
Abstract
In the present study we report the development of an effective and relatively efficient protocol for protoplast-to-plant regeneration of parsnip (Pastinaca sativa L.) via indirect somatic embryogenesis. The regenerative potential of three open-pollinated and four hybrid cultivars was assessed. The protoplast isolation efficiency after digestion of source material in an enzyme mixture consisted of 1% cellulase Onozuka R-10 and 0.1% pectolyase Y-23 reached on average 3.6 × 106 of cells per g of fresh mass. Protoplasts embedded in an alginate matrix and cultured in parsnip protoplast culture medium with phytosulfokine-α and putrescine reconstructed their cell wall and re-entered mitotic divisions. After the release from alginate, microcallus proliferated continuously on Gamborg B5 medium with vitamins supplemented with 100 nM of phytosulfokine-α. Indirect somatic embryogenesis occurred during the callus culture of cultivar ‘Półdługi biały’. The regenerated and acclimatized plants were morphologically similar to their donors and displayed no variation in the ploidy level.
Key message
The main objective of this study was to develop the protoplast-to-plant regeneration protocol for parsnip that could be exploited as a platform for production of somatic hybrids via protoplast fusion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants have a remarkable regeneration potential, which ensures their adaptational success, as they have to cope with the damage caused by both abiotic and biotic environmental conditions. This regeneration potential has attracted scientific interest initially focused on the plant response to wounding and formation of proliferating cell masses (Sugiyama 2015). It was soon discovered that phytohormones are essential for in vitro plant cell proliferation, and with the appropriate auxin to cytokinin ratio regeneration of both shoot and root was possible (Skoog and Miller 1957). Certain somatic cells might gain pluripotency or even totipotency under specific conditions. This might lead to the formation of embryo-like structures, as described for carrot phloem suspension cultures (Steward 1958; Schmidt et al. 1997) tracked back somatic embryo formation to small cell clusters or even single cells producing proembryogenic mass (PEM) - an intermediate step toward somatic embryogenesis (SE). SE proved to be an interesting process for both basic and applied studies. Due to a considerable similarity to zygotic embryogenesis, SE has been extensively studied in order to describe morphophysiological, biochemical and molecular events occurring in the course of embryogenesis in Angiosperms and Gymnosperms (Tautorus et al. 1991; Zimmerman 1993; Dodeman et al. 1997; Quiroz-Figueroa et al. 2006). SE has also become an essential tool in plant biotechnology - not only is it a novel system for crop improvement that might aid conventional agricultural methods, but also might be used as an alternative to traditional mass propagation protocols (Kumar and van Staden 2017). Embryogenic tissue has the potential to produce embryos without genetic alternation for an extended period of time, as described for mango, banana or coriander (Ganapathi et al. 2001; Ara et al. 2004; Murthy et al. 2008). This feature shows prospects for a long-term preservation of plant genetic resources and production of artificial seeds (Rai et al. 2010; Rihan et al. 2017).
Protoplasts serve as a unique tool for a range of procedures focused on overcoming naturally occurring sexual incompatibility barriers and the efficient genetic transformation of plant cells. Related genera of cultivated crops possess large reservoirs of genes having various agronomically important traits such as increased resistance to abiotic and biotic stress, cytoplasmic male sterility (CMS) or phenotypic traits resulting in crop quality improvement (Sherraf et al. 1994; Cheng et al. 1995; Yu et al. 2013; Guo et al. 2013). Exploitation of protoplast technology coupled with their symmetrical or asymmetrical fusion can effectively contribute to the generation of novel germplasm for elite breeding of conventional crosses and promote crop improvement in existing cultivars (Carlson et al. 1972; Wang et al. 2013). The efficiency of protoplast-to-plant regeneration might be impacted by somaclonal variation occurring in in vitro cultures (Larkin and Scowgraft 1981). Undifferentiated cells, with protoplasts and protoplast-derived callus among them, are particularly prone to genetic changes triggered by unfavourable physicochemical conditions (Krishna et al. 2016). Somaclonal variation might result in ploidy changes, as a result of endoreduplication in callus cells (Ochatt et al. 2000) or lead to genetic variation attributed to single/multiple base substitution or indels, chromosome rearrangements or changes in the status of DNA methylation (Krishna et al. 2016). In commercial crop production based on micropropagation, somaclonal variation is generally considered as undesirable as it might lead to abnormal morphology less vigour or delayed flowering (Winkelmann et al. 2008). On the other hand, genetic variability obtained through protoplast regeneration might prove beneficial in enhancing resistance to both abiotic and biotic stress, as described for carrot (Grzebelus et al. 2013; Kiełkowska et al. 2019). Nonetheless, the implementation of protoplast fusion requires a well-established tissue culture system, including protoplast isolation, plant regeneration via SE or organogenesis and acclimatization to greenhouse or field conditions. It has to be tailored to the species of interest.
Parsnip (Pastinaca sativa L.) is a monocarpic perennial species typically grown as a biennial crop. It produces thick white-to-yellow, funnel-shaped taproot rich in fiber, calcium and manganese (Chappell and Dunford 2021). Wild parsnip most probably originated in the Caucasus Mountains – a centre for diversity of Pastinaca genus. It is thought to have spread throughout the world as a result of its domestication (Rubatzky et al. 1999). In some regions wild parsnip is considered an invasive species due to its ability to adapt to variable environmental conditions (Averill and DiTommaso 2007). Although this member of Apiaceae family has been grown for human consumption for centuries, it is still considered as a niche crop. Today parsnip is gaining popularity, mostly due to the increased product variety on the European market. The discovery of cytoplasmic male sterility in wild parsnip populations, coupled with its introduction into cultivated populations, facilitated the production of F1 hybrids. Hybrids represent the majority of seeds available on the market but older cultivars are mostly open-pollinated. Nowadays, the majority of genetic improvements, such as an increase of total yield or introgression of pest/disease resistance, are obtained through hybridization of germplasm with desirable phenotypic traits (Chappell and Dunford 2021). The availability of public resources from which genetic diversity could be exploited in parsnip breeding is quite limited. Currently there are also no reports of in vitro germplasm conservation of parsnip. Moreover, no work focused on somatic hybridization of Pastinaca sativa has been published to date.
