
Abstract
The feasibility of an additional ligand coordination at the 11th coordination site of actinium, lanthanum, and lutetium ions in 10-fold coordinated macropa complexes has been studied by means of density functional theory calculations. The study covered the two main macropa conformers, Δ(δλδ)(δλδ) and Δ(λδλ)(λδλ), favoured by larger (Ac3+, La3+) and smaller (Lu3+) ions, respectively. At the molecular level, the coordination of H2O is the most favourable to the largest Ac3+ while only slightly less to La3+. Protonation of the picoline arms enhances the coordination by shifting the metal ion closer to the open site of the ligand. The choice of macropa conformer has only a slight influence on the strength and bonding properties of the H2O coordination. Aqueous solution environment decreases considerably the energy gain of H2O coordination at the 11th coordination site.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Radioactive isotopes are used in medicine since 1936, when Dr. John Lawrence (brother of Nobel laureate Ernest Lawrence) began treating a 28-year-old leukaemia patient with radioactive 32P [1]. By now, the diagnostic and therapeutic applications of radioisotopes became routine in medicine. From the latter applications best known is the radiation therapy treatment of cancer and other cases of abnormal tissue growth, such as hyperthyroidism [2]. During this therapy, the patient’s tumour is bombarded with ionizing radiation, which results in breaks in the DNA molecules preventing in this way their replication.
The α-emitting actinium isotope, 225Ac, is a promising tool for treatment of a range of cancers by means of targeted α therapy (TAT) and is already at an advanced level of clinical tests [3,4,5,6]. This isotope showed very good preliminary results in curing metastatic castration-resistant prostate cancer [7,8,9], acute myeloid leukaemia [10, 11], and neuroendocrine tumours [12].
For administration of radioisotopes, generally their chelate complexes are used which are conjugated to a biological targeting vector (antibody or peptide), transporting them to the desired location in vivo. Hitherto, the tetraazamacrocycle 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) ligand proved to be the most suitable for linking of 225Ac and lanthanide ions [13, 14] to biomolecules. These chelate complexes passed successfully the first clinical tests [15]. However, while DOTA forms highly stable complexes with small ions (e.g., late lanthanides), the stability decreases considerably with the size of the ion. This property is a disadvantage for Ac3+, which is the largest 3+ ion in the periodic table.
Other types of macrocycles, based on the 1,7,10,16-tetraoxa-4,13-diazacyclooctadecane (D18C6) core, showed preference for large over small metal ions [16,17,18,19,20,21,22,23]. Recently, a D18C6-based macrocycle, N,N′-bis[(6-carboxy-2-pyridil)methyl]-4,13-diaza-18-crown-6 (called macropa), was reported to form rapidly a stable complex with 225Ac3+ [24]. Previously, macropa was found to selectively bind large lanthanide [25], p-block metal [26], and alkaline earth [26, 27] ions with respect to their smaller analogues. The selectivity for the large lanthanide ions was found to be superior to related diaza[18]crown-6 [16, 19] and diaza[15]crown-5 ligands [28, 29]. This rare selectivity for large metal ions makes macropa a distinguished candidate for medical applications. Clinical tests with 225Ac are in progress with promising results: recently a single dose of 225Ac-RPS-074, an 225Ac(macropa) bifunctional construct designed for treatment of prostate cancer, resulted in a complete tumour ablation in mice [30].
The complex formation of macropa (H2L) with metals was investigated in numerous studies [24,25,26,27, 31, 32]. Information on the structure of the complexes could be gained in the crystal state from X-ray diffraction [24, 27] while in the solution from 1H and 13C NMR measurements [25]. These studies indicated the importance of two macropa conformers, the larger metal ions preferring a different macropa conformer than the smaller ones. These two conformers are the Δ(δλδ)(δλδ) and Δ(λδλ)(λδλ) ones, where Δ defines the absolute configuration of the picoline pendant arms, whereas δ and λ the absolute configurations of the six five-membered chelate rings formed in the complex [33, 34]. (It should be noted that altogether 16 conformations, i.e. eight enantiomeric pairs of diastereoisomers, may be possible for macropa complexes. However, the other conformers were generally found higher in energy by calculations [25, 31, 32, 35]; therefore, they are omitted in the present study.) In both conformers, macropa binds with ten heteroatom (O, N) donors to the metals. In addition, for the large La and Ba ions, the coordination of a small solvent molecule (H2O to the La [24] and DMF to the Ba complex [27]) was observed in the crystal. A coordination at the 11th site was not reported in the NMR studies.
Quantum chemical calculations were used to uncover the bonding properties and various systematic trends in lanthanide (Ln) and actinide (An) macropa complexes [25, 31, 32]. In these cases, the gas-phase structure was modelled by the M(L)+ ion, while the effect of solvent was taken into account by using periodic boundary conditions with appropriate variants of the polarized continuum model (PCM). For the sake of consistency, an additional coordination at the 11th site (which may be relevant only for the larger metal ions, and not confirmed in the solution anyway) was neglected in these studies.
In the present work, we study the effect of an H2O ligand on the structural and bonding properties of macropa complexes with the f elements La, Lu, and Ac. They are characteristic representatives of macropa complexes with different properties: La(macropa) was shown to have an H2O ligand at the 11th coordination site in the crystal; Lu is bonded to a different macropa conformer as La and (explained by the small radius) no additional ligand was observed in the crystal; and Ac from the 5f row has the largest 3+ ionic radius in the periodic system and is of paramount importance for TAT applications. The Ac(L)+ complex was shown to have similar structural and energetic properties to those of La(L)+ [32].
