
Abstract
In the present study, a new category of 1,3,4-thiadiazoles was developed by submitting methyl 2-(4-hydroxy-3-methoxybenzylidene) hydrazine-1-carbodithioate to react with the appropriate hydrazonoyl halides in presence of few drops of diisopropyl ethyl amine. The chemical structures of the newly synthesized derivatives were inferred by means of their micro-analytical and spectral data. Utilizing combined molecular docking and molecular dynamics techniques, the binding affinities and features of the synthesized compounds were evaluated against four SARS-CoV-2 target enzymes, namely, main protease (Mpro), papain-like protease (PLpro), RNA-dependent RNA polymerase (RdRp), and receptor-binding domain (RBD) of the spike protein. Compound 7 demonstrated promising binding affinities with the target enzymes Mpro, PLpro, RdRp, and RBD with docking scores of −11.4, −9.4, −8.2, and −6.8 kcal/mol, respectively. In addition, compound 7 exhibited MM-GBSA//100 ns MD docking score of −35.9 kcal/mol against Mpro. Structural and energetic analyses revealed the stability of the 7-Mpro complex over 100 ns MD simulations. In addition, compound 7 obeyed Lipinski’s rule of five, as it has acceptable absorption, distribution, and oral bioavailability inside the body. Therefore, compound 7 is considered as a promising starting point for designing potential therapeutic agents against Covid-19.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A new strain for SARS-CoV-1 identified recently as SARS-CoV-2 in late December 2019 resulted in serious physical and psychological damages to the human health; a massive outbreak initially in Wuhan, China, and spread rapidly in different nations around the global in a short time [1, 2]. The World Health Organization (WHO) declared this highly infectious respiratory disease Covid-19 as a pandemic [3]. It is acceptable to think that a sufficient understanding of SARS-CoV-2 and the full clinical picture of the resulting Covid-19 disease will take some time. However, the first detected clinical sign of Covid-19 was pneumonia [4]. Recently, asymptomic infections and gastrrointestinal symptoms were also reported especially among young children [5, 6]. Pneumonia mostly appeared in the second or third week of the infection. Decreased oxygen saturation, blood gas deviations, and changes visible through chest X-rays are prominent signs of viral pneumonia. In addition, lymphopenia documented to be common, and inflammatory markers (Proinflammatory cytokines and CRP) are elevated. Consequently, investigation of anti-Covid-19 therapeutic agents became an urgent demand and attracted more interest recently owing to the lack of specific drugs for the treatment of Covid-19 [7, 8]. Nevertheless, several existing drugs are available only to overcome the clinical symptoms of Covid-19.
On the other hand, 1,3,4-thiadiazole moieties have been reported for their pharmaceutical properties. Many antiviral drugs like acetazolamide, besaglybuzole (glybuzole), and furidiazine (triafur) were reported to append the 1,3,4-thiadiazole in their constructors[9,10,11,12,13].
Urgent needs for develoment novel anti-Covid19 agents have directed us to synthesize some new bioactive heterocyclic molecules. In the present study, we aimed to identify potential Covid-19 inhibitors through a computer-based molecular docking and molecular dynamics techniques [14,15,16]. In addition, ADMET (absorption, distribution, metabolic, excretion, and toxicity) and pharmacokinetics parameters of the prepared ligand molecules were performed to identify their drug-likeness properties [17].
Results & discussion
Chemistry
Methyl 2-(4-hydroxy-3-methoxybenzylidene)hydrazine-1-carbodi-thioate (2) acts as key molecule for the design of new desired 1,3,4-thiadiazole compounds. It is allowed to react with a selected group of hydrazonoyl halide derivatives by grinding method “grindstone chemistry” under solvent-free conditions with the addition of catalytic amount of DIPEA (diisopropyl ethyl amine) to afford the target molecules 3–7, (Scheme 1).

Synthetic procedures of the desired 1,3,4-thiadiazoles 3–7
The chemical structures of all newly prepared molecules are affirmed by spectral and elemental data. For instance, IR spectrum of the target molecule 7 revealed a strong broad absorption band at v 3337 assigned for NH group. Additionally, it showed a strong sharp absorption band at v 1681 attributed to the carbonyl group. Meanwhile, 1H-NMR spectrum exhibited singlet signal at δ 3.83 ppm represented the methoxy group along with multiplet signal at δ 6.86–7.85 ppm for aromatic protons. Also, it revealed doublet signal at δ 7.75 ppm attributed to the aromatic hydrogen and doublet signal at δ 8.15 ppm represented the aromatic hydrogen. Moreover, it showed three singlet signals at δ 8.36, 9.65, and 10.68 ppm for CH = N, OH and NH, respectively as illustrated in Fig. 1.