In the present study, we investigated protoplast isolation efficiency and regeneration ability in a set of seven open-pollinated and hybrid cultivars of parsnip. We aimed at the development of an effective and relatively efficient protocol for the regeneration of plants, exploiting the advantages of somatic embryogenesis. Considering the growing economic importance of cultivated parsnip, the developed protoplast-to-plant re-generation protocol might aid traditional breeding programs. It is also a crucial premise for further attempts at protoplast fusion. It could be exploited as a tool for broadening available germplasm collections and for the transfer of cytoplasmic male sterility into other male-fertile Apiaceae species.
Materials and methods
Material sources and culture conditions
Seven parsnip accessions were used as a source of protoplasts, including three open-pollinated and four hybrid cultivars Selected cultivars were characterised by a considerable variability within the most important agronomical traits such as earliness, length and shape of the storage root, yield or canker resistance (Table 1). Protoplasts were isolated from in vitro grown plants produced from surface sterilized seeds. Seeds were sterilized as follows: first, seeds were incubated in a water bath at 40 °C for 30 min, then transferred to 0.2% (v/v) solution of fungicide Gwarant (active compound: chlorothalonil; UPL, PL) and placed on a gyratory shaker (180 rpm) for 30 min. Finally, the seeds were immersed in 20% (w/v) solution of chloramin T (Chempur, PL) for 30 min. After each step, the seeds were rinsed in 70% ethanol for 30–60 s. After three washes with sterile water for 5 min each, the seeds were air-dried on a sterile filter paper. Dry seeds were transferred onto a 9 cm Petri dish with solid germination medium (MS30; Table 2). Cultures were kept in the dark, a 24-hour cycle comprised of a 16-hour incubation at 20 °C and an 8-hourincubation at 30 °C. After 21 days seedlings were transferred into jars with fresh MS30 medium and incubated at 24 ± 2 ºC with a 16/8 h (light/dark) photoperiod (a light intensity of 55 µmol m–2 s–1; fluorescent lamps Sylvania Gro-lux T8, USA).
Protoplast isolation and culture
Protoplasts of parsnip accessions were isolated from expanded leaves with petioles of 2-week-old in vitro grown plants. Tissue (approximately 1 g) was placed on a 90 × 15 mm Petri dish with 8 ml of PSII solution (Table 2) and immediately cut into fine pieces and then incubated 1 h in the dark at 26 ± 2 °C for cell plasmolysis. Then the mannitol solution was replaced by an enzyme solution (Table 2). Tissue was digested on a gyratory shaker (28 rpm; Rotamax 120 Heidolph Instruments, DE) for 14–16 h, in the dark at 26 ± 2 °C. The released protoplasts were separated from undigested tissue by filtration through a 100 μm nylon sieve (Merck Millipore) and centrifuged at 100 g for 5 min (MPW-223e, MPW Med Instruments, PL; rotor type: MPW no 12,485). The pellet was resuspended in 8 ml of sucrose/MES buffer (Table 2), very slowly overlaid with 2 ml of W5 salt solution (Table 2), and centrifuged at 145 g for 10 min. Viable protoplasts localized at the interface between two solutions of different concentrations as a green band were transferred into a fresh tube and washed twice by centrifugation at 100 g for 5 min in one of the filter-sterilized protoplast culture media: coriander protoplast culture medium (CPC), fennel protoplast culture medium (FPC) or parsnip protoplast culture medium (PPC6; Table 2). CPC and FPC media were adapted from Ali et al. (2018b; Miura and Tabata (1986), respectively, with some modifications. PPC6 medium was developed on the basis of carrot protoplast culture medium (Grzebelus et al. 2012a), with some modifications. After the purification step, protoplast yield was determined by cell counting, using Fuchs Rosenthal hemocytometer chamber. The working density of protoplasts was adjusted to 8 × 105 protoplasts per ml.
Equal volumes of protoplast suspension in either CPC, FCP or PPC6 medium and sodium alginate solution (Table 2) were mixed carefully. Aliquots (app. 400 µl) of protoplasts/alginate mixture were spread onto Ca-agar medium (Table 2) in 60 × 15 mm Petri dishes and one thin alginate layer per dish was formed as described by Grzebelus et al. (2012a). After a one-hour incubation at room temperature, solidification of the alginate matrix with embedded protoplasts occurred. Solid thin alginate layers with immobilized protoplasts were gently transferred to 60 × 15 mm Petri dishes containing 4 ml of CPC, FCP or PPC6 medium (one alginate layer per one Petri dish). Additionally, two variants of PPC6 medium were used: PPC6 supplemented with 100 nM phytosulfokine-α (PSK; PeptaNova GmbH, DE) and PPC6 supplemented with 100 nM PSK and 8 mg ml−1 putrescine (Pu; Duchefa Biochemie). In order to maintain aseptic conditions of the cultures, 400 mg ml−1 cefotaxime (Polfa Tarchomin SA, PL) was added to all media. Cultures were incubated at 26 ± 2 °C in the dark. Culture medium with all supplements was renewed after 10 days of culture.
