl-derived protoplast cultures of common buckwheat and tested ten different compositions of PGRs and detected cell divisions after four days. In our investigation, a rich mineral-organic KM medium was applied, and the first cell divisions occurred after five days only after additional supplementation with PSK. We suggest the time differences for the first cell divisions might be due to the genotype used, tissue age, and composition of the culture medium. Applying mineral- and organic-rich media based on the KM formula even affected maintenance of the higher viability rate of Beta vulgaris protoplasts in contrast to the MS salt-based media (Grzebelus et al. 2012b). For Kalanchöe, the protoplast divisions were noted only for the KM medium; for the MS medium, the authors did not observe cell division (Cui et al. 2019).
A widespread way to enhance the mitotic divisions in protoplast cultures involves supplementing the culture medium with surfactants, polyamines, or artificial gases. This study shows the stimulating effect of the peptidyl growth factors, that is, PSK, on protoplast plating efficiency. For the first time, the positive effect of PSK was reported on Asparagus officinalis cell proliferation (Matsubayashi and Sakagami 1996). This sulphated peptide has been found to be effective for promoting cell division in suspension cultures of Oryza sativa (Matsubayashi et al. 1997) and protoplast cultures of Beta vulgaris (Grzebelus et al. 2012b), Daucus species (Mackowska et al. 2014), Brassica oleracea (Kiełkowska and Adamus 2017, 2019) and Fagopyrum tataricum (personal communication). Moreover, Grzebelus et al. (2012b) noted that PSK is able to reverse the recalcitrant behaviour of mesophyll protoplasts originating from Beta vulgaris. Apart from PSK, the polyamine PUT was tested. Polyamines regulate DNA replication, transcription, and translation, affecting cell division and differentiation (Davey et al. 2005b). They protect cells from the oxidative stresses generated during protoplast isolation and culture (Kiełkowska and Adamus 2021; Mackowska et al. 2014). However, in this study, applying PUT was not found to have a significant effect on protoplast plating efficiency. Papadakis and Roubelakis-Angelakis (2005) noted PUT improves cell viability and plating efficiency and prevents the programmed cell death of protoplasts by decreasing the accumulation of superoxide. Huhtinen et al. (1982) demonstrated that the protoplast cultures of Alnus glutinosa and A. incana supported cell division and cell colony formation after the application of ornithine and PUT. Also, Kiełkowska and Adamus (2021) noted the increase in mitotic activity and shoot regeneration in protoplast cultures of Brassica oleracea. Similar to PUT, the application of CPPU, a urea-type synthetic cytokinin, did not increase the number of cell aggregates formed. It was noted that CPPU stimulates cell expansion and division during the development of the fruits of Cucumis sativus (Li et al. 2017) and Actinidia arguta (Kim et al. 2006). Moreover, the application of CPPU affects direct and secondary somatic embryogenesis (Bogdanovic et al. 2021; Murthy and Saxena 1994; Zhang et al. 2005). As phenolic compounds may negatively affect protoplast development, PVP was applied. However, no effect of PVP on the plating efficiency was observed in the present studies. Similar results were noted by Saxena and Gill (1986) and Reustle and Natter (1994). They did not see the apparent effect of PVP on guar and grapevine protoplast plating efficiency. To summarise, our data indicate that PSK is a powerful additional supplement enabling the development of common buckwheat protoplasts.
This study achieved the regeneration of common buckwheat from protoplasts isolated from different donor materials (MC and hypocotyls). Adachi et al. (1989) first attempted to isolate protoplasts from the hypocotyls of common buckwheat and reported abnormal regenerated plants after 18 months of callus culture. Likewise, Gumerova (2004) used the same protoplast source material and noted poor plant regeneration after nine months of culture. In both studies, regeneration was successful, but the yield was low, and the callus obtained from the protoplasts had a low regenerative ability. Compared with those research results, the procedures applied in this study resulted in faster plant regeneration since it took only three to five months. Like Adachi et al. (1989) and Gumerova (2004), we performed the protoplast-derived callus multiplication step on a medium supplemented with auxin and cytokinin. Such a combination has also been well studied for callus induction in other species, such as Lycopersicon esculentum, Nigella damascena and Salvia moorcroftiana (Bano et al. 2022; Chaudhry et al. 2007; Klimek-Chodacka et al. 2020). We followed the same scheme and used a medium supplemented with 2,4-D and KIN. It should be noted that, in our case, the callus multiplication medium (CM_MS1) was additionally supplemented with PSK. Undoubtedly, this medium stimulated callus growth for plant materials originating from all the protoplast sources tested. Although, in the case of the callus originating from hypocotyl- and L1-derived protoplasts, the histological sections revealed the abundance of parenchymatous cells and lack of or small presence of meristematic-like cells (Fig. 4b), which explains the long lasting and poor regeneration rate compared to the NL2 line.
