
Abstract
Here, we present new cryogenic infrared spectra of the (Imidazole)\(_{n}\mathrm{H}^{+}\) (n=1,2,3) ions. The data was obtained using helium tagging infrared predissociation spectroscopy. The new results were compared with the data obtained by Gerardi et al. (Chem. Phys. Lett. 501:172–178, 2011) using the same technique but with argon as a tag. Comparison of the two experiments, assisted by theoretical calculations, allowed us to evaluate the preferable attachment positions of argon to the (Imidazole)\(_{n}\mathrm{H}^{+}\) frame. Argon attaches to nitrogen-bonded hydrogen in the case of the (Imidazole)H\(^+\) ion, while in (Imidazole)\(_{2}\mathrm{H}^{+}\) and (Imidazole)\(_{3}\mathrm{H}^{+}\) the preferred docking sites for the argon are in the center of the complex. This conclusion is supported by analyzing the spectral features attributed to the N–H stretching vibrations. Symmetry adapted perturbation theory (SAPT) analysis of the non-covalent forces between argon and the (Imidazole)\(_{n}\mathrm{H}^{+}\) (n=1,2,3) frame revealed that this switch of docking preference with increasing complex size is caused by an interplay between induction and dispersion interactions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Intermolecular (or non-covalent) interactions are ubiquitous in real-life chemical systems. For example, these interactions are responsible for non-ideality of gases, they govern the three-dimensional structures of essential biomacromolecules, such as proteins, they determine the packing of the molecules in molecular crystals [1–5]. However, the accurate description of the subtle energetics of those interactions is a task that requires specialized instruments [1–4, 6, 7]. A rather popular model system for the study of weak non-covalent interactions are complexes of noble gas atoms with aromatic systems due to the absence of the possibility to form covalent bonds with the substrate [8–11].

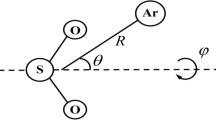
Structures of the protonated imidazole (\(\mathrm{ImH^+}\)) and its clusters (\(\mathrm{Im}_n\mathrm{H^+}, n=2,3\)) [12]
In this work, we investigate how argon attaches to protonated imidazole (\({\mathrm{ImH}^+}\)) and its clusters (\({\mathrm{Im}_n\mathrm{H}^+}, n=2,3\)). The structures of these species are given in Fig. 1. Imidazole (Im) itself is an aromatic five-membered aromatic heterocycle with two nitrogen atoms. This molecule is a basic building block of many biologically important molecules, e.g., histamine and histidine. The imidazole units in the complexes we consider (\({\mathrm{Im_{2}H}^+}\) and \({\mathrm{Im_{3}H}^+}\)) are held together by \(\mathrm {N-H\ldots N}\) hydrogen bonds, which makes them the shortest imidazole wires: chains of imidazoles connected via N\(\cdots\)H−N hydrogen bonds. These wires have gained recent attention in theoretical and experimental studies in both gaseous and condensed phases due to their ability to conduct protons via the Grotthuss mechanism (i.e., proton migration via the hydrogen bond) [12–19].
The identification of the argon docking positions in the \({\mathrm{Im}_n\mathrm{H}^+}\) (n=1,2,3) complexes is made by comparing the IR vibrational predissociation spectra of these species obtained using argon tagging in Ref. [12] with the spectra described in the present work using helium as a tag. Helium is a less polarizable and less perturbing tag than argon. Therefore, the comparison of the spectra taken with the help of different noble gas atoms facilitates the isolation of the spectral features arising from the presence of argon. The theoretical calculations explain the observed spectral features. The docking preference of argon in the \({\mathrm{ImH}^+}\) – \({\mathrm{Im_2H}^+}\) – \({\mathrm{Im_3H}^+}\) sequence is rationalized by analyzing the intermolecular forces performed using the symmetry-adapted perturbation theory (SAPT) approach [3, 4].
Computational methods
General considerations
The quantum chemical calculations were performed using Orca 4 [20], Turbomole [21], Psi4 [22], and Gaussian [23] program suites. In order to reduce computational time, all the computations, except for those done in Gaussian, have applied the resolution of identity (RI) technique [24–30]. The def2-TZVPP, def2-TZVP, def2-SVPD, def2-SV(P), and aug-cc-pVTZ basis sets with a corresponding auxiliary basis were used [31–34]. The DFT calculations were using the B3LYP [35–37] exchange-correlation functional with D3 dispersion correction with Becke–Johnson (BJ) damping [6]. The intermolecular interactions of the \(\mathrm{Im}_{n}\mathrm{H}^+\) (n=1,2,3) complexes with argon were analyzed at the SAPT2+\(\delta\)MP2/aug-cc-pVTZ//B3LYP-D3/def2-TZVPP level of theory [3, 4, 38, 39]. Here and further, the notation X//Y will indicate that the energy of the molecule was obtained with method X at the geometry optimized with method Y.
Computations for ImH+ . . . X (X = He, Ar)
In the case of \(\mathrm{ImH^+}\) and the \(\mathrm{ImH^+ \ldots X}\) (X = He, Ar) complexes we have performed geometry optimizations at the B3LYP-D3/def2-SVPD, B3LYP-D3/def2-TZVPP, and MP2(full)/def2-SVPD levels of theory followed by harmonic frequency calculations. Geometries optimized at the B3LYP-D3/def2-TZVPP level were used for single point energy calculations at the DLPNO-CCSD(T) [40, 41], CCSD(T) with the def2-SVPD and def2-TZVPP basis sets, SAPT2+\(\delta\)MP2/aug-cc-pVTZ and SAPT2+3\(\delta\)MP2/aug-cc-pVTZ levels of theory [3, 4].