Plant regeneration
After about 8 weeks of protoplast culture, visible protoplast-derived callus microcolonies were separated from alginate layers by incubation in 8 ml of sodium citrate solution (Table 2) for 30 min to one hour. The alginate residues and citrate solution were removed by centrifugation (5 min at 100 g) and the pellet was carefully washed in the CPPD medium (Dirks et al. 1996; Table 2). Colonies derived from one alginate layer were resuspended in 4 ml of the CPPD medium and plated in 2 ml aliquots on filter paper placed in 90 × 15 mm Petri dish with the BI + PSK medium (Table 2). Cultures were maintained in the dark at 26 ± 2 °C and subcultured on the fresh BI + PSK medium every 30 days until the formation of proembryogenic mass (PEM) and the development of somatic embryos. Developing embryos were transferred into plastic jars with the R medium (Table 2) and incubated for 8 weeks at 24 ± 2 °C and 16 h photoperiod (a light intensity of 55 µmol m–2 s–1; fluorescent lamps Sylvania Gro-lux T8, USA), with one transfer onto fresh R medium after 4 weeks of culture. Regenerated plants were transferred to peat substrate at 20 °C and kept in the SANYO MLR-352 H climatic chambers (Sanyo Electric Biomedical Co. Ltd., JP) at 90% humidity for 2 weeks and then 70% for the next 2–3 weeks. Then they were transferred into 16 cm pots filled with universal potting soil (Hartmann, PL) and grown under normal greenhouse conditions (18–26 °C, 16/8 photoperiod, light intensity of 40 µmol m−2 s−1 - sodium lamps Lucalox LU600W/PSL, HU, optimally irrigated and fertilized). The ploidy level of regenerants was determined by flow cytometry, as described by Kiełkowska and Adamus (2010), using leaf samples from greenhouse-grown plants. The nuclear suspensions were measured for the relative nuclear DNA content using Partec CyFlow Ploidy Analyser (Sysmex,JP). Leaves of diploid (2n = 2x = 18) plants of parsnip were used as a reference standard.
Data collection and statistical analysis
Isolation yield was determined using a hemocytometer (Heinz Herenz, DE) and presented as the number of protoplasts per gram of fresh mass (FM). The viability of protoplasts was estimated by staining with fluorescein diacetate (FDA) one hour after immobilization in alginate matrix, and expressed as a percentage of protoplasts with green fluorescence out of total observed cells. The protocol for protoplast staining was as follows: 15 µl of 0.3% filter-sterilized FDA-acetone stock solution was dissolved in 1 ml of PPC6 medium to prepare FDA working solution. 100 µl of that solution was added to the culture of immobilized protoplasts. Plating efficiency, expressed as a percentage of the number of cell colonies per total number of observed objects (i.e. cell aggregates and undivided cells), was assessed in the 20-day-old cultures. All microscopic observations were performed under an inverted Leica DMi8 microscope (Leica Microsystems, Germany) with suitable filter set for visualization of fluorescein fluorescence (λEx = 460–500 nm, λEm = 512–542 nm).
The regeneration potential of PEM was then estimated as the ratio of developed somatic embryos to fully regenerated, morphologically intact plants produced after 8 weeks of culture on R medium. The rate of successful acclimatization of regenerated plants to ex vitro conditions was estimated 6 weeks after the transfer to the greenhouse.
As repetitions, three to six independent protoplast isolation experiments were carried out. Each single treatment was represented by three Petri dishes. Microscopic observations were performed on 300–400 cells per Petri dish. The overall effect of treatments was determined using multivariate analysis of variance (ANOVA) in Statistica ver. 13.0 (StatSoft. Inc.) at p ≤ 0.05. Tukey HSD post-hoc test for an unequal sample size was used for the separation of means.
Results
Yield and viability of isolated protoplasts
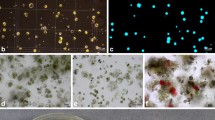
The leaves from in vitro grown plantlets (Fig. 1a) of all cultivars were an effective source of tissue for protoplast isolation, releasing on average 3.6 ± 0.3 × 106 cells per g of FM (Figs. 1b and 2a). The mean protoplast yield for ‘Gladiator F1’ was approximately twice as high (5.1 ± 1.4) as the yield obtained for the least efficient accession ‘Victor F1’ (2.3 ± 0.8). The quality of alginate embedded protoplasts, determined by staining with FDA, was high regardless of accession and varied from 67.0 ± 1.6 for ‘Gladiator F1’ to 89.5 ± 1.0 for ‘Countess F1’ (Fig. 2b). The viable protoplasts were round in structure with no tendency to shrinking (Fig. 1c, d).

Protoplast-to-plant regeneration of parsnip. a Twenty-one day-old seed-derived donor plant for protoplast isolation; b mesophyll-derived protoplasts; c fluorescein diacetate (FDA) stained protoplast one hour after alginate embedding; d apple-green fluorescence of viable protoplast after FDA staining; e plasmolysis of protoplast in a 10-day-old culture in the CPC medium; f first mitotic division of protoplast-derived cell in the PPC6 medium; g, h multicellular protoplast-derived aggregates; i alginate layer fully overgrown with callus; j non-embryogenic callus proliferating on BI + PSK medium; k proembryogenic mass developed in the fourth month of callus culture on BI + PSK medium, l protoplast-derived proembryogenic mass with globular somatic embryos; m cotyledonary somatic embryo; n complex of protoplast-derived somatic embryos at different stages of development; o protoplast-derived plant ready for ex vitro acclimatization. Scale bars: 50 μm (b–h), 1 mm (k–m), 1 cm (a, i–j, o)

Effect of the applied protocol of protoplast isolation, purification and culture on the yield, quality and plating efficiency in parsnip protoplast cultures. a The yield of protoplast isolations per 1 g of fresh mass (n = 3–6); b protoplast viability 1 h after isolation (n = 3–6); c the effect of the culture medium on plating efficiency in the 20-day-old cultures (n = 3); d the effect of PPC6 medium supplementation on plating efficiency in the 20-day-old cultures (n = 3–6); CPC coriander protoplast culture medium, FCP fennel protoplast culture medium, PPC6 parsnip protoplast culture medium, PSK phytosulfokine (100 nM); Pu – putrescine (8 mg ml-1). Bars represent standard error. Means denoted with different letters were significantly different ( p ≤ 0.05)
Formation of multicellular aggregates on different culture media
Alginate embedded protoplasts derived from seven accessions were cultured in a liquid CPC, FPC and PPC6 medium supplemented with PSK and PSK in combination with Pu. Cultures in CPC medium, FPC medium and PPC6 medium with no PSK and Pu supplementation were treated as a control. During first 20 days of culture no mitotic activity was observed in the CPC and FPC cultures, either with or without the PSK and Pu supplementation and in the control (Fig. 2c). Observed protoplasts were severely damaged, with disrupted cell wall integrity and progressing plasmolysis (Fig. 1e). After 6 days of culture the first mitotic divisions were observed in PPC6 medium supplemented with both PSK and PSK + Pu (Fig. 1f), whereas no mitotic activity was observed until the 12th day of culture in PPC6 control cultures. The mean plating efficiency (i.e. number of cell aggregates forming) assessed in the 20th day of protoplast culture varied significantly between the analysed accessions (from 7.0 ± 1.5% for ‘Countess F1’ to 23.9 ± 2.6% for ‘Półdługi biały’; Table 3). The exogenously applied PSK and PSK + Pu had a beneficial effect on the division rate of the protoplast-derived parsnip cells that resulted in the formation of large multicell aggregates (Fig. 1g, h). On the 20th day of culture only 6.2 ± 0.9% of cells cultured on the control PPC6 medium underwent division and formed multicell aggregates. The number of cells forming aggregates observed in PSK and PSK + Pu supplemented media was 2.5 to 3-fold higher (p < 0.01) in comparison with the control (16.8 ± 1.9% and 19.1 ± 2.0%, respectively, Table 4). No significant differences in the stimulation of cell division were observed between PPC6 cultures supplemented with PSK and PSK combined with Pu (Table 3). The beneficial effect of exogenously applied PSK on cell divisions was universal and accession-independent, i.e., protoplast cultures of each cultivar showed a significant increase in the plating efficiency in PPC6 medium supplemented with both PSK and PSK + Pu (Fig. 2d).