Besides the characteristics of the source material, the culture medium’s composition directly affects tissue regeneration (Adedeji et al. 2020). We used two variants of the MS regeneration medium: RM_MS3 and RM_MS4, which differed in cytokinin composition. The substitution of KIN for TDZ drastically changed the panorama of the experiment, showing an abundant growth of somatic embryos and rapid development of shoots (Fig. 1g). It is typical to use different combinations of PGRs during common buckwheat regeneration, especially cytokinins such as BAP + KIN (Woo et al. 2000), or auxins such as 2,4-D + NAA and IAA + IBA (Kumar and Saraswat 2018). Moreover, auxins, especially 2,4-D, promote the induction of common buckwheat somatic embryogenesis (Gumerova et al. 2001; Gumerova et al. 2003). However, Yang et al. (2012) state that using phenylurea derivatives, especially TDZ, in the RM, stimulates the development of embryogenic cells and, therefore, somatic embryogenesis. This indicates that TDZ may have an auxin effect. Besides, it has been shown that the use of TDZ in in vitro cultures of common buckwheat is more effective for shoot regeneration than traditional purine-type cytokinins (Guo et al. 1992). Berbec and Doroszewska (1999) noted similar results when testing different combinations of growth regulators during regeneration of two common buckwheat diploid (Kora and Hruszowska) and tetraploid cultivars (Emka). According to these authors, the frequency of shoot regeneration was higher after applying IAA + TDZ, even after using TDZ as the only phytohormone in the medium. In our study, the histological sections of the protoplast-derived callus originating from the NL2 line revealed a high presence of meristematic cells, which could be directly influenced by the composition of the culture medium and could explain the short time needed for induction of somatic embryos and regeneration.
To sum up, compared with earlier works related to the regeneration of plants from common buckwheat via protoplast cultures, the protoplast yield and the time to the first division of protoplast-derived cells do not differ much (Table 5). The plating efficiency was considerably higher than in previous research, especially for protoplasts isolated from the L1 line. However, our tremendous success was the time to achieve plant regeneration. Complete regenerated plants were obtained in a maximum of five months, four times faster than Adachi et al. (1989) reported.
Conclusions
The potential of the protoplast-to-plant system for the regeneration of common buckwheat plants using MC-derived from immature embryos as the protoplast source has been confirmed. The use of PSK during protoplast culture and hormonal supplementation (TDZ + KIN and BAP + KIN) during plant regeneration played a critical role. It was also verified that TDZ is efficient for stimulating somatic embryogenesis. This study showed a rapid and potential technique for common buckwheat propagation using in vitro cultures. It is also the basis for future research related to buckwheat crop improvement through genetic engineering or somatic hybridisation.
Data availability
All data generated or analysed during this study are included in this published article.