1H.−NMR spectrum of the target molecule 7
Figure 2 revealed the significant signals of the target molecule 7 which confirmed the formation of the compound.

13C-NMR spectrum of target molecule 7
The chemical composition of the target molecule 7 was affirmed also by the mass spectrum (m/z 445) [M+], which agrees with its molecular formula C23H19N5O3S, as represented in Fig. 3.

Mass spectrum of compound 7
Molecular docking calculations
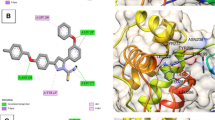
The molecular docking technique [11, 18] was used to predict the binding modes and affinities of the newly synthesized compounds with SARS-CoV-2 targets Mpro, PLpro, RdRp, and RBD of S-protein. The predicted docking scores are tabulated in Table 1. 2D (two dimensional) and 3D (three dimensional) representations of binding modes of best docked compound 7 inside the active site of Mpro, PLpro, RdRp, and RBD are displayed in Fig. 4. The representations of the rest docked compounds against the targets are shown as Figs. S1–S4, respectively, in the Supplementary file section.

2D representations of interactions between the reference drug and the targets. Blue stick models represented the docked compounds, and colored balls represented the active site region. H-bond interactions are shown in green and blue dotted lines. π-interactions are shown in orange lines
It is observed from the data in Table 1 that compound 7 exhibited binding affinities against all the selected targets better than the reference drug (Darunavir) (Fig. 4).
In addition, most of the synthesized compounds demonstrated promising binding affinities against Mpro with binding energies ranged from −11.4 to −6.4 kcal/mol. The high docking scores of the studied compounds with Mpro would be returned to their ability to form hydrogen bonds, hydrophobic and van der Waals interactions with the amino acid residues of active sites (Fig. 5).

(Left side) 2D and (right side) 3D representations of intermolecular interactions of compound 7 against the active sites of SARS-CoV-2 (Mpro), (PL.pro), (RdRp), and (RBD) of the spike protein. H-bond interactions are shown in green and blue dotted lines. π- interactions are shown in orange lines
Compared to Mpro, the examined compounds showed relatively weak binding affinities with PLpro, RdRp, and RBD, with docking scores ranged from −9.4 to −5.6, −8.2 to −5.2, and −6.8 to −5.3 kcal/mol, respectively. Interestingly, compound 7 displayed the highest binding affinity against all targets with docking scores of −11.4, −9.4, −8.2, and −6.8 kcal/mol, respectively. The promising binding affinity of 7 against the target Mpro is attributed to its ability to exhibit two hydrogen bond interactions with Thr24, and Ser144 at 2.17 and 2.91 Å, respectively (Fig. 4). Besides, inspecting the binding modes of compound 7 with PLpro, RdRp, and RBD unveiled its potentiality to form hydrogen bonds and Pi-stacking interactions, as presented in Fig. 4. The ligand molecule 7 docked with PLpro amino acid residues (Lys218, Tyr251, and Phe258) through pi-stacked interactions at distances 4.27, 5.51, and 5.58 Å, respectively. Further, compound 7 interacted with the residues Arg116 and Phe35 of the target RdRp through pi-stacked interactions at distances 5.89, and 3.92 Å, respectively. Finally, it docked with RBD through two hydrogen bonds and one pi–pi interaction with the amino acid residues Tyr385, Arg393, and Phe40.
Molecular dynamics (MD) simulations
Towards more reliable binding affinities, the molecular dynamics simulations were performed for all synthesized compounds in complex with SARS-CoV-2 targets. The binding energies (ΔGbinding) were then calculated using the molecular mechanics-generalized born surface area (MM-GBSA) approach based on the collected snapshots for Mpro, PLpro, RdRp, and RBD of spike protein over the production stage of 25 ns. The calculated MM-GBSA binding energies are listed in Table 2.
It is apparent from Table 2 that the examined compounds with Mpro showed higher binding affinities over 25 ns MD simulations than with PLpro, RdRp, and RBD. The calculated MM-GBSA binding energies were in line with the predicted docking scores, demonstrating the high potency of the examined ligand molecules with Mpro over the other SARS-CoV-2 targets.
Among the examined compounds, 7 exhibited the lowest binding energy with Mpro with a ΔGbinding value of −39.2 kcal/mol. Moreover, it showed weak binding energies of −15.8, −15.5, and −14.9 kcal/mol with PLpro, RdRp, and RBD, respectively. These results declared the selectivity of the compound 7 towards Mpro over PLpro, RdRp, and RBD. However, compound 4 demonstrated the lowest binding with PLpro, RdRp, and RBD with a ΔGbinding value of −27.9, −26.1, and −21.7 kcal/mol, respectively (Table 2). MD simulation for compound 7 in complex with Mpro and compound 4 complexed with PLpro, RdRp, and RBD were then elongated to 100 ns. Additionally, the corresponding MM-GBSA binding energy was calculated and was compared to the reference drug Darunavir (Fig. 6).