Plant regeneration
Protoplast-derived callus (Fig. 1i) released from the alginate matrix proliferated continuously on BI + PSK medium (Fig. 1j). Callus transferred onto BI PSK-free medium did not proliferate and showed signs of ageing, i.e., it turned brown and dried up. Development of proembryogenic mass was observed only for ‘Półdługi biały’ callus derived from protoplasts cultured in PPC6 medium supplemented with PSK, after four months of culture on BI + PSK medium in the dark (Fig. 1k). The embryos originated at the upper surface of the callus and were non-synchronous in development (Fig. 1l–n). The regeneration potential of PEM (the ratio of developed embryos to regenerated plants) was calculated at 6.7%.
In total, 82 properly developed plants, i.e. producing 10–15 true leaves and 4–5 primary roots with no apparent phenotypic alterations, ready for ex vitro acclimatization, were obtained (Fig. 1o). Fifty-seven plants (~ 70%) were successfully acclimatised to the greenhouse conditions. All of the analysed plants retained the ploidy of donor plants (2n = 22; Table 4). The whole protocol of protoplast-to-plant regeneration of parsnip took approximately 8–10 months to obtain plants that grew naturally in outdoor conditions.
Discussion
In this study, we developed an efficient protoplast isolation and culture method for several parsnip cultivars, along with the protocol for protoplast-to-plant regeneration via somatic embryogenesis for ‘Półdługi biały’ cultivar. To the best of our knowledge, this is the first ever observation of plant regeneration in Pastinaca sativa from protoplasts via somatic embryogenesis. The primary aim of this study was to establish a suitable protocol of protoplast isolation that can be used in somatic hybridization program in the future.
The efficiency of protoplast isolation and plant regeneration depends on several factors, including the genotype, source of donor tissue, culture system, and medium composition (Reed and Bargmann 2021). We used leaves and petioles of in vitro grown plants as a source of protoplasts, similarly to other studies carried out on the species of the Apiaceae family (Grzebelus et al. 2012a; Maćkowska et al. 2014; Kiełkowska et al. 2019). The use of such a tissue source for protoplast isolation, instead of often utilized suspension cultures (Cai and Kang 2014; Hu et al. 2015; Ali et al. 2018b), has certain advantages. Firstly, the long-term cell suspension cultures are prone to somaclonal variation that could possibly impact the regenerative ability of isolated protoplasts, as described in oil palm (Masani et al. 2013). Moreover, in commercial crop production, somaclonal variation is regarded as an undesirable phenomenon that might impact the transfer of agronomically important traits during the fusion of protoplasts. The use of cell suspensions might also pose some inconvenience due to higher maintenance requirements and costs when compared to in vitro seed germination and seedling culture. On the other hand, when the regenerative ability of protoplasts derived from somatic tissues is very low and limits further implementations, as in grapevine, the use of embryogenic callus can potentially improve regeneration success (Bertini et al. 2019).
Appropriate enzyme treatment and incubation time are crucial for obtaining viable protoplasts when working on diverse tissues of different plant species (Chamani et al. 2012). The 14–16 h incubation of tissue in the mixture of cell-wall degradation enzymes, 0.1% pectolyase and 1% cellulase, have made it possible to isolate protoplasts in satisfactory numbers, comparable or higher to those obtained in other studies carried out on species belonging to Apiaceae family, i.e., carrot, coriander, fennel or parsley (Dudits et al. 1980; Miura and Tabata 1986; Grzebelus et al. 2012a; Maćkowska et al. 2014; Mujib et al. 2014; Ali et al. 2018a; Kiełkowska et al. 2019). When large populations of protoplasts are required, e.g. for fusion, 105 to 107 viable cells should be released from 1 g of fresh mass (Davey et al. 2010). Even though the protoplast yield obtained in this study did not exceed 107 and was clearly shaped by the genotype, it was sufficient to perform further implementations such as plant regeneration. The viability of protoplasts released from the studied accessions was high (on average 77%) and proved that the used protocol of isolation was suitable for obtaining large quantities of high-quality material for further culture.