Abbreviations
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- BAP:
-
6-Benzylaminopurine
- BM:
-
Basal medium
- CM:
-
Callus multiplication medium
- CPPU:
-
N-(2-chloro-4-pyridyl)-N′-phenylurea
- FDA:
-
Fluorescein diacetate
- FW:
-
Fresh weight
- IBA:
-
Indole-3-butyric acid
- KIN:
-
Kinetin
- LMPA:
-
Low melting point agarose
- MC:
-
Morphogenic callus
- NAA:
-
1-Naphthalenacetic acid
- PGRs:
-
Plant growth regulators
- PSK:
-
Phytosulfokine-α
- PUT:
-
Putrescine
- PVP:
-
Polyvinylpyrrolidone
- RM:
-
Regeneration medium
- RT:
-
Room temperature
- TDZ:
-
Thidiazuron
References
Adachi T (1990) How to combine the reproductive system with biotechnology in order to overcome the breeding barrier in buckwheat. Fagopyrum 10(1):7–11
Adachi T, Yamaguchi A, Miike Y, Hoffmann F (1989) Plant regeneration from protoplasts of common buckwheat (Fagopyrum esculentum). Plant Cell Rep 8(4):247–250. https://doi.org/10.1007/bf00778544
Adedeji OS, Naing AH, Kim CK (2020) Protoplast isolation and shoot regeneration from protoplast-derived calli of Chrysanthemum cv. White ND Plant Cell Tiss Org 141(3):571–581. https://doi.org/10.1007/s11240-020-01816-3
Bano AS, Khattak AM, Basit A, Alam M, Shah ST, Ahmad N, Gilani SAQ, Ullah I, Anwar S & Mohamed HI (2022) Callus induction, proliferation, enhanced secondary metabolites production and antioxidants activity of Salvia moorcroftiana L. as influenced by combinations of auxin, cytokinin and melatonin. Braz Arch Biol Techn 65
Berbec A, Doroszewska T (1999) Regeneration in vitro of three cultivars of buckwheat (Fagopyrum esculentum Moench.) as affected by medium composition. Fagopyrum 16:49–52
Betekhtin A, Rojek M, Jaskowiak J, Milewska-Hendel A, Kwasniewska J, Kostyukova Y, Kurczynska E, Rumyantseva N & Hasterok R (2017) Nuclear genome stability in long-term cultivated callus lines of Fagopyrum tataricum (L.) Gaertn. PLoS ONE. https://doi.org/10.1371/journal.pone.0173537
Betekhtin A, Pinski A, Milewska-Hendel A, Kurczynska E, Hasterok R (2019) Stability and instability processes in the calli of Fagopyrum tataricum that have different morphogenic potentials. Plant Cell Tiss Org 137(2):343–357. https://doi.org/10.1007/s11240-019-01575-w
Bogdanovic MD, Cukovic KB, Subotic AR, Dragicevic MB, Simonovic AD, Filipovic BK, Todorovic SI (2021) Secondary somatic embryogenesis in Centaurium erythraea Rafn. Plants 10(2):199. https://doi.org/10.3390/plants10020199
Castelblanque L, García-Sogo B, Pineda B, Moreno V (2009) Efficient plant regeneration from protoplasts of Kalanchoe blossfeldiana via organogenesis. Plant Cell Tiss Org 100(1):107. https://doi.org/10.1007/s11240-009-9617-8
Cawoy V, Lutts S, Kinet JM (2006) Osmotic stress at seedling stage impairs reproductive development in buckwheat (Fagopyrum esculentum). Physiol Plantarum 128(4):689–700. https://doi.org/10.1111/j.1399-3054.2006.00801.x
Cawoy V, Ledent J-F, Kinet J-M, Jacquemart A-L (2009) Floral biology of common buckwheat (Fagopyrum esculentum Moench). Eur J Plant Sci Biotechnol 3(1):1–9
Chaudhry Z, Afroz A, Rashid H (2007) Effect of variety and plant growth regulators on callus proliferation and regeneration response of three tomato cultivars (Lycopersicon esculentum). Pak J Bot 39(3):857–869
Cui J, Kuligowska Mackenzie K, Eeckhaut T, Müller R, Lütken H (2019) Protoplast isolation and culture from Kalanchoë species: optimization of plant growth regulator concentration for efficient callus production. Plant Cell Tiss Org 138(2):287–297. https://doi.org/10.1007/s11240-019-01624-4
Davey MR, Anthony P, Power JB, Lowe KC (2005a) Plant protoplast technology: current status. Acta Physiol Plant 27(1):117–130. https://doi.org/10.1007/s11738-005-0044-0