Decomposition of MM-GBSA binding energies for the investigated inhibitors in complex with of SARS-CoV-2 a) Mpro, b) PL.pro, c) RdRp, and d) RBD of the spike protein throughout 100 ns MD simulations
MM-GBSA binding energy of compound 7-Mpro, compound 4-PLpro, compound 4-RdRp, and compound 4-RBD complexes was decomposed to explore the predominant interactions between the investigated inhibitors and target. According to the data, it was found that the docking energy was calculated by Evdw interactions with an average value of −47.2, −38.4, and −36.8 kcal/mol for investigated inhibitor with Mpro, PLpro, and RdRp, respectively (Fig. 6). For compound 4 complexed with RdRp, the docking energy was dominated by Eele interactions with an average value of −65.5 kcal/mol which was three times higher than that of Lopinavir and curcumin, with an average value of −42.5 kcal/mol (Fig. 6). Together these results demonstrated the promising binding affinity of compounds 7 and 4 with SARS-CoV-2 targets.
Post-dynamics analysis
The interaction nature and stability of compound 7 and Darunavir within the active site of Mpro was estimated using structural and energetic analyses. Structural and energetic analyses including energy per-frame, centre-of-mass distance (CoM), root-mean-square deviation (RMSD), and root-mean-square fluctuation (RMSF) were performed over 100 ns MD simulations.
Docking energy per frame
The stability of compound 7 and Darunavir in complex with SARS-CoV-2 Mpro was estimated using the correlation between the binding energy per-frame and time. MM-GBSA binding energy was subsequently evaluated per-frame for the most promising compound with each target and displayed in Fig. 7. The most interesting aspect of this graph is the overall stability of two identified compounds over 100 ns MD simulations with average values of −35.9, and −34.8 for compound 7-Mpro, and darunavir-Mpro complexes, respectively.

Variations in the MM-GBSA binding energies for compound 7 (in black), and Darunavir (in red) with SARS-CoV-2 M.pro during 100 ns MD simulations
Center-of-mass distance
Interestingly, investigating the center-of-mass (CoM) distance between the compound 7, and Darunavir and the residue Glu166 through the 100 ns MD simulations would reflect a strong indication of the high stability of the identified compounds inside the Mpro active site. The CoM distances were inspected over the 100 ns MD simulations and represented in Fig. 8. What stands out in Fig. 8 is the average CoM distance between the identified compounds and the key amino acid residue Glu166 was approximately constant, with average CoM distances of 5.7, and 12.1 Å, respectively. The current data revealed that compound 7 bound more tightly to the Mpro complex compared to Darunavir.

Centre-of-mass (CoM) distances between 7 (in black), and darunavir (in red) with GLU166 of SARS-CoV-2 M.pro throughout 100 ns MD simulations
Root-mean-square deviation
The structural changes of 7-Mpro and darunavir-Mpro complexes were evaluated using the root-mean-square deviation (RMSD). The conformational change of backbone atoms of the most promising three compounds in complex with SARS-CoV-2 Mpro has been compared with initial conformations over 100 ns MD simulations as shown in Fig. 9. As shown in Fig. 8, for 7- Mpro and darunavir-Mpro complexes, the distance was noticed to be below 0.25 nm and the overall stability of these compounds inside the SARS-CoV-2 Mpro active site. These results confirmed that compound 7 is tightly bonded in the active site and does not affect the overall topology of SARS-CoV-2 Mpro.

Root-mean-square-deviation (RMSD) of the backbone atoms from the initial structure for 7 (in black) and darunavir (in red) with the SARS-CoV-2 main protease (M.pro) over 100 ns MD simulations
ADMET and drug-likeness properties of the molecules
ADMET studies of the prepared molecules exhibited that they have acceptable absorption and distribution properties in the range of (91–80-98.76%) and (0.53–0.70), respectively. The physiochemical properties of the compounds exhibited acceptable values, as they have molecular weights and partition coefficients in the range of (256.35–445.49 g/mol), and < 5, respectively. Moreover, the molecules have no toxicity and carcinogenicity. All tested compounds showed good oral bioavailability within the body as they obeyed Lipinski's rule of five (Table 3).
Conclusion
In this study, a new series of 1,3,4-thiadiazole derivatives was synthesized, characterized, and theoretically evaluated as Covid-19 inhibitors against four SARS-CoV-2 targets namely, main protease (Mpro), papain-like protease (PLpro), RNA-dependent RNA polymerase (RdRp), and receptor-binding domain (RBD) of the spike protein. The molecular docking studies and molecular dynamics simulations exhibited the promising binding affinity of compound 7 with all targets. Therefore, it could be select as promising chemical moiety for designing of future inhibitors as anti-Covid-19 agents.