For calculations in the rigid monomer approximation, we have optimized the structure of the ImH\(^+\) species using the BP86/def2-TZVPP approach. Three unique grids \(24 \times 24 \times 20\) Bohr surrounding ImH\(^+\) with an increment of 1 Bohr (\(\approx 0.53\) Å) for possible He positions were computed at the B3LYP-D3/def2-TZVPP, MP2/def-SVPD, and DLPNO-CCSD(T)/def2-SVPD levels of theory. Additional single point energy computations of the ImH\(^+\) frame and inert gas atom were done to estimate the dissociation energies \(D_\mathrm {e}\) of this complex. The three-dimensional (3D) Schrödinger equation for the motion of the helium atom around the \(\mathrm{ImH^+}\) frame in the rigid monomer approximation was solved via an extended in-house Python script employing the discrete variable representation (DVR) method [42].
Computations for Im2H+ and Im2H+ . . . Ar
All the calculations for \(\mathrm{Im_2H^+}\) and \(\mathrm{Im_2H^+ \ldots Ar}\) were done at the B3LYP-D3/def2-TZVPP level. Geometry optimizations of Im\(_2\)H\(^+\) and four possible isomers of Im\(_2\)H\(^+ \ldots\)Ar (see Results section) were followed by harmonic frequency calculations and one-dimensional proton transfer scans along the proton transfer coordinate Q defined as
where \(r\mathrm {(N-H)}\) and \(r\mathrm {(N\ldots H)}\) are the two distances between hydrogen and the two nitrogens in the \(\mathrm{N-H \ldots H}\) part of \(\mathrm{Im_2H^+}\) (see Fig. 1).
The four possible isomers of \(\mathrm{Im_2H^+ \ldots}\) Ar were reoptimized at the B3LYP-D3/def2-SV(P) level of theory and used as starting points for NVE ab initio molecular dynamics (AIMD) simulations with Orca. Initial conditions were sampled from the Maxwell-Boltzmann distribution for \(T = 350\) K to induce the zero-point like motions in the vibrational modes with frequencies below 240 cm \(^{-1}\). For each of the isomers, a set of ten trajectories were produced. The simulation length was 6 ps with 1 fs time step, and the first 1 ps of each trajectory was ignored as an equilibration phase. The power spectra [43] for N–H stretching vibrations were computed using our in-house Python scripts employing frequency-shift correction, allowing large time steps without a loss of the accuracy for the high-frequency motions [44]. Argon distributions from AIMD trajectories were computed using the TRAVIS software [45].
Computations for Im3H+ and Im3H+ . . . Ar
The geometries of the four possible isomers of \(\mathrm{Im_3H^+\ldots Ar}\) were built by hand. They and the untagged \(\mathrm{Im_{3}H^+}\) were optimized at the B3LYP-D3/def2-TZVP level, followed by harmonic frequency calculations. The obtained optimized structures were also confirmed as the equilibrium structures at the B3LYP-D3/def2-TZVPP level of theory. The interaction energies of the \(\mathrm{Im_{3}H^+}\) complex with argon were computed at the SAPT2+ \(\delta\) P2/aug-cc-pVTZ//B3LYP-D3/def2-TZVPP level of theory.
Experimental methods
Experiments were carried out in an in-house tandem mass spectrometer at the EPFL (Lausanne) equipped with a cryogenic octupole ion trap described elsewhere [46]. We have produced protonated gas-phase ions by electrospraying imidazole (99% purity, Sigma Aldrich) dissolved in a mixture of water and methanol. The ions were focused in an electrodynamic ion funnel. The electrosprayed ions were accumulated in a hexapole ion trap and released in short ion packets that are mass-selected in a quadrupole mass filter and guided to a cryogenic octupole ion trap, maintained at 3.5 K. Multiple collisions with helium buffer gas in the octupole ion trap have led to trapping, cooling, and formation of weakly bound complexes with helium atoms. Then IR laser radiation excited the trapped ions in order to perform predissociation spectroscopy. If the helium-tagged ions absorb infrared light, the energy is redistributed in the cluster, leading to the removal of the tag, which is recorded as a depletion of ion yield in the mass spectra.
The vibrational spectra were taken in the range of 900–3500 cm\(^{-1}\); however, the laser power below 1200 cm\(^{-1}\) was insufficient to get a reasonable signal-to-noise ratio. Thus, we were not able to record proton transfer lines around 1100 cm\(^{-1}\) for \(\mathrm {Im_2H^+}\).
Results
General considerations
To explore the possible isomerism of the tag (He, Ar) attachment to the \(\mathrm {Im}_n\mathrm{H}^+\) ion, one has to consider the high IR frequency range corresponding to the stretching vibrations of N–H bonds (\(\ge\) 3000 cm\(^{-1}\)). These polarized bonds can serve as the attachment positions for inert gas atoms to the ions. Such attachment leads to shifts in the N–H stretching frequency. By comparing frequencies for different isomers with the ones predicted by computational chemistry, it is possible to conclude the structural motifs of the weak bonds in the van der Waals bonded complexes [47–50].
In order to do so, we use the following three sets of IR spectroscopic data (Table 1): the uncharged imidazole species (Im and Im\(_2\)) from Ref. [51] in helium droplets, and two sets of the ionic vibrational spectra for \(\mathrm{Im}_n\mathrm{H\ldots X}^+\) (n=1,2,3), measured with X=Ar reported in Ref. [12] and with X = He as described in the current work.