Davey MR, Anthony P, Power JB, Lowe KC (2005b) Plant protoplasts: status and biotechnological perspectives. Biotechnol Adv 23(2):131–171. https://doi.org/10.1016/j.biotechadv.2004.09.008
Deryckere D, Eeckhaut T, Van Huylenbroeck J, Van Bockstaele E (2012) Low melting point agarose beads as a standard method for plantlet regeneration from protoplasts within the Cichorium genus. Plant Cell Rep 31(12):2261–2269. https://doi.org/10.1007/s00299-012-1335-8
Dirks R, Sidorov V, Tulmans C (1996) A new protoplast culture system in Daucus carota L. and its applications for mutant selection and transformation. Theor Appl Genet 93(5–6):809–815. https://doi.org/10.1007/BF00224080
Duquenne B, Eeckhaut T, Werbrouck S, Van Huylenbroeck J (2007) Effect of enzyme concentrations on protoplast isolation and protoplast culture of Spathiphyllum and Anthurium. Plant Cell Tiss Org 91(2):165–173. https://doi.org/10.1007/s11240-007-9226-3
Eeckhaut T, Lakshmanan PS, Deryckere D, Van Bockstaele E, Van Huylenbroeck J (2013) Progress in plant protoplast research. Planta 238(6):991–1003. https://doi.org/10.1007/s00425-013-1936-7
Fehér A, Dudits D (1994) Plant protoplasts for cell fusion and direct DNA uptake: culture and regeneration system. In: Vasil IK, Thorpe TA (eds) Plant cell and tissue culture. Springer, Netherlands, Dordrecht, pp 71–118
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50(1):151–158. https://doi.org/10.1016/0014-4827(68)90403-5
Grosser JW, Calovic M & Louzada ES (2010) Protoplast fusion technology—somatic hybridization and cybridization. Plant Cell Culture 175–198
Grzebelus E, Szklarczyk M, Baranski R (2012a) An improved protocol for plant regeneration from leaf- and hypocotyl-derived protoplasts of carrot. Plant Cell Tiss Org 109(1):101–109. https://doi.org/10.1007/s11240-011-0078-5
Grzebelus E, Szklarczyk M, Gren J, Sniegowska K, Jopek M, Kacinska I & Mrozek K (2012b) Phytosulfokine stimulates cell divisions in sugar beet (Beta vulgaris L.) mesophyll protoplast cultures. Plant Growth Regul 67(1):93–100. https://doi.org/10.1007/s10725-011-9654-2
Gumerova EA (2004) Realisation of the morphogenic potential of common buckwheat (Fagopyrum esculentum Moench.) hypocotyls depending on the method of regeneration KIBB KSC Russian Academy of Science
Gumerova E, Gatina E, Chuenkova S, Rumyantseva N (2001) Somatic embryogenesis in common buckwheat Fagopyrum esculentum Moench. In: Proceedings of the 8th international symposium on Buckwheat, Chunchon, Korea, Citeseer, pp 377–381
Gumerova EA, Galeeva EI, Chuyenkova SA, Rumyantseva NI (2003) Somatic embryogenesis and bud formation on cultured Fagopyrum esculentum hypocotyls. Russ J Plant Physl+ 50(5):640–645. https://doi.org/10.1023/A:1025640107932
Guo F, Zhou J, Luo X, Ma H (1992) Plant regeneration of tetraploid plants of Fagopyrum esculentum Moench in tissue culture. In: Proceedings of the 5th international symposium on Buckwheat, pp 309–314
Holländer-Czytko H, Amrhein N (1983) Subcellular compartment of shikimic acid and phenylalanine in buckwheat cell suspension cultures grown in the presence of shikimate pathway inhibitors. Plant Sci Lett 29(1):89–96. https://doi.org/10.1016/0304-4211(83)90027-5
Hou SY, Sun ZX, Bin LH, Wang YG, Huang KS, Xu DM, Han YH (2015) Regeneration of buckwheat plantlets from hypocotyl and the influence of exogenous hormones on rutin content and rutin biosynthetic gene expression in vitro. Plant Cell Tiss Org 120(3):1159–1167. https://doi.org/10.1007/s11240-014-0671-5
Huhtinen O, Honkanen J, Simola L (1982) Ornithine- and putrescine-supported divisions and cell colony formation in leaf protoplasts of Alders (Alnus Glutinosa and A. Incana). Plant Sci Lett 28(1):3–9. https://doi.org/10.1016/S0304-4211(82)80003-5
Jones AMP, Shukla MR, Biswas GCG, Saxena PK (2015) Protoplast-to-plant regeneration of American elm (Ulmus americana). Protoplasma 252(3):925–931. https://doi.org/10.1007/s00709-014-0724-y