Material and methods
Instrumentation
All melting points were uncorrected and measured using electrothermal device. The IR spectra were recorded (KBr discs) using Shimadzu FT-IR 8201 PC spectrophotometer. 1H- and 13C-NMR spectra were recorded in (CD3)2SO solutions on a BRUKER 500 FT-NMR system spectrometer, and chemical shifts are expressed in ppm units using TMS as an internal reference. Mass spectra were recorded on a GC–MS QP1000 EX Shimadzu. Elemental analyses were carried out at the Microanalytical Center of Cairo University.
Synthetic procedures of the target molecules (3–7)
A mixture of compound 2 (1.28 gm, 5 mmol) and the selected derivative of the hydrazonoyl halides (5 mmol) and 2–3 drops of DIPEA as a catalyst, were ground well in an open mortar with a pestle for 5–7 min. at RT till the mixture turned into melt. The grinding was continued for approximately 5–10 min, and the reaction was monitored by TLC. The solid was collected and washed with (water/ethanol) the recrystallized from the proper solvent to give the desired derivatives 3–7, respectively.
“Ethyl 5-(4-hydroxy-3-methoxybenzylidene)hydrazono)-4-phenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate” (3)
Yellow crystals (95%); m.p. 172–174 °C, FT-IR (KBr, cm−1): v 1554 (C = C), 1599 (C = N), 1712 (C = O), 3471 (OH); 1H-NMR (DMSO-d6): δ 9.65 (s, 1H, OH), 8.29 (s, 1H, CH), 7.90 (d, 2H, J = 10 Hz, ArH), 7.33–7.45 (m, 4H, ArH), 7.17 (d, 1H, J = 10 Hz, ArH), 6.83 (d, 1H, J = 10 Hz, ArH), 4.15 (q, 2H, CH2CH3) 3.82 (s, 3H, OCH3),1.29 (t, 3H, CH2CH3);13C-NMR (100 MHz, DMSO-d6): δ 13.97 (CH3), 55.54 (OCH3), 62.75 (CH2), 110.18 (Ar.), 115.61(Ar.), 122.40 (Ar.), 122.56 (Ar.), 125.51 (Ar.), 127.32 (Ar.), 127.92 (Ar.), 129.07 (Ar.), 138.61 (Ar.), 142.22 (Ar.), 147.92 (Ar.), 149.51(CH), 156.12 (Ar.), 158.07 (Ar.), 163.75 (C = O); MS m/z (%): 398 (M+, 60). Anal. Calcd. for “C19H18N4O4S” (398): C, 57.28; H, 4.55; N, 14.06. Found: C, 57.34; H, 4.51; N, 14.01%.
“Ethyl 5-(4-hydroxy-3-methoxybenzylidene)hydrazono)-4-(p-tolyl)-4,5-dihydro-1,3,4-thiadiazole-2-carboxylate” (4)
Yellow crystals (92%); m.p. 180–182 oC, FT-IR: v 1550 (C = N), 1600 (C = N), 1705 (C = O), 3502 (OH); 1H-NMR: δ 9.65 (s, 1H, OH), 8.29 (s, 1H, CH), 7.76 (d, 2H, J = 10 Hz, Ar–H), 7.31–7.33 (m, 3H, Ar–H), 7.18 (d, 1H, J = 10 Hz, Ar–H), 6.84 (d, 1H, J = 10 Hz, Ar–H), 4.33 (q, 2H, CH2, CH2CH3), 3.81 (s, 3H, OCH3), 2.36 (s, 3H, CH3),1.30 (t, 3H, CH2CH3);13C-NMR: δ 13.97 (CH3), 20.64 (CH3), 55.53 (OCH3), 62.72 (CH2), 110.19 (Ar.), 115.58 (Ar.), 122.35 (Ar.), 122.56 (Ar.), 125.51 (Ar.), 129.46 (Ar.), 136.09 (Ar.), 136.91(Ar.), 141.88 (Ar.), 147.91 (Ar.), 149.55 (CH), 155.94 (Ar.), 158.08 (Ar.), 163.81 (C = O); MS m/z (%):412 (M+, 30). Anal. Calcd. for “C20H20N4O4S” (412): C, 58.24; H, 4.89; N, 13.58. Found: C, 58.19; H, 4.83; N, 13.53%.