The osmotic conditions of the solutions and media in protoplast-based culture systems are critically important. Several osmotic compounds such as mannitol, glucose, or sucrose are frequently added to the solutions and media used for protoplast isolation and culture (Reed and Bargmann 2021). In the presented protocol of isolation, mannitol was used in concentrations described by Grzebelus et al.(2012) as optimal for the isolation of protoplasts from carrot. However, due to very low viability of protoplasts, the osmotic pressure of the culture medium was considerably increased by the addition of 0.6 M of glucose (compared to 0.4 M for carrot protoplast culture medium), as the major osmotic regulator. This change provided suitable conditions for the re-formation of the cell wall and first mitotic divisions in the protoplast cultures of parsnip.
The applied concentrations of plant growth regulators in the liquid culture medium promoted divisions of embedded protoplasts within the first 6 days of culture. Davey et al. (2010) concluded that cell wall synthesis and mitotic divisions in early phases of protoplast culture are crucial in the successful development of plant regeneration systems. Protoplasts of ‘Półdługi biały’ and ‘Gladiator F1’ were characterized by the highest frequency of divisions and therefore produced more microcallus colonies during the culture. Medium supplementation with a plant peptide phytosulfokine-α, a signal molecule involved in many development processes, including cell-to-cell communication (Matsubayashi 2013), cell growth and expansion (Kutschmar et al. 2009) and adventitious root formation (Yamakawa et al. 1998), greatly improved the rate of cell divisions in parsnip protoplast cultures. The protoplast-derived cells of all tested accessions divided more frequently in the presence of 2,4-D and zeatin coupled with PSK than it the control medium containing both hormones but no PSK. A similar effect was observed for several species such as asparagus, beet, cabbage, carrot and rice (Matsubayashi and Sakagami 1996; Matsubayashi et al. 1997; Grzebelus et al. 2012a, b; Kiełkowska and Adamus 2017). Our study shows that both undertaking of mitotic divisions by protoplast-derived cells and proliferation of callus might be dependent on the presence of exogenous PSK in the culture medium. The oxidative stress generated during protoplast isolation, purification and subsequent culture might prevent cells from exhibiting further differentiation, leading to organogenesis and/or somatic embryogenesis (Watanabe et al. 2002). It was postulated that the addition of supplements such as silver nitrate, polyvinylpyrrolidone, activated charcoal or amino acids might aid protoplast divisions and the formation of microcallus (Reed and Bargmann 2021). Among these supplements, polyamines (PAs), low-molecular aliphatic amines, are proved to have a promotive effect on cell divisions and the formation of somatic embryos in many species, e.g., carrot, grapevine, ginseng and sweet orange (Fienberg et al. 1984; Faure et al. 1991; Kevers et al. 2000; Wu et al. 2009). To assess the influence of PAs on the stimulation of mitotic divisions and possible formation of somatic embryos in parsnip, protoplast culture medium (PPC6 + PSK) was supplemented with putrescine (Pu) – a common PA present in plants. Although the mean increase in plating efficiency of approximately 2% was observed for all accessions, the differences were not significant enough to implement Pu in the protoplast culture protocol.
The continuous presence of PSK might be a prerequisite for the development of proembryogenic callus in various species. It was reported that PSK promoted somatic embryogenesis in cell suspension cultures of Japanese cedar and carrot (Hanai et al. 2000; Igasaki et al. 2003). In faba bean, PSK induced SE, however, no embryos converted into plants. Plant formation from somatic embryos was observed in exogenous PSK enriched pea callus cultures (Ochatt et al. 2018). In parsnip, we observed highly genotype-dependent effect of PSK efficacy in fostering development of proembryogenic callus, whereas the presence of exogenous PSK was essential for the induction and maintenance of divisions of protoplast-derived cells in all the studied accessions. Moreover, no promoting effect of Pu on the formation of somatic embryos was observed, as all the regenerated plants originated from embryos developed from the callus cultured on the medium supplemented only with PSK. The limited regeneration potential observed in protoplast-derived callus of parsnip might be attributed to a somaclonal variation that arose during protoplast culture and or/ protoplast-derived callus culture. It is a common phenomenon described for many species and its occurrence can be very high, e.g. in the range of 6–90% of off-type plantlets in in vitro cultures of banana (Smith 1988, Sahijram et al.2003). In Apiaceae, somaclonal variation of protoplast regenerants was also observed. Its potentially beneficial effect was described for carrot. Regenerants obtained from protoplast cultures subjected to biotic stress, i.e. Alternaria radicina fungal culture filtrates, showed lower susceptibility to this pathogen (Grzebelus et al. 2013). Similarly, application of salt stress to the protoplast cultures of carrot resulted in higher survival rate of regenerated plants in saline soil (Kiełkowska et al. 2019). Nonetheless, the oxidative stress generated during culture initiation and subsequent subcultures can lead to changes in chromosome numbers, likely due to endoreduplication within callus cells. The change in the ploidy level of regenerants was observed for several species within Apiaceae, i.e. carrot and celariac (Bruznican et al. 2019, Grzebelus et al. 2012, Kiełkowska et al. 2019). In parsnip such changes in chromosome number might impact the regenerative ability of protoplast-derived callus and inhibit the development of proembryogenic mass. Also, changes in the level of DNA methylation resulting from the presence of free radicals in in vitro cultures might contribute to reduced regeneration ability (Krishna et al. 2016). To assess the impact of the oxidative stress on inhibition of SE in parsnip and to improve the efficiency of SE, further studies implementing exogenous polyamines (e.g. spermine, spermidine) and/or antioxidants in the callus culture medium should be carried out. The regenerated and acclimatized to ex vitro conditions plants were morphologically similar to the donor and displayed no variation in the level of ploidy. This indicates that culture conditions applied in the described protocol for protoplast-to-plant regeneration of parsnip, particularly for cultivar Półdługi Biały, did not increase the level of somaclonal variation beyond the point of irreversible intra-nuclear rearrangements.
To our knowledge, this is the first publicly available report of a successful plant regeneration from leaf-derived protoplast cultures of parsnip. The established protocol produced sufficient yield of protoplasts that may provide a platform for production of somatic hybrids via protoplast fusion. We demonstrated that an adequate efficiency of cell colony formation and plant regeneration could be induced after supplementation of the culture medium with PSK in ‘Półdługi biały’ accession. The embryo-regenerated plants were morphologically similar to the parent plant and retained its ploidy.