Computational details
Previous theoretical studies on Ln and An macropa complexes used density functional theory (DFT) calculations, in particular the TPSSh meta-hybrid exchange-correlation functional [36] in conjunction with 4f/5f-in-core quasirelativistic pseudopotentials (LCPP) and valence double-zeta basis sets [37,38,39]. This level of theory provided earlier a good description of the geometry and energetic properties of related complexes [31, 40, 41]. In the present study, we preferred 4f/5f-in-valence basis sets, which should give a more reliable description of the bonding interactions with La and Ac than LCPP, particularly the donation from the ligand to the valence f and d orbitals of the metal.
The first set of calculations were performed with the Amsterdam density functional package (ADF2018 [42, 43]). Due to its better SCF convergence properties, for the present study, the B3PW91 exchange-correlation functional was chosen [44, 45], which provided a performance comparable to TPSSh on Ln complexes [36]. The B3PW91 functional was taken from the LibXC library version 4.2.1 [46, 47] and included optimized D3 dispersion parameters by Grimme et al. [48]. The scalar relativistic effects were taken into account using the zero-order regular approximation (ZORA) [49]. The TZP all-electron basis consisted of uncontracted sets of Slater-type orbitals (STOs) optimized for use with ZORA [50]. No frozen-core approximation was applied. Further advantage of this basis set was its consistency for all the three metals (La, Lu, Ac). An auxiliary set of s, p, d, f, and g STOs was used to fit the molecular density and to represent the Coulomb and exchange potentials accurately in each SCF cycle. Due to the closed-shell character of the complexes, they were treated using the spin-restricted formalism. This theoretical level is denoted as B3PW91/ZORA.
The effect of spin-orbit (SO) interactions [51, 52] was probed on the Ac(H2L)H2O3+ and Lu(L)H2O+ complexes. The effects were quite small for the Ac complexes (the largest being a 0.003 Å decrease of the weak Ac–O5 bond and 6 kJ/mol increase of the formation energy) while negligible for the Lu complexes. Therefore, we decided to neglect the SO effects throughout the present study. The energy decomposition [53, 54] and quantum theory of atoms in molecules (QTAIM [55,56,57]) analyses were performed also with ADF2018 [42].
The consistency of the results was probed with pseudopotential (PP) basis sets using the Gaussian 09 code [58]. Utilizing the advantage of analytical second derivatives and the larger choice of solvent models, the thermodynamic contributions and solvent effects were evaluated with this code. The B3PW91 exchange-correlation functional was coupled with small-core pseudopotentials (SCPP) of the Stuttgart-Cologne group [59] for La and Lu in conjunction with triple-zeta 4f-in-valence basis sets [60] and the 6-311G(d,p) basis sets for the light ligand atoms. Unfortunately, calculations on the Ac complexes with the related 5f-in-valence pseudopotential [61,62,63] failed because of severe SCF convergence problems. Therefore, for Ac, the 5f-in-core pseudopotential (LCPP) was applied with a [7s6p5d2f1g]/[6s5p4d2f1g] valence basis set [39]. The dispersion interactions were taken into account by using the empirical D3 parameters of Grimme et al. [48]. This theoretical level (denoted as B3PW91/PP) resulted in deviations from the B3PW91/ZORA geometries generally below 0.01 Å in the bond distances of the M(macropa) moieties, while a few hundredths of Å in the weak M–OH2 bonds. The largest deviations were observed for the Ac complexes where the 5f-in-core approximation had to be used.
The minimum characters of the optimized structures were confirmed by frequency calculations. The translational, rotational, and vibrational contributions to the absolute energies at standard conditions (25 °C, 1 atm) were obtained by means of statistical thermodynamics. The neglect of electronic contributions is expected to cause negligible errors in the formation energies due to the similar electronic structure of the complex species involved in the formation reactions.
The solvent effects in water were estimated by single-point calculations on the optimized geometries using the polarizable continuum model (PCM) [64, 65] with radii and non-electrostatic terms from Truhlar and co-workers’ SMD solvation model [66]. For the La3+, Lu3+, and Ac3+ ions, the PCM radii 1.874, 1.659 [31], and 1.933 Å [32], respectively, were used without scaling (α = 1.0). This approach has been successfully applied for the interpretation of relative stabilities of Ln(macropa) complexes in aqueous solution [31].
Selection of model structures
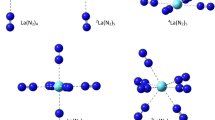
The computed model structures of the complexes are shown in Fig. 1. The M(H2L)H2O3+ model (Fig. 1a) is designed on the basis of the crystal structure of La(macropa), which has the composition La(HL)H2O · (ClO4)2 [24]. The main characteristic features in the crystal are the 11-fold coordination of La (the usual 10-fold one to macropa plus to H2O) and the protonated carboxylate group on one of the picolinate arms of the macropa ligand (HL−). These features are missing from the crystal structure of the Lu(macropa) complex, Lu(L)(ClO4) [24].

Model structures (a) M(H2L)H2O3+ and (b) M(L)H2O+
The hydrogen of the COOH group in the crystal of La(HL)H2O · (ClO4)2 forms a strong intermolecular hydrogen bond (HB) with the C=O moiety of the COO− group in a neighbouring complex. This intermolecular HB is very strong, as evidenced by the short O…O distance of 2.4 Å in the hydrogen bridge, by the lengthened HB acceptor C=O bond (1.270 Å) compared to the related C=O bond in the Lu complex with no HB (1.212/1.240 Å) and by the close HB acceptor C=O and HB donor C–OH distances of 1.270 and 1.294 Å, respectively. The strong HB smoothens the differences between the donor COOH and acceptor COO− groups in La(HL)H2O · (ClO4)2 and inverts the bond orders within the COO− group: the considerably lengthened HB acceptor C=O bond (1.270 Å) has rather a single-bond character, while the short C–O− bond coordinating to La (1.227 Å) rather a double-bond one. The above-described close parameters of the COOH and COO− groups are complemented by the very close O-La distances of the C=O…La and C–O−…La coordinative interactions (2.552 and 2.527 Å, respectively).