Kang HH, Naing AH, Kim CK (2020) Protoplast isolation and shoot regeneration from protoplast-derived callus of Petunia hybrida Cv. Mirage Rose Biol 9(8):228. https://doi.org/10.3390/biology9080228
Kao KN, Michayluk MR (1975) Nutritional requirements for growth of Vicia Hajastana cells and protoplasts at a very low population density in liquid media. Planta 126(2):105–110. https://doi.org/10.1007/Bf00380613
Kawai M, Katsumata R, Tsuruta T, Shimura G, Suga Y, Samejima H (1979) The disintegration of soybean by a cellulase preparation from Irpex lacteus Fr. and the enzyme relating to it. Agr Biol Chem Tokyo 43(9):1855–1862. https://doi.org/10.1080/00021369.1979.10863737
Kiełkowska A, Adamus A (2017) Early studies on the effect of peptide growth factor phytosulfokine-α on Brassica oleracea var. capitata L. protoplasts. Acta Soc Bot Pol.https://doi.org/10.5586/asbp.3558
Kiełkowska A, Adamus A (2019) Peptide growth factor phytosulfokine-α stimulates cell divisions and enhances regeneration from B. oleracea var. capitata L. protoplast culture. J Plant Growth Regul 38(3):931–944. https://doi.org/10.1007/s00344-018-9903-y
Kiełkowska A, Adamus A (2021) Exogenously applied polyamines reduce reactive oxygen species, enhancing cell division and the shoot regeneration from Brassica oleracea L. var. capitata protoplasts. Agronomy 11(4):735. https://doi.org/10.3390/agronomy11040735
Kim JG, Takami Y, Mizugami T, Beppu K, Fukuda T, Kataoka I (2006) CPPU application on size and quality of hardy kiwifruit. Sci Hortic 110(2):219–222. https://doi.org/10.1016/j.scienta.2006.06.017
Klimek-Chodacka M, Kadluczka D, Lukasiewicz A, Malec-Pala A, Baranski R, Grzebelus E (2020) Effective callus induction and plant regeneration in callus and protoplast cultures of Nigella damascena L. Plant Cell Tiss Org 143(3):693–707. https://doi.org/10.1007/s11240-020-01953-9
Kumar M, Saraswat R (2018) Plant regeneration and genetic transformation in buckwheat (Fagopyrum spp.), a multipurpose gluten free crop of high nutraceutical importance: a critical review. Ann Plant Sci 7:1954–1962. https://doi.org/10.21746/aps.2018.7.1.7
Kwon S-J, Han M-H, Huh Y-S, Roy SK, Lee C-W, Woo S-H (2013) Plantlet regeneration via somatic embryogenesis from hypocotyls of common buckwheat (Fagopyrum esculentum Moench.). Korean J Crop Sci 58(4):331–335
Lachmann SA (1990) Callus regeneration from hypocotyl protoplast of tartary buckwheat (Fagopyrum tataricum Gaertn.). Fagopyrum 10:62–64
Lachmann S, Kishima Y, Adachi T (1994) Protoplast fusion in buckwheat: preliminary results on somatic hybridization. Fagopyrum 14:7–12
Li J, Xu J, Guo Q-W, Wu Z, Zhang T, Zhang K-J, Cheng C-y, Zhu P-y, Lou Q-F, Chen J-F (2017) Proteomic insight into fruit set of cucumber (Cucumis sativus L.) suggests the cues of hormone-independent parthenocarpy. Bmc Genomics 18(1):896. https://doi.org/10.1186/s12864-017-4290-5
Mackowska K, Jarosz A, Grzebelus E (2014) Plant regeneration from leaf-derived protoplasts within the Daucus genus: effect of different conditions in alginate embedding and phytosulfokine application. Plant Cell Tiss Org 117(2):241–252. https://doi.org/10.1007/s11240-014-0436-1
Matsubayashi Y, Sakagami Y (1996) Phytosulfokine, sulfated peptides that induce the proliferation of single mesophyll cells of Asparagus officinalis L. Proc Natl Acad Sci USA 93(15):7623–7627. https://doi.org/10.1073/pnas.93.15.7623
Matsubayashi Y, Takagi L, Sakagami Y (1997) Phytosulfokine-α, a sulfated pentapeptide, stimulates the proliferation of rice cells by means of specific high- and low-affinity binding sites. Proc Natl Acad Sci USA 94(24):13357–13362. https://doi.org/10.1073/pnas.94.24.13357