Data availability
The datasets supporting the conclusions of this article are included within the article or are available from corresponding authors on reasonable request.
Abbreviations
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- CPC:
-
Coriander protoplast culture medium
- FDA:
-
Fluorescein diacetate
- FPC:
-
Fennel protoplast culture medium
- PEM:
-
Proembryogenic mass
- PSK:
-
Phytosulfokine-α
- Pu:
-
Putrescine
- SE:
-
Somatic embryogenesis
References
Ali M, Mujib A, Zafar N, Tonk D (2018a) Somatic embryogenesis, biochemical alterations and synthetic seed development in two varieties of coriander (Coriandrum sativum L.). Adv Hortic Sci 32:239–248. https://doi.org/10.13128/ahs-22287
Ali M, Mujib A, Zafar N, Tonk D (2018b) Protoplast isolation and plant regeneration in two cultivated coriander varieties, Co-1 and RS. Biotechnologia 99:345–355. https://doi.org/10.5114/bta.2018.79965
Ara H, Jaiswal U, Jaiswal VS (2004) An improved method of proliferation of proembryogenic calli of Mangifera indica L. var. Amrapali for scale-up of somatic embryo production. Indian J Biotechnol 3:229–234
Averill KM, DiTommaso A (2007) Wild parsnip (Pastinaca sativa): a troublesome species of increasing concern. Weed Technol 21:279–287. https://doi.org/10.1614/wt-05-186.1
Bertini E, Tornielli GB, Pezzotti M, Zenoni S (2019) Regeneration of plants from embryogenic callus-derived protoplasts of Garganega and Sangiovese grapevine (Vitis vinifera L.) cultivars. Plant, Cell Tissue Organ Cult 138:239–246. https://doi.org/10.1007/s11240-019-01619-1
Bruznican S, Eeckhaut T, de Clerq H et al (2019) Celeriac protoplast regenerated plants phenotypic and genotypic characterization. Acta Hortic 1264:123–128. https://doi.org/10.17660/ACTAHORTIC.2019.1264.14
Cai X, Kang XY (2014) Plant regeneration from cell suspension-derived protoplasts of Populus × beijingensis. Vitro Cell Dev Biol Plant 50:92–98. https://doi.org/10.1007/s11627-013-9540-x
Carlson PS, Smith HH, Dearing RD (1972) Parasexual interspecific plant hybridization. Proc Natl Acad Sci USA 69:2292–2294. https://doi.org/10.1073/PNAS.69.8.2292
Chamani E, Tahami SK, Zare N et al (2012) Effect of different cellulase and pectinase enzyme treatments on protoplast isolation and viability in Lilium ledebeourii Bioss. Not Bot Horti Agrobot Cluj Napoca 40:123–128. https://doi.org/10.15835/nbha4028055
Chappell LH, Dunford AJ (2021) Parsnip Pastinaca sativa L breeding for the future. In: Dennis V Kayri J, Jain, Shri M, Johnson (eds.). Advances in plant breeding strategies: vegetable crops. Springer Nature, Berlin. pp 239–273
Cheng J, Saunders JA, Sinden SL (1995) Colorado potato beetle resistant somatic hybrid potato plants produced via protoplast electrofusion. In Vitro Cell Dev Biol Plant 31:90–95. https://doi.org/10.1007/BF02632243
Davey MR, Anthony P, Patel D, Power JB (2010) Plant protoplasts: isolation, culture and plant regeneration. In: Davey MR, Anthony P (eds) Plant Cell Culture: essential methods. John Wiley & Sons, Ltd, pp 153–173
Dirks R, Sidorov V, Tulmans C (1996) A new protoplast culture system in Daucus carota L. and its applications for mutant selection and transformation. Theor Appl Genet 93:809–815. https://doi.org/10.1007/BF00224080
Dodeman VL, Ducreux G, Kreis M (1997) Zygotic embryogenesis versus somatic embryogenesis. J Exp Bot 48:1493–1509. https://doi.org/10.1093/jxb/48.8.1493
Dudits D, Fejér O, Hadlaczky G et al (1980) Intergeneric gene transfer mediated by plant protoplast fusion. Molec Gen Genet 179:283–288. https://doi.org/10.1007/BF00425455
Faure O, Mengoli M, Nougarede A, Bagni N (1991) Polyamine pattern and biosynthesis in zygotic and somatic embryo stages of Vitis vinifera. J Plant Physiol 138:545–549. https://doi.org/10.1016/S0176-1617(11)80238-5
Fienberg AA, Choi JH, Lubich WP, Sung ZR (1984) Developmental regulation of polyamine metabolism in growth and differentiation of carrot culture. Planta 162:532–539. https://doi.org/10.1007/BF00399919
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–158. https://doi.org/10.1016/0014-4827(68)90403-5
Ganapathi TR, Srinivas L, Suprasanna P, Bapat VA (2001) Regeneration of plants from alginate-encapsulated somatic embryos of banana cv. Rasthali (Musa spp. AAB group). In Vitro Cell Dev Biol Plant 37:178–181. https://doi.org/10.1007/s11627-001-0031-0
Grzebelus E, Szklarczyk M, Baranski R (2012a) An improved protocol for plant regeneration from leaf- and hypocotyl-derived protoplasts of carrot. Plant Cell Tissue Organ Cult 109:101–109. https://doi.org/10.1007/s11240-011-0078-5
Grzebelus E, Szklarczyk M, Greń J et al (2012b) Phytosulfokine stimulates cell divisions in sugar beet (Beta vulgaris L.) mesophyll protoplast cultures. Plant Growth Regul 67:93–100. https://doi.org/10.1007/s10725-011-9654-2
Grzebelus E, Kruk M, Macko-Podgórni A, Grzebelus D (2013) Response of carrot protoplasts and protoplast-derived aggregates to selection using a fungal culture filtrate of Alternaria radicina. Plant Cell, Tissue Organ Cult 115:209–222. https://doi.org/10.1007/s11240-013-0353-8