The above shown geometrical characteristics already imply that the La(HL)H2O2+ unit from the crystal structure containing one COOH, and one COO− group would not be a suitable model for the metal coordination in the crystal. Indeed, test calculations on this La(HL)H2O2+ structure resulted in a difference of 0.37 Å between the two types of O-La distances, deviating considerably from the 0.03 Å difference found in the crystal (vide supra). In contrast, a reasonable structure model for the solid-state La complex can be the La(H2L)H2O3+ ion, with both carboxylate groups protonated. This structure has C2 symmetry. The optimized geometry reproduced well the geometrical parameters from the crystal structure. Exceptions were the M–O2 and M–O5 bonds (overestimated by ca. 0.1 Å), which are the most affected by the H2L approximation.
The quality of the La(H2L)H2O3+ model for the description of bonding in the complex can be assessed by comparing the La–O2 and La–O5 bonds. The computed B3PW91/ZORA La-O5 distance was shorter by ca. 0.01 Å than the La-O2 one, while in the crystal, it is longer by ca. 0.02 Å. Hence, in the chosen structure model, the H2O coordination is slightly overestimated with respect to the coordination of the macropa ligand. Test calculations at other DFT levels (changing the functional and/or the basis set) retained the computed trend. We note that in the deviations between computations and experiment, the neglect of crystal packing effects may play also some role. Moreover, in the other possible model structures, La(HL)H2O2+ and La(H2L)H2O2+, the errors of the La-O5 distances were much larger. Nevertheless, we can assume a systematic error of the M(H2L)H2O3+ model for the three f metals, La, Lu, and Ac. Therefore, it can be suitable for a qualitative comparison of the nature of H2O coordination in their complexes with macropa.
Scarce experimental information is available on the structures present in solution. A single NMR study indicated that macropa complexes of 4f elements prefer different structures depending on the size of the Ln3+ ions: La3+ was shown to coordinate with the Δ(δλδ)(δλδ) while the small Yb3+ with the Δ(λδλ)(λδλ) conformation of macropa [25]. On the basis of the close ionic radii and DFT calculations, the latter conformation was accepted for Lu3+ too [25, 31, 32]. The formation of inter-macropa HB is not probable in diluted aqueous solution, and depending on the pH, the protonation of the COO− groups is challenged by solute-solvent interactions. The pKa of picolinic acid was reported to be 5.40 [67], which means that in solution at pH = 7, the amount of picolinic acid is ca. 1% of that of the picolinate anion. On this basis, under the mild physiological conditions, only a marginal hydrolysis of the macropa L2− form can be expected. Accordingly, in previous DFT studies, the M(L)+ ions were used in modelling the relative stabilities measured in solution [25, 31, 32]. The selection of that structure is supported by the fact that coordination of H2O to the 11th site of Ln was not reported from the solution study [25].
Thus, our second model structure is the M(L)H2O+ ion (Fig. 1b), by which we could compare the feasibilities and consequences of H2O addition to the core M(L)+ complexes. In order to provide information for solution conditions too, the energetic stabilization of M(L)H2O+ species by solvation was estimated using single-point PCM/SMD calculations.
Structural characteristics
The H2O hydrogens can form intramolecular HBs with the heteroatoms of macropa. Based on the spatial conditions, in our model structures, this can happen with O3 and O4 atoms of macropa. In the search for global minima, we find two such HB schemes: HB with the opposite lying O3 and O3´ atoms (see Fig. 1; most such computed structures have C2 symmetry), and HB with the O3 and O4 atoms, positioned at the same side of the macropa crown. The former structures were characteristic on the Ac, La, and the Lu Δ(δλδ)(δλδ) complexes, while the latter asymmetric structure proved to be the global minimum of the Lu complexes with Δ(λδλ)(λδλ) conformation. In the former cases, the isomer with HB to O3 and O4 was obtained either at slightly higher energy or converged to the isomer with HB to O3 and O3´. The marginal energy differences between the HB isomers indicated that the orientation of the H2O molecule is of minor importance for the stability of the complexes.
Selected geometrical data from B3PW91/ZORA calculations are shown in Table 1 and Figs. 2, 3, and 5. The values presented in the figures as well as the Cartesian coordinates of the optimized structures are given in the Supplementary Material.

M–O2 bond lengths (Å) in the studied complexes

Variation of the O3…O3´ distances (Å) in the studied complexes
The geometrical parameter most characteristic on H2O coordination is the M–O5 bond distance (Table 1). They are shorter by 0.1–0.2 Å in the M(H2L)H2O3+ model structures as compared to the M(L)H2O+ ones, indicating a considerably stronger M…OH2 interaction in the protonated structures. The protonation of the COO− groups promoted this stronger interaction because it resulted in a more favourable position of M in the macropa cage. In terms of the Lewis model, the anionic (–O2/2′−) oxygens in the M(L)H2O+ structures change to neutral oxo (=O2/2′) groups in the M(H2L)H2O3+ complexes. Consequently, the latter =O2/2′…M interactions become weaker (the M-O2 distances increase by ca. 0.2 Å upon protonation, cf. Figure 2), and M is shifted towards the open site (crown) of the macropa ligand, where the H2O ligand coordinates. The weaker =O2/2′…M interactions (meaning less donation to M) result in a somewhat larger positive charge of M in the protonated complexes (vide infra in Table 2); thus, it becomes both electronically and sterically more suitable for an additional 11th coordination.
Parameter d in Table 1 characterizes the distance of M from the open site of L. It corresponds to the distance of M from the middle point of the O3…O3´ line. Because of the largest ion radius and the consequently largest Ac-O2/2′ distances, Ac is the closest to the open site of the ligand. The distance of La from the open site is slightly larger, and Lu is deeper inside of the L cage (i.e. closer to the picolinate arms) by 0.15–0.2 Å.