Im2H+ . . . X (X=He,Ar)
According to previous investigations [53–55], the ion \(\mathrm{ImH^+}\) offers two attachment positions for the weakly bound tag X (Table 2), namely the hydrogen connected to nitrogen, denoted as \(\mathrm{ImH^+ \ldots X}(\sigma )\), and on top of the \(\pi\)-aromatic system of imidazole, which will be denoted as \(\mathrm{ImH^+ \ldots X}(\pi )\). In the case of the \(\mathrm {ImH^+ \ldots Ar}\) complex, all the applied quantum-chemical methods indicate a prevalence of the ImH\(^+ \ldots\)Ar(\(\sigma\)) isomer over ImH\(^+ \ldots\)Ar(\(\pi\)). Due to the large mass of argon, we can assume the harmonic approximation to be valid for estimating the difference between the zero-point vibrational energies (ZPVE) of these structures. At the B3LYP-D3/def2-TZVPP level of theory, the ZPVE of the ImH\(^+ \ldots\)Ar(\(\pi\)) isomer is higher than the ZPVE of ImH\(^+ \ldots\)Ar(\(\sigma\)) by 10 cm\(^{-1}\). Therefore, the inclusion of the ZPVE correction does not change the conclusions drawn from the single-point energy calculations.
The bonding motif in the \(\mathrm {ImH^+ \ldots Ar}\) complex is confirmed by comparison with IR spectra from Ref. [12]. In the experiment there is an observed splitting between N–H stretching vibrations of 45 cm\(^{-1}\) (see Table 1). The ImH\(^+ \ldots\)Ar(\(\sigma\)) isomer shows a similarly large splitting, while the splitting between N–H stretching frequencies does not exceed 7 cm\(^{-1}\) for the ImH\(^+ \ldots\)Ar(\(\pi\)) (see Fig. 2). The resulting frequencies themselves are also in favor of ImH\(^+ \ldots\)Ar(\(\sigma\)) for all three computational methods. This fully agrees with the conclusions about the ImH\(^+ \ldots\)Ar structure made in Ref. [12].

Experimental vibrational spectra of ImH\(^+\ldots\)Ar (Ref. [12]) and ImH\(^+\ldots\)He (this work) for N–H stretching vibration and their theoretical analogs computed at the B3LYP-D3/def2-TZVPP level of theory in the scaled harmonic oscillator (sHO) approximation. The scaling factor used was 0.9657 [56]. The gray area in the ImH\(^+\ldots\)He signal denotes the ± RMSD (root-mean-square deviation) of the background noise level measurements
The analogous comparison of the computed frequencies for ImH\(^+ \ldots\)He and ImH\(^+\) shows no dependence of the N–H stretching frequencies on the helium atom position or even of the helium's presence. All three species (ImH\(^+\), ImH\(^+ \ldots\)He(\(\sigma\)), and ImH\(^+ \ldots\)He(\(\pi\))) agree equally well with our experimental data (Table 1). Such similarity in vibrational frequencies is due to the smaller polarizability of the helium tag compared to argon. This is confirmed by comparing the experimental data from ImH\(^+ \ldots\)Ar and ImH\(^+ \ldots\)He (Table 1). An attachment of Ar to the N–H bond mainly increases the reduced mass of the N–H vibration, thus lowering its frequency. Therefore the larger frequency in ImH\(^+ \ldots\)Ar (3469 cm\(^{-1}\)) corresponds to the free N–H stretching, and the smaller one (3424 cm\(^{-1}\)) comes from N–H\(\ldots\)Ar. The frequencies of N–H stretching in ImH\(^+ \ldots\)He (3468 cm\(^{-1}\)) and of the analogous vibrations in ImH\(^+ \ldots\)Ar (3469 cm\(^{-1}\)) are equal within the step size in our spectra. Thus, they correspond to the N–H vibration in ImH\(^+\) free of the tag.
Helium is a less perturbative tag than argon, and the potential of its interaction with the monomer has only shallow minima. Calculations (Table 2) suggest that the energy difference between the two forms ImH\(^+ \ldots\)He(\(\sigma\)) and ImH\(^+ \ldots\)He(\(\pi )\) is small, therefore the helium atom in the ImH\(^+ \ldots\)He cluster’s ground vibrational state may be delocalized over the ImH\(^+\) unit [8, 57]. In this case, ImH\(^+ \ldots\)He cannot be described as a single isomeric structure with a certain attachment point. To investigate this possibility, we performed calculations describing helium’s delocalization over ImH\(^+\) at a rigid monomer approximation. ImH\(^+\) was frozen at the geometry optimized at the BP86/def2-TZVPP level of theory, and the potential energy surface (PES) for movement of He around this monomer was computed at the DLPNO-CCSD(T)/def2-SVPD level of theory. A 3D Schrödinger equation describing the He motion was solved on the grid using the discrete variable representation (DVR) method [42]. The wavefunctions corresponding to the four lowest vibrational levels at PES from DLPNO-CCSD(T)/def2-SVPD are shown in Fig. 3.

Wavefunctions of the lowest vibrational states of He motion around \(\mathrm {ImH^+}\) in the rigid monomer approximation obtained for the PES from DLPNO-CCSD(T)/def2-SVPD calculation. The density of the dots corresponds to the density of the wavefunction describing helium’s position, and the color of the dots represents the wavefunction’s sign (positive is green, negative is magenta). Values E are the energies of each of the eigenstates, numbers #n (\(n=0\)-3) indicate the energy ordering of these states. The arrows indicate the pairs of tunneling levels, \(\Delta E\) are the energy differences of these levels and \(\tau =h/\Delta E\) is the corresponding tunneling timescale
The result shows that, in the lowest vibrational states, the helium tag in the ImH\(^+ \ldots\)He particle is localized in the ImH\(^+ \ldots\)He(\(\pi )\) conformation. This supports localization preferences predicted by CCSD(T)/def2-TZVPP and SAPT2+3\(\delta\)MP2/aug-cc-pVTZ calculations (see Table 2). However, since the two vibrational levels differ only by one wavenumber, coupling with ImH\(^+\) degrees of freedom will probably delocalize helium over the molecule.