Guo WW, Xiao SX, Deng XX (2013) Somatic cybrid production via protoplast fusion for citrus improvement. Sci Hortic 163:20–26. https://doi.org/10.1016/j.scienta.2013.07.018
Hanai H, Matsuno T, Yamamoto M et al (2000) A secreted peptide growth factor, phytosulfokine, acting as a stimulatory factor of carrot somatic embryo formation. Plant Cell Physiol 41:27–32. https://doi.org/10.1093/pcp/41.1.27
Hu X, Yin Y, He T (2015) Plant regeneration from protoplasts of Gentiana macrophylla pall. Using agar-pool culture. Plant Cell Tissue Organ Cult 121:345–351. https://doi.org/10.1007/s11240-014-0705-z
Igasaki T, Akashi N, Ujino-Ihara T et al (2003) Phytosulfokine stimulates somatic embryogenesis in Cryptomeria japonica. Plant Cell Physiol 44:1412–1416. https://doi.org/10.1093/pcp/pcg161
Kao KN, Michayluk MR (1975) Nutritional requirements for growth of Vicia hajastana cells and protoplasts at a very low population density in liquid media. Planta 126:105–110. https://doi.org/10.1007/BF00380613
Kevers C, le Gal N, Monteiro M et al (2000) Somatic embryogenesis of Panax ginseng in liquid cultures: a role for polyamines and their metabolic pathways. Plant Growth Regul 31:209–214. https://doi.org/10.1023/A:1006344316683
Kiełkowska A, Adamus A (2010) In vitro culture of unfertilized ovules in carrot (Daucus carota L.). Plant Cell Tissue Organ Cult 3:309–319. https://doi.org/10.1007/S11240-010-9735-3
Kiełkowska A, Adamus A (2017) Early studies on the effect of peptide growth factor phytosulfokine-α on Brassica oleracea var. capitata L. protoplasts. Acta Societatis Botanicorum Poloniae 86:3558. https://doi.org/10.5586/asbp.3558
Kiełkowska A, Grzebelus E, Lis-Krzyścin A, Maćkowska K (2019) Application of the salt stress to the protoplast cultures of the carrot (Daucus carota L.) and evaluation of the response of regenerants to soil salinity. Plant, Cell Tissue Organ Cult 137:379–395. https://doi.org/10.1007/s11240-019-01578-7
Krishna H, Alizadeh M, Sign D et al (2016) Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 6:1–18. https://doi.org/10.1007/S13205-016-0389-7
Kumar V, van Staden J (2017) New insights into plant somatic embryogenesis: an epigenetic view. Acta Physiol Plant 39:1–17. https://doi.org/10.1007/s11738-017-2487-5
Kutschmar A, Rzewuski G, Stührwohldt N et al (2009) PSK-α promotes root growth in Arabidopsis. New Phytol 181:820–831. https://doi.org/10.1111/j.1469-8137.2008.02710.x
Larkin P, Scowcroft W (1981) Somaclonal variation - a novel source of variability from cell cultures for plant improvement. Theor Appl Genet 60:197–214. https://doi.org/10.1007/BF02342540
Linsmaier EM, Skoog F (1965) Organic growth factor requirements of tobacco tissue cultures. Physiol Plant 18:100–127. https://doi.org/10.1111/J.1399-3054.1965.TB06874.X
Maćkowska K, Jarosz A, Grzebelus E (2014) Plant regeneration from leaf-derived protoplasts within the Daucus genus: effect of different conditions in alginate embedding and phytosulfokine application. Plant. Cell Tissue Organ Cult 117:241–252. https://doi.org/10.1007/s11240-014-0436-1
Masani MY, Noll G, Parveez GK et al (2013) Regeneration of viable oil palm plants from protoplasts by optimizing media components, growth regulators and cultivation procedures. Plant Sci 210:118–127. https://doi.org/10.1016/j.plantsci.2013.05.021
Matsubayashi Y (2013) Phytosulfokine. In: Kastin AJ (ed) Handbook of biologically active peptides, 2nd edn. Elsevier Inc, Amsterdam, pp 35–39
Matsubayashi Y, Sakagami Y (1996) Phytosulfokine, sulfated peptides that induce the proliferation of single mesophyll cells of Asparagus officinalis L. Proc Natl Acad Sci USA 93:7623–7627. https://doi.org/10.1073/pnas.93.15.7623
Matsubayashi Y, Takagi L, Sakagami Y (1997) Phytosulfokine-alpha, a sulfated pentapeptide, stimulates the proliferation of rice cells by means of specific high- and low-affinity binding sites. Proc Natl Acad Sci USA 94:13357–13362. https://doi.org/10.1073/PNAS.94.24.13357
Menczel L, Nagy F, Kiss ZR, Maliga P (1981) Streptomycin resistant and sensitive somatic hybrids of Nicotiana tabacum + Nicotiana knightiana: correlation of resistance to N. tabacum plastids. Theor Appl Genet 59:191–195. https://doi.org/10.1007/BF00264975
Miura Y, Tabata M (1986) Direct somatic embryogenesis from protoplasts of Foeniculum vulgare. Plant Cell Rep 5:310–313. https://doi.org/10.1007/BF00269830
Mujib A, Tonk D, Ali M (2014) Plant regeneration from protoplasts in indian local Coriandrum sativum L.: scanning electron microscopy and histological evidences for somatic embryogenesis. Plant Cell Tissue Organ Cult 117:323–334. https://doi.org/10.1007/s11240-014-0442-3
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497. https://doi.org/10.1111/j.1399-3054.1962.tb08052.x
Murthy HN, Hahn EJ, Paek KY (2008) Recurrent somatic embryogenesis and plant regeneration in Coriandrum sativum L. Sci Hortic 118:168–171. https://doi.org/10.1016/j.scienta.2008.05.037
Ochatt S, Mousset-Declas C, Rancillac M (2000) Fertile pea plants regenerate from protoplasts when calluses have not undergone endoreduplication. Plant Sci 156:177–183. https://doi.org/10.1016/S0168-9452(00)00250-8