The following information on the steric conditions can be gained from the presented computed data:
-
(i)
While the M–O2 bond distances are generally close in the two conformers of the M(H2L)3+ and M(L)+ complexes, the d distances are considerably different (Table 1). In the Δ(δλδ)(δλδ) conformers, the metals are closer to the open site generally by 0.2–0.3 Å than in the Δ(λδλ)(λδλ) ones.
-
(ii)
The open site of the Δ(λδλ)(λδλ) conformers of the M(H2L)3+ and M(L)+ parents is considerably narrower than that of the Δ(δλδ)(δλδ) conformers (see the O3…O3´ distances in Fig. 3 as well as the structures in Fig. 4), making an attack of the H2O ligand sterically more difficult to the Δ(λδλ)(λδλ) conformer. In these conformers, the coordination of H2O increases the O3…O3´ distances considerably (Fig. 3). The distortion results also in a substantial opening of the N1-M-N1´ angles (up to ca. 40 degrees, cf. Fig. 5). These observed characteristics indicate that the Δ(δλδ)(δλδ) conformers are sterically more suitable for an additional (11th) coordination.
-
(iii)
The M-O5 distances are nearly the same in the Δ(δλδ)(δλδ) and Δ(λδλ)(λδλ) structures. This suggests that the strength of the M…OH2 interaction is close in the two conformers.
-
(iv)
Both the M–O2/2′ and M–O5 bonds follow the trend in the M3+ ionic radii.

Perspective view of the (a) Δ(δλδ)(δλδ) and b) Δ(λδλ)(λδλ) conformers of Lu(L)+ from the direction of the crown, demonstrating the stretched-narrower character of this site of the Δ(λδλ)(λδλ) conformer

Variation of the N1-M-N1´ angle (deg) in the studied complexes
Bonding
Selected data from the QTAIM and energy decomposition analyses are compiled in Table 2. The Bader charges show only small differences in the M and O charges. The smaller O2 charges in the protonated complexes are in good agreement with the above-described change in the character of these oxygens upon protonation of the COO− groups. On the other hand, the larger positive M charges in the latter complexes are in accordance with the smaller charge transfer from H2L due to the considerably larger M-O2 distances. The charge transfer from the 10 donor atoms of the macropa ligand amounts to ca. 0.7 e, which makes this ligand the dominant electron donor to M. In contrast, the donation by the single O atom of the H2O ligand is very weak in the M(H2L)H2O3+ structures, while negligibly small in the M(L)H2O+ ones. The donation from the macropa ligand increases slightly from Ac to Lu. Accordingly, the positive charge of the metals decreases in this direction.
The delocalization indices express the number of electrons forming the covalent (donor-acceptor) bonding between M and the donor atoms of the ligands. The values related to the most interesting donors, O2/2′ from macropa and O5 from H2O, are listed in Table 2. The most important observation is that the two donor-acceptor interactions are comparable in the M(H2L)H2O3+ structures, while in the M(L)H2O+ complexes, the M…O2/2′ interaction is dominant. These features correlate well with the related M-O distances (cf. Table 1). Another characteristic feature is that while both the M…O2/2′ and M…O5 interactions increase from Ac to Lu, the increase of the M…O5 interaction becomes quite significant in the Lu complexes.
The above-described characteristics are reflected also by the energy decomposition data (Table 2). Here, the interaction energy between the selected fragments, ΔEint, is defined as
where ΔVelst corresponds to the classical electrostatic interaction between the charge distributions of the isolated fragments after brought together in the complex, ΔEPauli is the repulsion between occupied orbitals (practically the steric repulsion), and ΔEoi is the orbital interaction energy between the fragments in the complex, accounting for electron pair bonding, charge transfer, and polarization [54]. In the present analysis, we focus on the total interaction energy as well as on the ratio of the ΔVelst and ΔEoi components upon H2O coordination in the two model structures.
The data were evaluated using three different fragmentation models. The model with the three fragments M3+, H2L, or L2− and H2O provided the total interaction energy between M and the two ligands (ΔETotal). In this model, the electrostatic interactions are somewhat overestimated, because in the complexes, the charge of M is lower than +3 (between +2 and +3 e, cf. Table 2), and the charges of the macropa ligands are somewhat reduced due to the charge donation to M. The second model with the M(macropa) and neutral H2O fragments facilitates the assessment of the energetics of H2O coordination to the parent M(macropa) complexes. Because the charge donation from H2O is marginal (cf. Table 2), this model with neutral H2O gives more reliable information on the electrostatic contributions than the above three-fragment model. The third fragmentation model covers the M(L)+ and M(H2L)3+ parents, consisting of the M3+ metal and H2L or L2− macropa fragments. It is used for the assessment of the M…macropa interaction in the absence of the H2O ligand.
In the data compiled in Table 2, the following trends could be observed (additional data are given in the Supplementary Material):
-
(i)
In agreement with the charged character of the L2− ligand, in the M(L)H2O+ complexes, the electrostatic contribution of the M…macropa bonding is very large compared to the M(H2L)H2O3+ complexes with the formally neutral H2L ligand. The ratio of the electrostatic and covalent contributions in M(L)H2O+ was obtained to be around 67% compared to ca. 52% in M(H2L)H2O3+. In fact, this feature originates from the parent M(macropa) complexes. The considerably larger electrostatic contribution is the main source of the twice larger ΔETotal in the M(L)H2O+ complexes.
-
(ii)
The interaction energies (also the contributions, given in the Supplementary Material) increase gradually from Ac to Lu. The trend follows qualitatively the change in the ionic radii (like the M-O2 distances), i.e. the large increase from La to Lu is in good agreement with the larger decrease of the ionic radius between the latter ions.
-
(iii)
In general, the ΔETotal interaction energies in Table 2 are somewhat larger in the Δ(λδλ)(λδλ) conformers. This larger interaction energy can compensate for the less favourable steric conditions (vide supra). We note that ΔEint in Eq. (1) means the interaction energy between the fragments with frozen geometry from the complex; therefore, it should not be mistaken with the formation energies from relaxed reactants. The frozen macropa structures suffer from substantial strain energy.