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plantarum 15(3):473–497. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Murthy BNS, Saxena PK (1994) Somatic embryogenesis in peanut (Arachis hypogaea L.): stimulation of direct differentiation of somatic embryos by forchlorfenuron (CPPU). Plant Cell Rep 14(2):145–150 doi:https://doi.org/10.1007/BF00233779
Nešković M, Vujičić R, Budimir S (1987) Somatic embryogenesis and bud formation from immature embryos of buckwheat (Fagopyrum esculentum Moench.). Plant Cell Rep 6:423–426. https://doi.org/10.1007/bf00272773
Ning Y, Hu B, Yu H, Liu X, Jiao B, Lu X (2022) Optimization of protoplast preparation and establishment of genetic transformation system of an arctic-derived fungus Eutypella sp. Front Microbiol 13:769008. https://doi.org/10.3389/fmicb.2022.769008
Noguchi S, Shimura G, Kawai M, Suga Y, Samejima H (1978) Properties of partially purified cellulolytic and plant tissue macerating enzymes of Irpex lacteus Fr. in special reference to their application Agric Biol Chem 42(2):339–345. https://doi.org/10.1271/bbb1961.42.339
Papadakis AK, Roubelakis-Angelakis KA (2005) Polyamines inhibit NADPH oxidase-mediated superoxide generation and putrescine prevents programmed cell death induced by polyamine oxidase-generated hydrogen peroxide. Planta 220(6):826–837. https://doi.org/10.1007/s00425-004-1400-9
Rahmani M-S, Pijut PM, Shabanian N (2016) Protoplast isolation and genetically true-to-type plant regeneration from leaf- and callus-derived protoplasts of Albizia julibrissin. Plant Cell Tiss Org 127(2):475–488. https://doi.org/10.1007/s11240-016-1072-8
Reed KM, Bargmann BOR (2021) Protoplast regeneration and its use in new plant breeding technologies. Front Genome Ed 3:734951. https://doi.org/10.3389/fgeed.2021.734951
Reustle G, Natter I (1994) Effect of polyvinylpyrrolidone and activated charcoal on formation of microcallus from grapevine protoplasts (Vitis sp.). Vitis 33(3):117–121. https://doi.org/10.5073/vitis.1994.33.117-121
Robertson D, Earle ED (1986) Plant regeneration from leaf protoplasts of Brassica oleracea var. italica CV Green Comet broccoli. Plant Cell Rep. 5(1):61–64.
Rumyanzeva NI, Lozovaya VV (1988) Isolation and culture of buckwheat (Fagopyrum esculentum Moench.) callus protoplasts. In: Puite KJ, Dons JJM, Huizing HJ, Kool AJ, Koornneef M, Krens FA (eds) Progress in plant protoplast research: proceedings of the 7th international protoplast symposium, Wageningen, the Netherlands, December 6–11, 1987. Springer Netherlands, Dordrecht, pp 45–46
Rumyantseva NI, Sal’nikov VV, Lebedeva VV (2005) Structural changes of cell surface in callus of Fagopyrum esculentum Moench. during induction of morphogenesis. Russ J Plant Physl+ 52(3):381–387. https://doi.org/10.1007/s11183-005-0057-y
Sakamoto S, Matsui K, Oshima Y, Mitsuda N (2020) Efficient transient gene expression system using buckwheat hypocotyl protoplasts for large-scale experiments. Breed Sci 70(1):128–134. https://doi.org/10.1270/jsbbs.19082
Saxena PK, Gill R (1986) Removal of browning and growth enhancement by polyvinylpolypyrrolidone in protoplast cultures of Cyamopsis tetragonoloba L. Biol Plant 28(4):313–315. https://doi.org/10.1007/BF02902302
Shaikh N, Guan L, Adachi T (2001) Ultrastuctural analyses on breeding barriers in post-fertilization of interspecific hybrids of buckwheat. In: Proceding of the VIII internatiol an symposium on Buckwheat, pp 319–329
Slawinska J, Obendorf RL (2001) Buckwheat seed set in planta and during in vitro inflorescence culture: evaluation of temperature and water deficit stress. Seed Sci Res 11(3):223–233
Slawinska J, Kantartzi SK, Obendrof RL (2009) In vitro organogenesis of Fagopyrum esculentum Moench (Ploygonaceae) as a method to study seed set in buckwheat. J Plant Sci Biotechnol 3(1):73–78