Im2H+ . . . X (X=He,Ar)

1D PES for proton transfer in Im\(_2\)H\(^+\) and Im\(_2\)H\(^+\ldots\)Ar at B3LYP-D3/def2-TZVPP with scaled harmonic frequencies of the N–H stretching vibration computed in the local minima. The given values correspond to the vibrations with the highest intensity in the dipole approximation. The proton transfer coordinate Q is given by Eq. (1)
Based on the results for ImH\(^+ \ldots\)He, we can assume that the helium tag has a negligible influence on the N–H stretching frequencies, and it is impossible to assign a certain isomer of the \(\mathrm {Im_2H^+\ldots He}\) particle. However, we can expect to determine the docking position of Ar in \(\mathrm {Im_2H^+ \ldots Ar}\). In the case of the imidazole dimer, the variety of attachment positions for the Ar tag is increased: \(\mathrm {Ar(\sigma ) \ldots ImH^+ \cdots Im}\), \(\mathrm {Ar(\sigma ) \ldots Im \cdots ImH^+}\), \(\mathrm {Ar(\pi ) \ldots ImH^+ \cdots Im}\), and \(\mathrm {Ar(\pi ) \ldots Im \cdots ImH^+}\) structures are possible (see Fig. 4). However, proton transfer will interconvert two of these isomers into each other. This can be represented by the reaction \(\mathrm {Ar \ldots ImH^+\cdots Im \leftrightarrow Ar \ldots Im \cdots ImH^+}\). Thus similar to ImH\(^+ \ldots\)Ar, only two isomeric forms \(\mathrm {Im_2H^+\ldots Ar(\sigma )}\) and \(\mathrm {Im_2H^+\ldots Ar(\pi )}\) are possible for \(\mathrm {Im_2H^+\ldots Ar}\). For these two isomers, we have computed a relaxed PES for proton transfer at the B3LYP-D3/def2-TZVPP level of theory. For the geometries at the local minima, additional optimizations were performed followed by harmonic frequency calculations. The results are available in Fig. 4. The \(\mathrm {Im_2H^+\ldots Ar(\pi )}\) conformer has a symmetric PES similar to the \(\mathrm {Im_2H^+}\) ion, because argon does not influence the energies of the system in this configuration. On the contrary, \(\mathrm {Im_2H^+\ldots Ar(\sigma )}\) has a non-symmetric PES, where the minimum \(\mathrm {Ar(\sigma ) \ldots ImH^+ \cdots Im}\) is favored over \(\mathrm {Ar(\sigma )\ldots Im\cdots ImH^+}\) by approx. 120 cm\(^{-1}\).

Experimental vibrational spectra of Im\(_2\)H\(^+\ldots\)Ar (Ref. [12]) and Im\(_2\)H\(^+\ldots\)He (this work) for N–H stretching vibration and their theoretical analogs computed at the B3LYP-D3/def2-TZVPP level of theory in the scaled harmonic oscillator (sHO) approximation as well as scaled power spectrum from AIMD at the B3LYP-D3/def2-SV(P) level. Harmonic spectra for the argon isomers are given for the most stable conformers. The scaling factor for harmonic frequencies was 0.9657 [56], the maximum of AIMD peak was scaled to be at the frequency of 3492 cm\(^{-1}\). The gray area in the Im\(_2\)H\(^+\ldots\)He signal denotes the ± RMSD of the background noise level measurements
A comparison of theoretical frequencies with the experimental data is given in Fig. 5, where the harmonic vibrational frequencies were scaled by 0.9657 [56] and the AIMD power spectrum averaged over all trajectories was scaled with its peak matching the 3492 cm\(^{-1}\) frequency. Theoretical calculations for \(\mathrm {Im_2H^+\ldots Ar(\sigma )}\) predict two peaks 50 cm\(^{-1}\) apart, while for the \(\mathrm {Im_2H^+\ldots Ar(\pi )}\), they predict two peaks separated by about 10 cm\(^{-1}\). These peaks have the same positions as for the pure \(\mathrm {Im_2H^+}\) cluster. Therefore, the results of the harmonic calculations favor the \(\mathrm {Im_2H^+\ldots Ar(\pi )}\) to be the species observed.
The shape of the peak was reproduced by AIMD. The trajectories show prevalence of the \(\mathrm {Im_2H^+\ldots Ar(\pi )}\) structure to \(\mathrm {Im_2H^+\ldots Ar(\sigma )}\) as 2-to-1. If we consider only the trajectories initiating from the \(\mathrm {Im_2H^+\ldots Ar(\sigma )}\) conformer, the ratio becomes 1-to-1. The argon distribution indicates strong conversion from \(\mathrm {Im_2H^+\ldots Ar(\sigma )}\) to \(\mathrm {Im_2H^+\ldots Ar(\pi )}\). The trajectories that start from the \(\mathrm {Im_2H^+\ldots Ar(\pi )}\) conformer show an even larger ratio of 3-to-1 for the initial conformer to stay. This preference of \(\mathrm {Im_2H^+\ldots Ar(\pi )}\) in the simulations can be visualized through the distribution of the argon atom around the \(\mathrm {Im_2H^+}\) core given in Fig. 6. No matter which structure was the initial one (\(\mathrm {Im_2H^+\ldots Ar(\sigma )}\) or \(\mathrm {Im_2H^+\ldots Ar(\pi )}\)), both resulting distributions show significant probability of argon to be present near the center of the \(\mathrm {Im_2H^+}\) fragment. Thus, we can state that the AIMD simulations support the hypothesis of \(\mathrm {Im_2H^+\ldots Ar}\) existing primarily in the form of \(\mathrm {Im_2H^+\ldots Ar(\pi )}\). This form is observed to show only a minor influence on the proton transfer motion (see Fig. 4).