-
(iv)
The ratio of the electrostatic and covalent contributions differs only marginally between the Δ(δλδ)(δλδ) and Δ(λδλ)(λδλ) conformers.
-
(v)
The interaction energies from the three-fragment analysis (ΔETotal) are larger by one order of magnitude than the M(macropa)…H2O interaction energies (ΔEM-H2O). This large difference is readily explained by the 10-fold coordination of macropa to M compared with the single-fold one of H2O.
-
(vi)
In terms of the interaction energies, the H2O coordination is considerably weaker in the M(L)H2O+ complexes than in the M(H2L)H2O3+ ones (cf. Figure 6a). This is in good agreement with the structural characteristics due to the stronger capture of the metal by the anionic L2− ligand and the resulting considerably longer M-O5 distances in these complexes (vide supra).
-
(vii)
The major contribution in the H2O-coordination is the electrostatic interaction (60–70%, Fig. 6b). The covalent contribution is somewhat stronger in the Lu complexes than in the Ac and La ones.

Energy decomposition results on the M(macropa)…H2O interaction: (a) interaction energy; (b) ratio of electrostatic and covalent contributions to the bonding
Thermodynamic stability
The scarce experimental [24, 25] and the previous theoretical studies on M(L)+ complexes [25, 31, 32] indicated the energetic preference of the Δ(δλδ)(δλδ) conformer for larger M (Ac, La), whereas the preference of Δ(λδλ)(λδλ) for M = Lu. The energetic advantage of Δ(δλδ)(δλδ) decreased with the ionic size (Ac > La). This energetic order is retained upon protonation of the COO− groups in the M(H2L)3+ structures in terms of the currently calculated electronic energies (see Supplementary Material). However, assessment of our calculated data shows that the protonation is somewhat more favourable for the Δ(λδλ)(λδλ) conformers, lowering their energies with respect to the Δ(δλδ)(δλδ) ones. Because the thermal contributions are also more favourable for the Δ(λδλ)(λδλ) conformers, already for La(H2L)3+, the Δ(λδλ)(λδλ) conformer became the more stable one in terms of Gibbs free energies.
The energy consequences of H2O coordination obtained at the B3PW91/PP level are given in Table 3. Gibbs free energies including the thermal and solvation effects are also included. The results in terms of absolute energies from the B3PW91/ZORA calculations are given in the Supplementary Material. The two theoretical levels agree in the trends in the relative and complex formation energies, while the ΔEf values are underestimated by the B3PW91/ZORA level with respect to B3PW91/PP. From the data, the following main conclusions can be drawn:
-
(i)
At the isolated molecule models the coordination of H2O proved to be quite exotherm, exceptions are the M(L)H2O+ Δ(λδλ)(λδλ) conformers with M = La and Lu (cf. ΔGf in Table 3).
-
(ii)
The coordination of H2O is promoted by protonation of the COO− groups, the ΔGf values being larger by ca. 20% in the M(H2L)H2O3+ complexes than in the respective M(L)H2O+ ones. This computed result agrees with the above outlined structural and bonding characteristics as well as with the experimental information on the crystalline complexes: coordinated H2O is found only in complexes with singly protonated HL− ligand [24, 27].
-
(iii)
The formation energies support the conclusion based on the above steric analysis: the Δ(δλδ)(δλδ) conformers are more suitable for coordinating a H2O molecule at the 11th coordination site of M than the Δ(λδλ)(λδλ) conformers.
-
(iv)
As expected on the basis of the larger ionic size, H2O-coordination is generally the most feasible by the largest Ac3+ ion and the least by the smallest Lu3+.
-
(v)
Polar solvent has been shown to stabilize the Δ(λδλ)(λδλ) conformer [32], which character was transferred also to the present H2O-coordinated complexes (cf. Table 3). The PCM/SMD solvation model calculations predicted a decreased propensity of H2O coordination in aqueous media. The very small solvation-corrected ΔGf,solv energies support the experimental information on the lack of H2O coordination in aqueous solution [25].
Altogether, the above results support the feasibility of H2O coordination at the 11th coordination site of Ac in both the Δ(δλδ)(δλδ) and Δ(λδλ)(λδλ) conformers in the isolated and solid states, similarly to the observed case of the Δ(δλδ)(δλδ) form of the crystalline La derivative (La(HL)H2O · (ClO4)2 [24]). In contrast, H2O coordination in polar solvents is not probable.
Conclusions
The present theoretical work was aimed to uncover the structural and bonding conditions of an additional ligand (H2O) at the 11th coordination site in macropa complexes of f elements. The experimental hint for the possibility of such a coordination was provided by the crystal structure of the La(macropa) complex (La(HL)H2O · (ClO4)2 [24]). In contrast, in solid state, the related Lu complex is only 10-fold coordinated, having no extra H2O ligand (Lu(L)(ClO4) [24]). These observations raise interesting questions about the competition of ligands, the effect of the metal size, the conformation, and the character of bonding in such donor-acceptor complexes.
The first step was to find the right model structures for crystal and solution phases. On the basis of the available experimental information, we chose the M(H2L)H2O3+ and M(L)H2O+ models, respectively. We investigated the two important macropa conformers, Δ(δλδ)(δλδ) and Δ(λδλ)(λδλ), known to be favoured by larger (Ac3+, La3+) and smaller (Lu3+) ions, respectively. Including the M(H2L)3+ and M(L)+ parents, altogether 24 molecules were included in the study. The applied theoretical level is hitherto the highest one on macropa complexes with heavy elements.
Protonation of the picoline arms enhanced the coordination by moving the metal ion closer to the open site of the ligand. The choice of macropa conformer had no large influence on the M-O5 distance and bonding properties of the H2O coordination. On the other hand, the Δ(δλδ)(δλδ) conformers are sterically more suitable for H2O coordination due to the wider slot of the macropa crown. This explains the significantly smaller formation energies of the Δ(λδλ)(λδλ) complexes.