Takahata Y & Jumonji E (1985) Plant regeneration from hypocotyl section and callus in buckwheat (Fagopyrum esculentum Moench.). Ann Rep Fac Educ Iwate Univ 45(1)
Taylor DP, Obendorf RL (2001) Quantitative assessment of some factors limiting seed set in buckwheat. Crop Sci 41(6):1792–1799. https://doi.org/10.2135/cropsci2001.1792
Tomasiak A, Zhou M, Betekhtin A (2022) Buckwheat in tissue culture research: current status and future perspectives. Int J Mol Sci 23(4):2298. https://doi.org/10.3390/ijms23042298
Woo S-H, Adachi T, Jong SK, Campbell CG (1999) Isolation of protoplasts from viable sperm cells of common buckwheat (Fagopyrum esculentum Moench.). Can J Plant Sci 80:583–585
Woo SH, Nair A, Adachi T, Campbell CG (2000) Plant regeneration from cotyledon tissues of common buckwheat (Fagopyrum esculentum Moench). In Vitro Cell Dev-Pl 36(5):358–361. https://doi.org/10.1007/s11627-000-0063-x
Woo S, Ohmoto T, Campbell C, Adachi T, Jong S (2001) Pre-and post-fertilization to backcrossing in interspecific hybridization between Fagopyrum esculentum and F. homotropicum with F. esculentum. In: Proceedings of the 8th international symposium on Buckwheat, Chunchon, Korea. Citeseer, pp 450–455
Woo S, Takaoka M, Kim H, Park C, Adachi T, Jong S (2004) Plant regeneration via shoot organogenesis from leaf callus culture of common buckwheat (Fagopyrum esculentum Moench.). In: Proceedings of the 9th international symposium on Buckwheat, pp 61–65
Woo S-H, Kamal AM, Tatsuro S, Campbell CG, Adachi T, Yun Y-H, Chung K-Y, Choi J-S (2010) Buckwheat (Fagopyrum esculentum Moench.): concepts, prospects and potential. Eur J Plant Sci Biotechnol 4(1):1–16
Yamane Y (1974) Induced differentiation of buckwheat plants from subcultured calluses in vitro. Jpn J Genetics 49(3):139–146
Yang X, Lü J, da Silva JAT, Ma G (2012) Somatic embryogenesis and shoot organogenesis from leaf explants of Primulina tabacum. Plant Cell Tiss Org 109(2):213–221. https://doi.org/10.1007/s11240-011-0087-4
Zhang Q, Chen J, Henny RJ (2005) Direct somatic embryogenesis and plant regeneration from leaf, petiole, and stem explants of Golden Pothos. Plant Cell Rep 23(9):587–595. https://doi.org/10.1007/s00299-004-0882-z
Zhang G, Xu Z, Gao Y, Huang X, Zou Y, Yang T (2015) Effects of germination on the nutritional properties, phenolic profiles, and antioxidant activities of buckwheat. J Food Sci 80(5):H1111–H1119. https://doi.org/10.1111/1750-3841.12830
Funding
This research was funded by the National Science Centre, Poland. Research project OPUS-19 (No. Reg. 2020/37/B/NZ9/01499 awarded to AB).
Author information
Authors and Affiliations
Contributions
Conceptualisation: AB, EG; Methodology: MZ, RP-P, AM-H, AB, EG; Formal analysis: MZ, RP-P, AM-H, AB, EG; Investigation: MZ, RP-P, AM-H, AB; Resources: AB, EG; Writing—original draft: MZ, RP-P, AB; Writing—review and editing: MZ, RP-P, AM-H, AB, EG; Visualisation: MZ, RP-P; Supervision: AB, EG; Project administration: AB, EG; Founding acquisition: AB. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
The use of all plant materials in this study complies with relevant institutional, national, and international guidelines and legislation. Seeds of F. esculentum cultivar Panda are commercially available and were purchased from the Malopolska Plant Breeding company (Poland).
Additional information
Communicated by Wagner Campos Otoni.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zaranek, M., Pérez-Pérez, R., Milewska-Hendel, A. et al. Efficient and rapid system of plant regeneration via protoplast cultures of Fagopyrum esculentum Moench. Plant Cell Tiss Organ Cult 154, 673–687 (2023). https://doi.org/10.1007/s11240-023-02542-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-023-02542-2