Argon atom distribution around the \(\mathrm {Im_2H^+}\) core as observed in AIMD simulations at the B3LYP-D3/def2-SV(P) level. Labels indicate the initial structure of the trajectory
Additional support for this hypothesis on the \(\mathrm {Im_2H^+\ldots Ar}\) structure can be obtained from comparison with other experimental frequencies. In the spectra of \(\mathrm {Im_2H^+\ldots He}\), the N–H stretching frequency is 3495 cm\(^{-1}\), reproducing the frequency observed in \(\mathrm {Im_2H^+\ldots Ar}\) within 5 cm\(^{-1}\). Using the analogy with ImH\(^{+}\), we can conclude that there is no perturbation in frequency by the argon attachment to the hydrogen in the N–H bond; therefore, argon is most likely not attached to this bond, but is present somewhere else. The final argument can be obtained from the quasi-equilibrium chemical reaction describing proton transfer: \(\mathrm {ImH^+ \ldots Im \leftrightarrow Im \ldots ImH^+}\). Two forms of imidazole (Im and ImH\(^+\)) coexist and interconvert into each other, therefore the N–H stretching frequency should be an average of the corresponding values in Im (3518 cm\(^{-1}\)) and ImH\(^+\) (3468 cm\(^{-1}\)) [58]: \(0.5 \times (3518 + 3468) = 3493\) cm\(^{-1}\). This value is strikingly close to the one observed in the experiment (Table 1), and this is possible only if the symmetric proton transfer is allowed, which happens only in the \(\mathrm {Im_2H^+\ldots Ar(\pi )}\) form. Taking all these arguments together, we can conclude that the argon tag in the \(\mathrm {Im_2H^+\ldots Ar}\) ion exists predominantly in the form of \(\mathrm {Im_2H^+\ldots Ar(\pi )}\).
Im3H+ . . . X (X=He,Ar)
Previous studies determined that the protonated imidazole trimer exists in the form of an imidazole chain [12, 13]. Both IR spectra of \(\mathrm {Im_3H^+\ldots He}\) and \(\mathrm {Im_3H^+\ldots Ar}\) have only one N–H stretching frequency, 3505 and 3503 cm\(^{-1}\), respectively. \(\mathrm {Im_3H}^+\), in contrast to \(\mathrm {Im_2H}^+\), has no proton transfer. Such an intramolecular reaction would require a chemical equilibrium of the form
i.e., the transfer can happen between equivalent structures, where the proton is located on the outer imidazoles in the chain. However, previous calculations performed have shown that the symmetric configuration \(\mathrm {Im\cdots ImH^+ \cdots Im}\) is the lowest energy structure with an energy preference of around 3000 cm\(^{-1}\) [13]. Therefore, this ion is \(\mathrm {ImH^+}\) solvated by two imidazoles. By comparing N–H stretching frequencies for pure imidazole (3518 cm\(^{-1}\)) with the ones obtained for \(\mathrm {Im_3H^+\ldots He}\) and \(\mathrm {Im_3H^+\ldots Ar}\), we can see that the frequencies differ only by 13 and 15 cm\(^{-1}\), respectively.

Total interaction energies of argon with \(\mathrm{Im_3H^+}\) for different isomers of \(\mathrm{Im_3H^+ \ldots Ar}\) at the SAPT2+\(\delta\)MP2/aug-cc-pVTZ//B3LYP-D3/def2-TZVP level of theory. All values are in cm\(^{-1}\)
We have manually constructed four isomers of the \(\mathrm {Im_3H^+\ldots Ar}\) to cover the possible docking variety of the argon atom: #1 and #2, where argon is docked to the \(\pi\)-system on either of the two sides of the central \(\mathrm {ImH^+}\) fragment; #3, where argon is docked at the Im fragment’s \(\pi\)-system; and #4, where argon is attached to the N–H of the Im, imitating the \(\mathrm {Im\ldots Ar(\sigma )}\) fragment. Since both N–H bonds of the central \(\mathrm {ImH^+}\) fragment are blocked by the hydrogen-bonded Im fragments, no other \(\mathrm {Ar(\sigma )}\) isomers are possible. All these structures were fully optimized at the B3LYP-D3/def2-TZVP level of theory, and at these geometries, the SAPT2+\(\delta\)MP2/aug-cc-pVTZ dissociation energies of the argon from the \(\mathrm {Im_3H^+}\) moiety were computed. The results are given in Fig. 7, where we see that the \(\mathrm {Im_3H^+\ldots Ar(\pi )}\) structures (#1, #2, and #3) are far more preferable in energy than the \(\mathrm {Im_3H^+\ldots Ar(\sigma )}\) structure (#4). Extrapolation of the discussion for \(\mathrm {ImH^+\ldots Ar}\) and \(\mathrm {Im_2H^+\ldots Ar}\), together with a single observable N–H stretching band and the SAPT results, leads to the conclusion that the argon atom attaches to the \(\pi\)-system of the central imidazole but not to the proton of the outer N–H bonds.
This conclusion showcases the tendency of argon attachment in the row \(\mathrm {Im}_n\mathrm{H}^+\) (n=1,2,3). Although the bonding motif is different for \(\mathrm {ImH}^+\) with respect to \(\mathrm {Im}_n\mathrm{H}^+\) (n=2,3), argon, in general, tends to stick as close to the excessive charge of the ion as possible. When we try to solidify this result by comparison of theory and experiment, we will find that the spectra predicted for the lowest isomers are too similar to each other (see Fig. 8). The significant shift observed in the ImH\(^+ \ldots\)Ar (see Fig. 2) is not present for Im\(_3\)H\(^+\ldots\)Ar because the highest isomer has argon attached in-between the N–H and C–H bonds; therefore, its influence on the N–H stretching frequencies is less prominent.