The interaction energy between the H2O ligand and the parent M(macropa) complex was found to be smaller by an order of magnitude than that between M and macropa. The major bonding contribution of the M…H2O interaction is the electrostatic one (ca. 65%). The coordination of H2O proved to be significantly exothermic in the solid-state model, being the most favourable to the largest Ac3+ ion, while only slightly less to La3+ and considerably less to Lu3+. In contrast, water solvent reduced considerably the energy gain of H2O coordination at the 11th coordination site. According to our model calculations, the coordination in solution is less likely, in good agreement with the available experimental information.
Data availability
Additional data on the optimized geometries (tables with geometrical parameters and list of Cartesian coordinates), bonding properties and energy data from B3PW91/PP and B3PW91/ZORA calculations are available as Electronic Supplementary Material.
References
Williams JE (1999) Donner laboratory: the birthplace of nuclear medicine. J Nucl Med 40:16N–20N
Stewart AJ, Cormack RA, Held KD (2015) Radiobiologic concepts for brachytherapy. In: Devlin P, Cormack RA, Holloway CL, Stewart AJ (eds) Brachytherapy. Applications and techniques. Springer Publishing Company, New York, pp 37–52. https://doi.org/10.1891/9781617052613.0002
Geerlings MW, Kaspersen FM, Apostolidis C, Van Der Hout R (1993) The feasibility of 225Ac as a source of α-particles in radioimmunotherapy. Nucl Med Commun 14(2):121–125. https://doi.org/10.1097/00006231-199302000-00009
Miederer M, Scheinberg DA, McDevitt MR (2008) Realizing the potential of the Actinium-225 radionuclide generator in targeted alpha particle therapy applications. Adv Drug Deliv Rev 60(12):1371–1382. https://doi.org/10.1016/j.addr.2008.04.009
Thiele NA, Wilson JJ (2018) Actinium-225 for targeted α therapy: coordination chemistry and current chelation approaches. Cancer Biother Radiopharm 33(8):336–348. https://doi.org/10.1089/cbr.2018.2494
Morgenstern A, Apostolidis C, Kratochwil C, Sathekge M, Krolicki L, Bruchertseifer F (2018) An overview of targeted alpha therapy with 225Actinium and 213Bismuth. Curr Radiopharm 11(3):200–208. https://doi.org/10.2174/1874471011666180502104524
Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F, Kopka K, Apostolidis C, Haberkorn U, Morgenstern A (2016) 225Ac-PSMA-617 for PSMA-targeted α-radiation therapy of metastatic castration-resistant prostate cancer. J Nucl Med 57(12):1941–1944. https://doi.org/10.2967/jnumed.116.178673
Kratochwil C, Bruchertseifer F, Rathke H, Bronzel M, Apostolidis C, Weichert W, Haberkorn U, Giesel FL, Morgenstern A (2017) Targeted α-therapy of metastatic castration-resistant prostate cancer with 225Ac-PSMA-617: dosimetry estimate and empiric dose finding. J Nucl Med 58(10):1624–1631. https://doi.org/10.2967/jnumed.117.191395
Hammer S, Hagemann UB, Zitzmann-Kolbe S, Larsen A, Ellingsen C, Geraudie S, Grant D, Indrevoll B, Smeets R, von Ahsen O, Kristian A, Lejeune P, Hennekes H, Karlsson J, Bjerke RM, Ryan OB, Cuthbertson AS, Mumberg D (2020) Preclinical efficacy of a PSMA-targeted thorium-227 conjugate (PSMA-TTC), a targeted alpha therapy for prostate cancer. Clin Cancer Res 26(8):1985–1996. https://doi.org/10.1158/1078-0432.CCR-19-2268
Jurcic JG, Rosenblat TL, McDevitt MR, Pandit-Taskar N, Carrasquillo JA, Chanel SM, Zikaras K, Frattini MG, Maslak PM, Cicic D, Larson SM, Scheinberg DA (2013) Targeted alpha-particle nano-generator actinium-225 (225Ac)-lintuzumab (anti-CD33) in acute myeloid leukemia (AML). Clin Lymphoma, Myeloma Leuk 13:S379–S380
Jurcic JG, Ravandi F, Pagel JM, Park JH, Douglas Smith B, Douer D, Yair Levy M, Estey E, Kantarjian HM, Earle D, Cicic D, Scheinberg DA (2015) Phase I trial of α-particle therapy with actinium-225 (225Ac)-lintuzumab (anti-CD33) and low-dose cytarabine (LDAC) in older patients with untreated acute myeloid leukemia (AML.). J Clin Oncol 33:7050–7050. https://doi.org/10.1200/jco.2015.33.15_suppl.7050
Kratochwil C, Bruchertseifer F, Giesel F, Apostolidis C, Haberkorn U, Morgenstern A (2015) Ac-225-DOTATOC - an empiric dose finding for alpha particle emitter based radionuclide therapy of neuroendocrine tumors. J Nucl Med 56(Suppl. 3):1232
Cacheris WP, Nickle SK, Sherry AD (1987) Thermodynamic study of lanthanide complexes of 1,4,7-triazacyclononane-N,N,N-triacetic acid and 1,4,7,10-tetraazacyclododecane-N,N,N,N-tetraacetic acid. Inorg Chem 26(6):958–960. https://doi.org/10.1021/ic00253a038
Clarke ET, Martell AE (1991) Stabilities of trivalent metal ion complexes of the tetraacetate derivatives of 12-, 13- and 14-membered tetraazamacrocycles. Inorg Chim Acta 190(1):37–46. https://doi.org/10.1016/S0020-1693(00)80229-7
Morgenstern A, Apostolidis C, Bruchertseifer F (2020) Supply and clinical application of actinium-225 and bismuth-213. Semin Nucl Med 50(2):119–123. https://doi.org/10.1053/j.semnuclmed.2020.02.003