“1-(5(-4-hydroxy-3-methoxybenzylidene)hydrazono)-4-phenyl-4,5-dihydro-1,3,4-thiadiazol-2-yl)ethan-1-one” (5)
Orange crystals (82%); mp. 212–214 oC, FT-IR: v 1550 (C = C), 1600 (C = N), 1678 (C = O), 3417 (OH); 1H-NMR: δ 9.65 (s, 1H, OH), 8.31 (s, 1H, CH), 7.34–7.97 (m, 6H, ArH), 7.16 (d, 1H, J = 10 Hz, ArH), 6.81(d, 1H, J = 10 Hz, ArH), 3.80 (s, 3H, OCH3), 2.48 (s, 3H, CH3); 13C-NMR: δ 25.14 (CH3), 55.54 (OCH3), 110.01 (Ar.), 115.59 (Ar.), 122.60 (Ar.), 125.49 (Ar.), 127.43 (Ar.), 129.13 (Ar.), 138.54 (Ar.), 147.87 (Ar.), 149.55 (CH), 150.17 (Ar.), 156.33 (Ar.), 164.04 (C = O); MS m/z (%): 368 (M+, 40). Anal. Calcd. for “C18H16N4O3S” (368): C, 58.68; H, 4.38; N, 15.21. Found: C, 58.63; H, 4.32; N, 15.16%.
“1-(5-(4-hydroxy-3-methoxybenzylidene)hydrazono)-4-(p-tolyl)-4,5-dihydro-1,3,4-thiadiazol-2-yl)ethan-1-one” (6)
Orange solid (81%); mp.191–193 °C, FT-IR: v 1541 (C = C), 1600 (C = N), 1681 (C = O), 3502 (OH); 1H-NMR: δ 9.64 (s, 1H, OH), 8.27 (s, 1H, CH), 7.80 (d, 2H, ArH), 7.30–7.32 (m, 3H, ArH), 7.13 (d, 1H, J = 10 Hz), 2.34 (s, 3H, CH3), 6.80 (d, 1H, J = 10 Hz, ArH), 3.79 (s, 3H, OCH3), 2.51 (s, 3H, CH3);13C-NMR: δ 20.66 (CH3), 24.95 (CH3), 55.52 (OCH3), 109.97(Ar.), 115.58 (Ar.), 122.53 (Ar.), 125.54 (Ar.), 129.48 (Ar.), 136.22(Ar.), 136.97 (Ar.), 147.94(Ar.), 149.53 (CH), 149.95 (Ar.), 156.07 (Ar.), 164.13 (C = O); MS m/z (%): 382 (M+, 15)%. Anal. Calcd. for “C19H18N4O3S” (382): C, 59.67; H, 4.74; N, 14.65. Found: C, 59.73; H, 4.70; N, 14.62%.
“5-(-4-hydroxy-3-methoxybenzylidene)hydrazono)-N,4-diphenyl-4,5-dihydro-1,3,4-thiadiazole-2-carboxamide” (7)
Yellow solid, m.p. 251–253 °C; yield (95%); FT-IR: v 1539 (C = C), 1600 (C = N), 1681 (C = O), 3337 (NH, OH);1H-NMR: δ 10.68 (s, 1H, NH), 9.65 (s,1H, OH), 8.36 (s, 1H, CH), 8.15 (d, 1H, J = 10 Hz, ArH), 7.75 (d, 1H, J = 10 Hz, ArH), 6.86–7.85 (m, 11H, ArH), 3.83 (s, 3H, OCH3);13C-NMR: δ 55.5 (OCH3), 110.05 (Ar.), 115.61(Ar.), 120.96 (Ar.), 122.34 (Ar.), 122.43 (Ar.), 124.70 (Ar.), 125.58 (Ar.), 126.94 (Ar.), 128.82 (Ar.), 128.93 (Ar.), 137.54 (Ar.), 138.94 (Ar.), 147.33 (Ar.), 147.95 (Ar.), 149.54 (CH), 155.89 (Ar.), 156.26 (Ar.), 164.16 (C = O); MS m/z [%]: 445 (M+), 444 (75), 317 (30), 281 (28), 255(80), 151(52), 127(48); Anal. Calcd. for “C23H19N5O3S” (445): C, 62.02; H, 4.30; N, 15.72%. Found: C, 62.06; H, 4.27; N, 15.65%.
Computational methodology
Target identification
The crystal structures of SARS-CoV-2 main protease (Mpro; PDB code: 6LU7) [19], papain-like protease (PLpro; PDB code: 6W9C) [20], RNA-dependent RNA polymerase (RdRp; PDB code: 6M71) [21], and receptor-binding domain (RBD) of spike protein (S-protein; PDB code: 6M0J) [22] were selected as templates for docking studies and molecular dynamics calculations. The water molecules, ions, and co-crystalized ligands if existing were removed [23]. Besides, the H + + server was utilized to investigate the protonation states of Mpro, PLpro, RdRp, and RBD of S-protein, and all missing hydrogen atoms were added [24].