Experimental vibrational spectra of Im\(_3\)H\(^+\ldots\)Ar (Ref. [12]) and Im\(_3\)H\(^+\ldots\)He (this work) for N–H stretching vibration and their theoretical analogs computed at the B3LYP-D3/def2-TZVP level of theory in the scaled harmonic oscillator (sHO) approximation. Numbering of the \(\mathrm {Im_3H\ldots Ar}^+\) conformers corresponds to the energy ordering in Fig. 7. The scaling factor used was 0.9671 [56]. The gray area in the Im\(_3\)H\(^+\ldots\)He signal denotes the ±RMSD of the background noise level measurements. The Im\(_3\)H\(^+\ldots\)Ar isomers are numbered according to Fig. 7
Bonding motif of argon and ImnH+ (n=1,2,3) complexes

SAPT2+\(\delta\)MP2/aug-cc-pVTZ//B3LYP-D3/def2-TZVPP analysis of the interaction energies between the argon atom with the \(\mathrm{Im}_n\mathrm{H^+}\) (n=1,2,3) frame
The observed docking preference of the argon atom for an increasing imidazole chain is puzzling:
-
in the case of the monomer (\(\mathrm{ImH^+}\)), argon attaches to the hydrogen involved in the N–H bond leading to the \(\mathrm{ImH^+\ldots Ar(\sigma )}\) structure;
-
in the case of the dimer (\(\mathrm{Im_2H^+}\)) and trimer (\(\mathrm{Im_3H^+}\)), argon prefers to be placed in the middle of the complex leaving the N–H bonds free, and thus leading to an \(\mathrm{ImH^+\ldots Ar(\pi )}\) structure to be observed.
We have performed SAPT2+\(\delta\)MP2/aug-cc-pVTZ single point energy calculations based on the B3LYP-D3/def2-TZVPP optimized molecular structures to rationalize this behavior of the complexes. The results are given in Fig. 9. SAPT disentangles the interaction energy in four terms [4]:
-
exchange (\(E_\mathrm {exch}\)), which is the repulsion of electronic densities of two fragments,
-
electrostatic interactions (\(E_\mathrm {el}\)), which is the interaction energy between static charges of two fragments,
-
induction (\(E_\mathrm {ind}\)), which appears due to polarization in one of the fragments by the static charges in another one,
-
dispersion (\(E_\mathrm {disp}\)), which appears due to the cross-polarization of the two fragments.
The \(\mathrm{Im}_n\mathrm{H^+\ldots Ar}(\pi )\) (n=1,2,3) complexes are all mainly stabilized by dispersion interactions for the following reason: when argon is located above the imidazole ring plane, it is in contact with as many atoms as possible. By increasing the size of the complex, more atoms come into contact with argon, increasing the magnitude of the dispersion interaction of this atom with the \(\mathrm{Im}_n\mathrm{H^+}\) frame, and thus compensating the decrease in the induction energy, which arises from delocalization of the charge between imidazole fragments.
The \(\mathrm{Im}_n\mathrm{H^+\ldots Ar}(\sigma )\) structures show a more complicated trend of energies. In the smallest cluster, \(\mathrm{ImH^+\ldots Ar(\sigma )}\), the main stabilization comes from induction, because argon is in direct contact with the proton, which is the source of the excessive charge. However, the larger the complex becomes, the larger is the delocalization of the charge. Such an effect leads to a decrease in the induction component of the interaction energy. To some extent, the loss of induction is being compensated by the increase of dispersion due to the increased size of the \(\mathrm{Im}_n\mathrm{H^+}\) core. However, this increase is smaller compared to that in the \(\mathrm{Im}_n\mathrm{H^+\ldots Ar}(\pi )\) structures, which leads to the change of the preferred position of the argon with respect to the \(\mathrm{Im}_n\mathrm{H^+}\) frame.
Conclusions
We characterized the protonated imidazole monomer, dimer, and trimer (\(\mathrm{Im}_n\mathrm{H^+}\), n=1,2,3) complexed with argon. This analysis is based on thorough examination of the vibrational spectra from the literature and our vibrational predissociation spectroscopy data for the \(\mathrm{Im}_n\mathrm{H^+}\) species combined with theoretical computations. Based on the experimental vibrational frequencies for N–H stretching vibrations and their computed analogs, we found the binding motifs of the argon tag to \(\mathrm{Im}_n\mathrm{H^+}\) (n=1,2,3) ions. Argon docking preference changes with an increase in the size of the \(\mathrm{Im}_n\mathrm{H^+}\) fragment: in the monomer, it attaches to the hydrogen connected to the nitrogen, while in the larger clusters, argon relocates to the top of the imidazole \(\pi\)-system. This change of the binding motif is dictated by the interplay between induction and dispersion interactions at different docking positions. This study contributes to a general understanding of how the different non-covalent interactions influence supramolecular structures in the presence of the ions, which may be relevant for the design of the molecular mechanics force fields.
Data availability
The experimentally obtained vibrational spectra and computational data sets (optimized geometries and SAPT results) are available in the supporting information.
Code availability
The used scripts for solving of the 3D Schrödinger equation and for analyzing the AIMD results are available from the Git repository: https://gitlab.com/madschumacher/imnhxplus.