Chang CA, Rowland ME (1983) Metal complex formation with 1,10-diaza-4,7,13,16-tetraoxacyclooctadecane-N,N'-diacetic acid. An approach to potential lanthanide ion selective reagents. Inorg Chem 22(26):3866–3869. https://doi.org/10.1021/ic00168a010
Damu KV, Shaikjee MS, Michael JP, Howard AS, Hancock RD (1986) Control of metal ion selectivity in ligands containing neutral oxygen and pyridyl groups. Inorg Chem 25(22):3879–3883. https://doi.org/10.1021/ic00242a010
Hancock RD, Bhavan R, Wade PW, Boeyens JCA, Dobson SM (1989) Ligand design for complexation in aqueous solution. 1. Neutral oxygen donor bearing groups as a means of controlling size-based selectivity for metal ions. Inorg Chem 28(2):187–194. https://doi.org/10.1021/ic00301a007
Brücher E, Györi B, Emri J, Solymosi P, Sztanyik LB, Varga L (1993) 1,10-Diaza-4,7,13,16-tetraoxacyclooctadecane-1,10-bis(malonate), a ligand with high Sr2+/Ca2+ and Pb2+/Zn2+ selectivities in aqueous solution. J Chem Soc Chem Commun 6:574–575. https://doi.org/10.1039/C39930000574
Zhang XX, Bordunov AV, Bradshaw JS, Dalley NK, Kou X, Izatt RM (1995) A new highly selective macrocycle for K+ and Ba2+: effect of formation of a pseudo second macroring through complexation. J Am Chem Soc 117(46):11507–11511. https://doi.org/10.1021/ja00151a014
Su N, Bradshaw JS, Zhang XX, Song H, Savage PB, Xue G, Krakowiak KE, Izatt RM (1999) Syntheses and metal ion complexation of novel 8-hydroxyquinoline-containing diaza-18-crown-6 ligands and analogues. J Org Chem 64(24):8855–8861. https://doi.org/10.1021/jo991081o
Jensen MP, Chiarizia R, Shkrob IA, Ulicki JS, Spindler BD, Murphy DJ, Hossain M, Roca-Sabio A, Platas-Iglesias C, de Blas A, Rodríguez-Blas T (2014) Aqueous complexes for efficient size-based separation of americium from curium. Inorg Chem 53(12):6003–6012. https://doi.org/10.1021/ic500244p
Regueiro-Figueroa M, Barriada JL, Pallier A, Esteban-Gómez D, Ad B, Rodríguez-Blas T, Tóth É, Platas-Iglesias C (2015) Stabilizing divalent europium in aqueous solution using size-discrimination and electrostatic effects. Inorg Chem 54(10):4940–4952. https://doi.org/10.1021/acs.inorgchem.5b00548
Thiele NA, Brown V, Kelly JM, Amor-Coarasa A, Jermilova U, MacMillan SN, Nikolopoulou A, Ponnala S, Ramogida CF, Robertson AKH, Rodríguez-Rodríguez C, Schaffer P, Williams Jr C, Babich JW, Radchenko V, Wilson JJ (2017) An eighteen-membered macrocyclic ligand for actinium-225 targeted alpha therapy. Angew Chem Int Ed 56(46):14712–14717. https://doi.org/10.1002/anie.201709532
Roca-Sabio A, Mato-Iglesias M, Esteban-Gómez D, Tóth É, de Blas A, Platas-Iglesias C, Rodríguez-Blas T (2009) Macrocyclic receptor exhibiting unprecedented selectivity for light lanthanides. J Am Chem Soc 131:3331–3341. https://doi.org/10.1021/ja808534w
Ferreirós-Martínez R, Esteban-Gómez D, Tóth E, de Blas A, Platas-Iglesias C, Rodríguez-Blas T (2011) Macrocyclic receptor showing extremely high Sr(II)/Ca(II) and Pb(II)/ Ca(II) selectivities with potential application in chelation treatment of metal intoxication. Inorg Chem 50(8):3772–3784. https://doi.org/10.1021/ic200182e
Thiele NA, MacMillan SN, Wilson JJ (2018) Rapid dissolution of BaSO4 by macropa, an 18-membered macrocycle with high affinity for Ba2+. J Am Chem Soc 140(49):17071–17078. https://doi.org/10.1021/jacs.8b08704
Roca-Sabio A, Mato-Iglesias M, Esteban-Gómez D, de Blas A, Rodríguez-Blas T, Platas-Iglesias C (2011) The effect of ring size variation on the structure and stability of lanthanide(III) complexes with crown ethers containing picolinate pendants. Dalton Trans 40(2):384–392. https://doi.org/10.1039/c0dt00746c
Chang CA, Ochaya VO (1986) Potential lanthanide ion selective reagents. 3. Metal complex formation with 1,7-diaza-4,10,13-trioxacyclopentadecane-N,N'-diacetic acid. Inorg Chem 25(3):355–358. https://doi.org/10.1021/ic00223a025
Kelly JM, Amor-Coarasa A, Ponnala S, Nikolopoulou A, Williams C, Thiele NA, Schyler D, Wilson JJ, DiMagno SG, Babich JW (2019) A single dose of 225Ac-RPS-074 induces a complete tumor response in a LNCaP xenograft model. J Nucl Med 60(5):649–655. https://doi.org/10.2967/jnumed.118.219592
Regueiro-Figueroa M, Esteban-Gómez D, de Blas A, Rodríguez-Blas T, Platas-Iglesias C (2014) Understanding stability trends along the lanthanide series. Chem Eur J 20:3974–3981. https://doi.org/10.1002/chem.201304469
Kovács A (2020) Theoretical study of actinide complexes with macropa. ACS Omega 5:26431–26440. https://doi.org/10.1021/acsomega.0c02873
Corey EJ, Bailar JC (1959) The stereochemistry of complex inorganic compounds. XXII Stereospecific Effects in Complex Ions. J Am Chem Soc 81(11):2620–2629. https://doi.org/10.1021/ja01520a006
Beattie JK (1971) Conformational analysis of tris(ethylenediamine) complexes. Acc Chem Res 4(7):253–259. https://doi.org/10.1021/ar50043a004