Inhibitor preparation
The chemical structures of the synthesized compounds were manually constructed, and their 3D structures were generated using Open Babel 2.4.1 tool [25,26,27]. All ligand molecules were then energetically minimized using the CHARMM Force Field [28].
Molecular docking
Molecular docking calculations [10, 12, 14, 18, 24, 29,30,31,32,33] were carried out using PyRx – virtual screening software [34]. The pdbqt files of Mpro, PLpro, RdRp, and RBD of S-protein targets were prepared according to PyRx protocol. The docking algorithms were conserved to their default values, except the number of genetic algorithms (GA) run and the maximum number of energy evaluation (eval). In the current study, GA and eval were set to 250 and 25,000,000, respectively. The docking grid was set to 25 Å × 25 Å × 25 Å with a spacing value of 0.375 Å [17, 35]. The grid center was positioned at the center of the active site of Mpro, PLpro, RdRp, and RBD of S-protein. The partial atomic charges of the examined compounds were estimated using the Gasteiger method [36]. The prediction of binding modes for each compound was handled using the built-in clustering analysis with an RMSD tolerance of 1.0 Å. Also, the lowest energy conformation from the largest cluster was picked out as a representative binding pose.
Molecular dynamics simulations
Molecular dynamics simulations were performed for the examined compounds in complex with the four studied SARS-CoV-2 targets using AMBER16 software. In MD simulations, the General AMBER force field (GAFF2) [37] and AMBER force field 14SB [38] were employed to describe the studied compounds and SARS-CoV-2 targets, respectively. The atomic partial charges of the examined compounds were calculated using the restrained electrostatic potential (RESP) approach at the HF/6 −31G* level with the help of Gaussian software [39]. Prior to RESP charge calculations, the studied compounds were first geometrically optimized at the B3LYP/6-31G* level of theory. The docked compound-target complexes were solvated in a cubic water box with 15 Å distances between the edges of the box and any atom of compound or compound-target complexes. The solvated compound-target systems were subsequently energy minimized for 5000 steps, gently annealed from 0 to 300 K over 50 ps, and equilibrated for 1 ns. The equilibrated systems were then simulated for 100 ns using periodic boundary conditions and NPT ensemble. The non-bonded cut-off distance was placed at 12 Å, and particle mesh Ewald (PME) method was applied to process long-range electrostatic interactions. The Langevin dynamics with the collision frequency gamma_ln set to 1.0 was used to conserve the temperature of the examined systems at 298 K. The pressure of the system was controlled using Berendsen barostat with a relaxation time of 2 ps. A time step of 2 fs and the SHAKE option to constrain all bonds involving hydrogen atoms were utilized. Coordinates and energy values were collected every 10 ps over the production stage for binding energy calculations and post-dynamics analyses. All MD simulations were conducted with the GPU of pmemd (pmemd.cuda) in AMBER16 on the CompChem GPU/CPU cluster (hpc.compchem.net). 2D and 3D visualization of the compound-targets interactions were performed using the Discovery studio software.
MM-GBSA binding energy
The binding free energies of the examined compounds with SARS-CoV-2 targets were estimated using molecular mechanical-generalized Born surface area (MM-GBSA) approach [40, 41]. For MM-GBSA calculations, uncorrelated snapshots were collected every 10 ps over the production stage. The MM-GBSA binding energy (\({\Delta \mathrm{G}}_{\mathrm{binding}}\)) can be conceptually summarized as:
where the energy term (G) is estimated as:
where Evdw and Eele are the van der Waals and electrostatic energies, respectively. GGB is the electrostatic solvation free energy calculated from the generalized Born equation, and GSA is the nonpolar contribution to the solvation free energy from the solvent-accessible surface area (SASA). Solute entropy contributions to binding energies were neglected, and a single-trajectory approach was employed, in which the coordinates of each molecule, receptor, and complex were isolated from a single trajectory.
ADMET analysis
The freely accessible online softwares such as admetSAR, SwissADME, and Mol inspiration are used to predict ADMET and drug-likeness properties of compounds.
Data availability
The data that support the findings of this study are included within the article and supplementary file.