References
Al-Hamdani YS, Tkatchenko A (2019) Understanding non-covalent interactions in larger molecular complexes from first principles. J Chem Phys 150(1):010901
Grimme S (2011) Density functional theory with london dispersion corrections. WIREs Computat Mole Sci 1(2):211–228
Jeziorski B, Moszynski R, Szalewicz K (1994) Perturbation theory approach to intermolecular potential energy surfaces of van der waals complexes. Chem Rev 94(7):1887–1930
Patkowski K (2020) Recent developments in symmetry-adapted perturbation theory. WIREs Comput Mole Sci 10(3)
Riley KE, Hobza P (2011) Noncovalent interactions in biochemistry. WIRE's Comput Mole Sci 1(1):3–17
Grimme S, Ehrlich S, Goerigk L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32(7):1456–1465
Grimme S, Hansen A, Brandenburg JG, Bannwarth C (2016) Dispersion-corrected mean-field electronic structure methods. Chem Rev 116(9):5105–5154
Lee S, Chung JS, Felker PM, López Cacheiro J, Fernández B, Bondo Pedersen T, Koch H (2003) Computational and experimental investigation of intermolecular states and forces in the benzene-helium van der waals complex. J Chem Phys 119(24):12956–12964. https://doi.org/10.1063/1.1628217
Makarewicz J, Shirkov L (2016) Character of intermolecular interaction in pyridine-argon complex: Ab initio potential energy surface, internal dynamics, and interrelations between sapt energy components. J Chem Phys 144(20):204115
Shirkov L, Makarewicz J (2015) The study of basis sets for the calculation of the structure and dynamics of the benzene-kr complex. J Chem Phys 142(20):204107
Shirkov L, Sladek V, Makarewicz J (2020) Ab initio relativistic potential energy surfaces of benzene-xe complex with application to intermolecular vibrations. J Chem Phys 152(11):114116
Gerardi H, Gardenier G, Viswanathan U, Auerbach S, Johnson M (2011) Vibrational predissociation spectroscopy and theory of ar-tagged, protonated imidazole (im) im1-3h+\(\cdot\)ar clusters. Chemical Physics Letters 501(4):172–178
Bua-ngern W, Chaiwongwattana S, Suwannakham P, Sagarik K (2016) Dynamics of proton transfer in imidazole hydrogen-bond chains. RSC Adv 6:99391–99403
Iannuzzi M (2006) Proton transfer in imidazole-based molecular crystals. J Chem Phys 124(20)
Iannuzzi M, Parrinello M (2004) Proton transfer in heterocycle crystals. Phys Rev Lett 93:025901
Kumar M, Venkatnathan A (2015) Quantum chemistry study of proton transport in imidazole chains. J Phys Chem B 119(7):3213–3222
Mangiatordi GF, Hermet J, Adamo C (2011) Modeling proton transfer in imidazole-like dimers: A density functional theory study. J Phys Chem A 115(12):2627–2634
Mangiatordi GF, Brémond E, Adamo C (2012) Dft and proton transfer reactions: A benchmark study on structure and kinetics. J Chem Theory Comput 8(9):3082–3088
Sugisawa H, Ida T, Krems RV (2020) Gaussian process model of 51-dimensional potential energy surface for protonated imidazole dimer. J Chem Phys 153(11):114101
Neese F (2012) The orca program system. Wiley Interdisciplinary Reviews: Comput Mole Sci 2(1):73–78. https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.81
TURBOMOLE V6.2 2010, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com
Parrish RM, Burns LA, Smith DGA, Simmonett AC, DePrince AE, Hohenstein EG, Bozkaya U, Sokolov AY, Di Remigio R, Richard RM, Gonthier JF, James AM, McAlexander HR, Kumar A, Saitow M, Wang X, Pritchard BP, Verma P, Schaefer HF, Patkowski K, King RA, Valeev EF, Evangelista FA, Turney JM, Crawford TD, Sherrill CD (2017) Psi4 1.1: An open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J Chem Theory Comput 13(7):3185–3197. https://doi.org/10.1021/acs.jctc.7b00174. pMID: 28489372
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16 Revision C.01. Gaussian Inc. Wallingford CT
Dunlap BI, Connolly JWD, Sabin JR (1979) On some approximations in applications of x\(\alpha\) theory. J Chem Phys 71(8):3396–3402
Izsák R, Neese F (2011) An overlap fitted chain of spheres exchange method. J Chem Phys 135(14)
Kossmann S, Neese F (2009) Comparison of two efficient approximate hartee-fock approaches. Chemical Physics Letters 481(4):240–243
Kossmann S, Neese F (2010) Efficient structure optimization with second-order many-body perturbation theory: The rijcosx-mp2 method. J Chem Theory Comput 6(8):2325–2338
Neese F (2003) An improvement of the resolution of the identity approximation for the formation of the coulomb matrix. J Comput Chem 24(14):1740–1747. https://onlinelibrary.wiley.com/doi/10.1002/jcc.10318
Neese F, Wennmohs F, Hansen A, Becker U (2009) Efficient, approximate and parallel hartree–fock and hybrid dft calculations. a “chain-of-spheres” algorithm for the hartree–fock exchange. Chem Phys 356(1):98–109. https://doi.org/10.1016/j.chemphys.2008.10.036. http://www.sciencedirect.com/science/article/pii/S0301010408005089, moving Frontiers in Quantum Chemistry:
Whitten JL (1973) Coulombic potential energy integrals and approximations. J Chem Phys 58(10):4496–4501
Dunning TH (1989) Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen. J Chem Phys 90(2):1007–1023. https://doi.org/10.1063/1.456153
Kendall RA, Dunning TH, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. systematic basis sets and wave functions. J Chem Phys 96(9):6796–6806. https://doi.org/10.1063/1.462569
Weigend F (2006) Accurate coulomb-fitting basis sets for h to rn. Phys Chem Chem Phys 8:1057–1065
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy. Phys Chem Chem Phys 7:3297–3305