Ochatt S, Conreux C, Mcolo MR et al (2018) Phytosulfokine-alpha, an enhancer of in vitro regeneration competence in recalcitrant legumes. Plant Cell Tissue Organ Cult 135:189–201. https://doi.org/10.1007/s11240-018-1455-0
Quiroz-Figueroa FR, Rojas-Herrera R, Galaz-Avalos RM, Loyola-Vargas VM (2006) Embryo production through somatic embryogenesis can be used to study cell differentiation in plants. Plant Cell Tissue Organ Cult 86:285–301. https://doi.org/10.1007/s11240-006-9139-6
Rai MK, Asthana P, Jaiswal VS, Jaiswal U (2010) Biotechnological advances in guava (Psidium guajava L.): recent developments and prospects for further research. Trees 24:1–12. https://doi.org/10.1007/s00468-009-0384-2
Reed KM, Bargmann BO (2021) Protoplast regeneration and its use in new plant breeding technologies. Front Genome Ed 3:20. https://doi.org/10.3389/fgeed.2021.734951
Rihan H, Kareem F, El-Mahrouk M, Fuller M (2017) Artificial seeds (principle, aspects and applications). Agronomy 7:71. https://doi.org/10.3390/agronomy7040071
Rubatzky VE, Quiros CF, Simon PW (1999) Carrots and related vegetable Umbelliferae. CAB International, New York
Sahijram Soneji K, Bollamma J (2003) Analyzing somaclonal variation in micropropagated bananas (Musa spp.). In Vitro Cell Dev Biol 39:551–556. https://doi.org/10.1079/IVP2003467/METRICS
Schmidt ED, Guzzo F, Toonen MA, de Vries SC (1997) A leucine-rich repeat containing receptor-like kinase marks somatic plant cells competent to form embryos. Development 124:2049–2062. https://doi.org/10.1242/dev.124.10.2049
Sherraf I, Tizroutine S, Chaput MH et al (1994) Production and characterization of intergeneric somatic hybrids through protoplast electrofusion between potato (Solanum tuberosum) and Lycopersicon pennellii. Plant, Cell Tissue Organ Cult 37:137–144. https://doi.org/10.1007/BF00043607
Skoog F, Miller CO (1957) Chemical regulation of growth and organ formation in plant tissues cultured in vitro. Symp Soc Exp Biol 11:118–130
Smith M (1988) A review of factors influencing the genetic stability of micropropagated bananas. Fruits 43:219–223
Steward FC (1958) Growth and organized development of cultured cells. III. Interpretations of the growth from free cell to carrot plant. Am J Bot 45:709. https://doi.org/10.2307/2439729
Sugiyama M (2015) Historical review of research on plant cell dedifferentiation. J Plant Res 128:349–359. https://doi.org/10.1007/s10265-015-0706-y
Tautorus TE, Fowke LC, Dunstan DI (1991) Somatic embryogenesis in conifers. Can J Bot 69:1873–1899. https://doi.org/10.1139/b91-237
Wang J, Jiang J, Wang Y (2013) Protoplast fusion for crop improvement and breeding in China. Plant, Cell Tissue Organ Cult 112:131–142. https://doi.org/10.1007/s11240-012-0221-y
Watanabe M, Suzuki K, Kawasaki H, Watanabe Y (2002) Differential responses of Brassica napus and Petunia hybrida to leaf protoplast isolation stress. Physiol Plant 114:645–651. https://doi.org/10.1034/j.1399-3054.2002.1140419.x
Winkelmann T, Prange A, Specht J et al (2008) Morphological characterization of plants regenerated from protoplasts of Cyclamen persicum Mill. Propag Ornam Plants 8:9–12
Wu XB, Wang J, Liu JH, Deng XX (2009) Involvement of polyamine biosynthesis in somatic embryogenesis of Valencia sweet orange (Citrus sinensis) induced by glycerol. J Plant Physiol 166:52–62. https://doi.org/10.1016/j.jplph.2008.02.005
Yamakawa S, Sakuta C, Matsubayashi Y et al (1998) The promotive effects of a peptidyl plant growth factor, phytosulfokine-α, on the formation of adventitious roots and expression of a gene for a root-specific cystatin in cucumber hypocotyls. J Plant Res 111:453–458. https://doi.org/10.1007/bf02507810
Yu Y, Ye W, He L et al (2013) Introgression of bacterial wilt resistance from eggplant to potato via protoplast fusion and genome components of the hybrids. Plant Cell Rep 32:1687–1701. https://doi.org/10.1007/s00299-013-1480-8
Zimmerman JL (1993) Somatic embryogenesis: a model for early development in higher plants. Plant Cell 5:1411–1423. https://doi.org/10.1105/tpc.5.10.1411
Acknowledgements
The authors wish to thank Urszula Czech for her excellent technical assistance in greenhouse experiments and Dagmara Więcek from Kutnowska Hodowla Buraka Cukrowego KHBC (Straszków, Poland) for the ploidy analyses of the regenerants.
Funding
The financial support of the Polish Ministry of Agriculture and Rural Development is acknowledged.
Author information
Authors and Affiliations
Contributions
Conceptualization, EG; methodology, KS and EG; validation, KS formal analysis, KS; investigation, KS; resources, EG; data curation, KS and EG; writing—original draft preparation, KS; writing—review and editing, KS and EG; visualization, KS; supervision, EG; project administration, EG; funding acquisition, EG All authors read, reviewed, and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Winkelmann.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stelmach, K., Grzebelus, E. Plant regeneration from protoplasts of Pastinaca sativa L. via somatic embryogenesis. Plant Cell Tiss Organ Cult 153, 205–217 (2023). https://doi.org/10.1007/s11240-023-02461-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-023-02461-2