References
Cubuk J, Alston JJ, Incicco JJ, Singh S, Stuchell-Brereton MD, Ward MD, Zimmerman MI, Vithani N, Griffith D, Wagoner JA (2021) The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat Commun 12:1–17
Bagherzadeh K, Daneshvarnejad K, Abbasinazari M, Azizian H (2020) In silico repositioning for dual inhibitor discovery of SARS-CoV-2 (COVID-19) 3C-like protease and papain-like peptidase. Preprints 2020, 2020040084 (https://doi.org/10.20944/preprints202004.0084.v1)
Velavan TP, Meyer CG (2020) The COVID-19 epidemic. Trop Med Int Heal 25:278–280. https://doi.org/10.1111/tmi.13383
Baloch S, Baloch MA, Zheng T, Pei X (2020) The coronavirus disease 2019 (COVID-19) pandemic. Tohoku J Exp Med 250:271–278
Cai X, Ma Y, Li S, Chen Y, Rong Z, Li W (2020) Clinical characteristics of 5 COVID-19 cases with non-respiratory symptoms as the first manifestation in children. 8:1–9. https://doi.org/10.3389/fped.2020.00258
Li B, Zhang S, Zhang R, Chen X, Wang Y, Zhu C (2020) Epidemiological and clinical characteristics of COVID-19 in children: a systematic review and meta-analysis. Front Pediatr 8:1–12. https://doi.org/10.3389/fped.2020.591132
Awadasseid A, Wu Y, Tanaka Y, Zhang W (2021) Effective drugs used to combat SARS-CoV-2 infection and the current status of vaccines. Biomed Pharmacother 111330
Liu C, Zhou Q, Li Y, Garner LV, Watkins SP, Carter LJ, Smoot J, Gregg AC, Daniels AD, Jervey S et al (2020) Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent Sci. https://doi.org/10.1021/acscentsci.0c00272
Shehadi IA, T Abdelrahman M, Abdelraof M, Rashdan HR (2022) Solvent-free synthesis, in vitro and in silico studies of novel potential 1, 3, 4-thiadiazole-based molecules against microbial pathogens. Molecules 27:342
Rashdan HR, Shehadi IA, Abdelmonsef AH (2021) Synthesis, anticancer evaluation, computer-aided docking studies, and ADMET prediction of 1,2,3-triazolyl-pyridine hybrids as human aurora b kinase inhibitors. ACS Omega 6:1445–1455. https://doi.org/10.1021/acsomega.0c05116
Rashdan HRM, Yousef TA, Abdelmonsef AH, Abou-krisha MM (2021) Synthesis and identification of novel potential thiadiazole based molecules containing 1,2,3-triazole moiety against COVID-19 main protease through structure-guided virtual screening approach. Biointerface Res Appl Chem 12:8258–8270. https://doi.org/10.33263/BRIAC126.82588270
Rashdan HR, Abdelmonsef AH, Abou-Krisha MM, Yousef TA (2021) Synthesis, identification, computer-aided docking studies, and ADMET prediction of novel benzimidazo-1,2,3-triazole based molecules as potential antimicrobial agents. Molecules 26
Mehta D, Taya P (2015) Neetu A review on the various biological activities of thiadiazole. Int J Pharm Pharm Sci 7:39–47
Abdelmonsef AH, Abdelhakeem MA, Mosallam AM, Temairk H, El-Naggar M, Okasha H, Rashdan HRM (2021) A search for antiinflammatory therapies: synthesis, in silico investigation of the mode of action, and in vitro analyses of new quinazolin-2,4-dione derivatives targeting phosphodiesterase-4 enzyme. J Heterocycl Chem 1:1–19. https://doi.org/10.1002/jhet.4395
Abo-Bakr AM, Alsoghier HM, Abdelmonsef AH (2022) Molecular docking, modeling, semiempirical calculations studies and in vitro evaluation of new synthesized pyrimidin-imide derivatives. J Mol Struct 1249:131548. https://doi.org/10.1016/j.molstruc.2021.131548
Hassan EA, Shehadi IA, Elmaghraby AM, Mostafa HM, Zayed SE, Abdelmonsef AH (2022) Synthesis, molecular docking analysis and in vitro biological evaluation of some new heterocyclic scaffolds-based indole moiety as possible antimicrobial agents. Front Mol Biosci 8:1–17. https://doi.org/10.3389/fmolb.2021.775013
Hussein AHM, El-Adasy A-BA, El-Saghier AM, Olish M, Abdelmonsef AH (2022) Synthesis, characterization, in silico molecular docking, and antibacterial activities of some new nitrogen-heterocyclic analogues based on a p- phenolic unit. RSC Adv 12:12607–12621. https://doi.org/10.1039/d2ra01794f
Rashdan HRM, El-Naggar M, Abdelmonsef AH (2021) Synthesis, molecular docking studies and in silico admet screening of new heterocycles linked thiazole conjugates as potent anti-hepatic cancer agents. Molecules 26:1–17. https://doi.org/10.3390/molecules26061705
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289–293. https://doi.org/10.1038/s41586-020-2223-y
Gao X, Qin B, Chen P, Zhu K, Hou P, Wojdyla JA, Wang M, Cui S (2021) Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm Sin B 11:237–245. https://doi.org/10.1016/j.apsb.2020.08.014
Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L et al (2020) Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368:779–782. https://doi.org/10.1126/science.abb7498
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L et al (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220. https://doi.org/10.1038/s41586-020-2180-5
Shehadi IA, Rashdan HR, Abdelmonsef AH (2020) Homology modeling and virtual screening studies of antigen MLAA-42Protein: Identification of noveldrugcandidates against leukemia-an in silico approach. Comput Math Methods Med 2020. https://doi.org/10.1155/2020/8196147
Haredi Abdelmonsef A (2019) Computer-aided identification of lung cancer inhibitors through homology modeling and virtual screening. Egypt J Med Hum Genet 20:1–14. https://doi.org/10.1186/s43042-019-0008-3
El-Naggar M, El-All A, Amira S, El-Naem SI, Abdalla MM, Rashdan HR (2020) New potent 5α- Reductase and aromatase inhibitors derived from 1,2,3-triazole derivative. Molecules 25. https://doi.org/10.3390/molecules25030672
Shehadi IA, Rashdan HR, Abdelmonsef AH (2020) Homology modeling and virtual screening studies of antigen MLAA-42 protein: identification of novel drug candidates against leukemia - An in silico approach. Comput Math Methods Med 2020
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: An Open chemical toolbox. J Cheminform 3:33. https://doi.org/10.1186/1758-2946-3-33
Brooks BR, Brooks C, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S et al (2009) CHARMM: Molecular dynamics simulation package. J Comput Chem 30:1545–1614. https://doi.org/10.1002/jcc.21287
Elnaggar DH, Abdel Hafez NA, Rashdan HR, Abdelwahed NA, Awad HM, Ali KA (2019) Synthesis, antimicrobial and antitumor evaluations of a new class of thiazoles substituted on the chromene scaffold. Mini Rev Med Chem 19. https://doi.org/10.2174/1389557519666190722123422
Abdelmonsef AH, Mosallam AM (2020) Synthesis, in vitro biological evaluation and in silico docking studies of new quinazolin‐2,4‐dione analogues as possible anticarcinoma agents. J Heterocycl Chem 1–18. https://doi.org/10.1002/jhet.3889
Noser AA, El-Naggar M, Donia T, Abdelmonsef AH (2020) Synthesis, in silico and in vitro assessment of new quinazolinones as anticancer agents via potential AKT inhibition. Molecules 25. https://doi.org/10.3390/molecules25204780
Noser AA, Abdelmonsef AH, El-Naggar M, Salem MM (2021) New amino acid schiff bases as anticancer agents via potential mitochondrial complex I-associated hexokinase inhibition and targeting AMP-protein kinases / mTOR signaling pathway. Molecules 1–28. https://doi.org/10.3390/molecules26175332
Haredi Abdelmonsef A, Eldeeb Mohamed M, El-Naggar M, Temairk H, Mohamed Mosallam A (2020) Novel quinazolin-2,4-dione hybrid molecules as possible inhibitors against malaria: synthesis and in silico Molecular docking studies. Front Mol Biosci 7:1–19. https://doi.org/10.3389/fmolb.2020.00105
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. In Chemical Biology; Springer 1263:243–250. ISBN 9780123944474
Ahmed A, Ibrahim A, Mosallam A, Taha M, Temairk H (2022) Synthesis and in silico docking study of some new quinazolin-2,4-diones targeting COVID-19 (SARS-Cov-2) main protease: a search for Anti-Covid19 drug candidates. Egypt J Chem. https://doi.org/10.21608/ejchem.2022.117407.5296
Bikadi Z, Hazai E (2009) Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform 1:1–16. https://doi.org/10.1186/1758-2946-1-15
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general Amber force field. J Comput Chem 25:1157–1174. https://doi.org/10.1002/jcc.20035
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11:3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
Stewart JJP (1989) Optimization of parameters for semiempirical methods I. Method J Comput Chem 10:209–220. https://doi.org/10.1002/jcc.540100208
Rashdan HRM, Shehadi IA, Abdelrahman MT, Hemdan BA (2021) Antibacterial activities and molecular docking of novel sulfone biscompound containing bioactive 1, 2, 3-triazole moiety. Molecules 26:4817
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10:449–461. https://doi.org/10.1517/17460441.2015.1032936
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
H.R.M.R. and A.H.A. made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rashdan, H.R.M., Abdelmonsef, A.H. In silico study to identify novel potential thiadiazole-based molecules as anti-Covid-19 candidates by hierarchical virtual screening and molecular dynamics simulations. Struct Chem 33, 1727–1739 (2022). https://doi.org/10.1007/s11224-022-01985-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-01985-1