Becke AD (1993) Density-functional thermochemistry. iii. the role of exact exchange. J Chem Phys 98(7):5648–5652. http://link.aip.org/link/JCP/98/5648/1
Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Vosko SH, Wilk L, Nusair M (1980) Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Canadian J Phys 58(8):1200–1211. https://doi.org/10.1139/p80-159
Hohenstein EG, Sherrill CD (2010) Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J Chem Phys 133(1)
Hohenstein EG, Sherrill CD (2012) Wavefunction methods for noncovalent interactions. WIREs Comput Mole Sci 2(2):304–326
Liakos DG, Neese F (2015) Domain based pair natural orbital coupled cluster studies on linear and folded alkane chains. J Chem Theory Comput 11(5):2137–2143
Liakos DG, Neese F (2015) Is it possible to obtain coupled cluster quality energies at near density functional theory cost? domain-based local pair natural orbital coupled cluster vs modern density functional theory. J Chem Theory Comput 11(9):4054–4063
Colbert DT, Miller WH (1992) A novel discrete variable representation for quantum mechanical reactive scattering via the S-matrix kohn method. J Chem Phys 96(3):1982–1991
Thomas M, Brehm M, Fligg R, Vöhringer P, Kirchner B (2013) Computing vibrational spectra from ab initio molecular dynamics. Phys Chem Chem Phys 15:6608–6622
Tikhonov DS (2016) Simple posterior frequency correction for vibrational spectra from molecular dynamics. J Chem Phys 144(17):174108
Brehm M, Kirchner B (2011) Travis - a free analyzer and visualizer for monte carlo and molecular dynamics trajectories. J Chem Inform Model 51(8):2007–2023
Svendsen A, Lorenz UJ, Boyarkin OV, Rizzo TR (2010) A new tandem mass spectrometer for photofragment spectroscopy of cold, gas-phase molecular ions. Rev Sci Instrum 81(7):073107
Bernhard D, Dietrich F, Fatima M, Perez C, Poblotzki A, Jansen G, Suhm MA, Schnell M, Gerhards M (2017) Multi-spectroscopic and theoretical analyses on the diphenyl ether-tert-butyl alcohol complex in the electronic ground and electronically excited state. Phys Chem Chem Phys 19:18076–18088
Medcraft C, Zinn S, Schnell M, Poblotzki A, Altnöder J, Heger M, Suhm MA, Bernhard D, Stamm A, Dietrich F, Gerhards M (2016) Aromatic embedding wins over classical hydrogen bonding - a multi-spectroscopic approach for the diphenyl ether-methanol complex. Phys Chem Chem Phys 18:25975–25983
Scutelnic V, Perez MAS, Marianski M, Warnke S, Gregor A, Rothlisberger U, Bowers MT, Baldauf C, von Helden G, Rizzo TR, Seo J (2018) The structure of the protonated serine octamer. J Am Chem Soc 140(24):7554–7560
Voronina L, Scutelnic V, Masellis C, Rizzo TR (2018) Can mutational analysis be used to assist structure determination of peptides? J Am Chem Soc 140(7):2401–2404
Choi MY, Miller RE (2006) Infrared laser spectroscopy of imidazole complexes in helium nanodroplets: monomer, dimer, and binary water complexes. J Phys Chem A 110(30):9344–9351
Forsting T, Zischang J, Suhm MA, Eckhoff M, Schröder B, Mata RA (2019) Strained hydrogen bonding in imidazole trimer: a combined infrared, raman, and theory study. Phys Chem Chem Phys 21:5989–5998
Adesokan AA, Chaban GM, Dopfer O, Gerber RB (2007) Vibrational spectroscopy of protonated imidazole and its complexes with water molecules: ab initio anharmonic calculations and experiments. J Phys Chem A 111(31):7374–7381
Andrei HS, Solcà N, Dopfer O (2005) Interaction of ionic biomolecular building blocks with nonpolar solvents: Acidity of the imidazole cation (im+) probed by ir spectra of im+-ln complexes (l = ar, n2; n ≤ 3). J Phys Chem A 109(16):3598–3607
Andrei HS, Solcà N, Dopfer O (2006) Microhydration of protonated biomolecular building blocks: Ir spectra of protonated imidazole-water complexes. ChemPhysChem 7(1):107–110
Kesharwani MK, Brauer B, Martin JML (2015) Frequency and zero-point vibrational energy scale factors for double-hybrid density functionals (and other selected methods): Can anharmonic force fields be avoided? J Phys Chem A 119(9):1701–1714
Bach A, Leutwyler S, Sabo D, Bačić Z (1997) Very large amplitude intermolecular vibrations and wave function delocalization in 2,3-dimethylnaphthalene\(\cdot\)he van der waals complex. J Chem Phys 107(21):8781–8793
Tatara W, Wójcik MJ, Lindgren J, Probst M (2003) Theoretical study of structures, energies, and vibrational spectra of the imidazole-imidazolium system. J Phys Chem A 107(39):7827–7831
Boys S, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. some procedures with reduced errors. Mole Phys 19(4):553–566. https://doi.org/10.1080/00268977000101561
Acknowledgements
This research was supported in part through the Maxwell computational resources operated at Deutsches Elektronen-Synchrotron DESY, Hamburg, Germany.
Funding
Open Access funding enabled and organized by Projekt DEAL. Benchmarking aspects of this work benefited from the environment provided by the local research training group BENCH (DFG-389479699/GRK2455).
Author information
Authors and Affiliations
Contributions
D.T., D.S., A.K., D.O. performed theoretical simulations. V.S. performed the experiments. D.T., D.S., and V.S. prepared figures. D.T., V.S., D.S., A.D., and M.S. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tikhonov, D.S., Scutelnic, V., Sharapa, D.I. et al. Structures of the (Imidazole)nH+ ... Ar (n=1,2,3) complexes determined from IR spectroscopy and quantum chemical calculations. Struct Chem 34, 203–213 (2023). https://doi.org/10.1007/s11224-022-02053-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-02053-4