Thiele NA, Woods JJ, Wilson JJ (2019) Implementing f-block metal ions in medicine: tuning the size selectivity of expanded macrocycles. Inorg Chem 58(16):10483–10500. https://doi.org/10.1021/acs.inorgchem.9b01277
Tao J, Perdew JP, Staroverov VN, Scuseria GE (2003) Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids (TPSSTPSS). Phys Rev Lett 91(14):146401. https://doi.org/10.1103/PhysRevLett.91.146401
Dolg M, Stoll H, Savin A, Preuss H (1989) Energy-adjusted pseudopotentials for the rare earth elements. Theor Chim Acta 75:173–194. https://doi.org/10.1007/BF00528565
Dolg M, Stoll H, Preuss H (1993) A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor Chim Acta 85:441–450. https://doi.org/10.1007/BF01112983
Moritz A, Cao X, Dolg M (2007) Quasirelativistic energy-consistent 5f-in-core pseudopotentials for trivalent actinide elements. Theor Chem Accounts 117:473–481. https://doi.org/10.1007/s00214-006-0180-7
Roca-Sabio A, Regueiro-Figueroa M, Esteban-Gómez D, de Blas A, Rodríguez-Blas T, Platas-Iglesias C (2012) Density functional dependence of molecular geometries in lanthanide(III) complexes relevant to bioanalytical and biomedical applications. Comput Theor Chem 999:93–104. https://doi.org/10.1016/j.comptc.2012.08.020
Huang P-W (2019) Understanding the stability trend along light lanthanide complexes with an ehtylenediamine-type ligand: a quantum chemical study. ChemistrySelect 4(42):12368–12374. https://doi.org/10.1002/slct.201902887
Amsterdam Density Functional Package (ADF2018). SCM Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands
te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJA, Snijders JG, Ziegler T (2001) Chemistry with ADF. J Comput Chem 22(9):931–967. https://doi.org/10.1002/jcc.1056
Becke AD (1993) Density-functional thermochemistry. III The Role of Exact Exchange. J Chem Phys 98:5648–5652. https://doi.org/10.1063/1.464913
Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B 45:13244. https://doi.org/10.1103/PhysRevB.45.13244
Marques MAL, Oliveira MJT, Burnus T (2012) Libxc: a library of exchange and correlation functionals for density functional theory. Comput Phys Commun 183(10):2272–2281. https://doi.org/10.1016/j.cpc.2012.05.007
Lehtola S, Steigemann C, Oliveira MJT, Marques MAL (2018) Recent developments in LibXC – a comprehensive library of functionals for density functional theory. SoftwareX 7 1(2018) 7:1–5. https://doi.org/10.1016/j.softx.2017.11.002
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132:154104. https://doi.org/10.1063/1.3382344
van Lenthe E, Baerends EJ, Snijders JG (1994) Relativistic total energy using regular approximations. J Chem Phys 101(11):9783–9792. https://doi.org/10.1063/1.467943
van Lenthe E, Baerends EJ (2003) Optimized slater-type basis sets for the elements 1-118. J Comput Chem 24:1142–1156. https://doi.org/10.1002/jcc.10255
van Lenthe E, Snijders J, Baerends E (1996) The zero-order regular approximation for relativistic effects: the effect of spin–orbit coupling in closed shell molecules. J Chem Phys 105:6505–6516. https://doi.org/10.1063/1.472460
van Lenthe E, van Leeuwen R, Baerends EJ, Snijders JG (1996) Relativistic regular two-component Hamiltonians. Int J Quantum Chem 57:281–293. https://doi.org/10.1002/(SICI)1097-461X(1996)57:3<281::AID-QUA2>3.0.CO;2-U
Ziegler T, Rauk A (1977) On the calculation of bonding energies by the Hartree Fock slater method. I. The transition state method. Theor Chim Acta 46:1. https://doi.org/10.1007/BF02401406
Bickelhaupt FM, Baerends EJ (2000) Kohn-sham density functional theory: predicting and understanding chemistry. In: Lipkowitz KB, Boyd DB (eds) Rev. Comput. Chem, vol 15. Wiley-VCH, New York, pp 1–86
Bader RFW (1990) Atoms in molecules. Oxford University Press, Oxford, A Quantum Theory
Rodríguez JJ, Köster AM, Ayers PW, Santos-Valle A, Vela A, Merino G (2009) An efficient grid-based scheme to compute QTAIM atomic properties without explicit calculation of zero-flux surfaces. J Comput Chem 30:1082–1092. https://doi.org/10.1002/jcc.21134
Rodríguez JJ, Bader RFW, Ayers PW, Michel C, Götz AW, Bo C (2009) A high performance grid-based algorithm for computing QTAIM properties. Chem Phys Lett 472:149–152. https://doi.org/10.1016/j.cplett.2009.02.081
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, revision D.01. Gaussian, Inc., Wallingford CT
Cao X, Dolg M (2001) Valence basis sets for relativistic energy-consistent small-core lanthanide pseudopotentials. J Chem Phys 115:7348–7355. https://doi.org/10.1063/1.1406535
Cao X, Dolg M (2002) Segmented contraction scheme for small-core lanthanide pseudopotential basis sets. J Mol Struct (THEOCHEM) 581:139–147. https://doi.org/10.1016/S0166-1280(01)00751-5
Küchle W, Dolg M, Stoll H, Preuss H (1994) Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J Chem Phys 100(10):7535–7542. https://doi.org/10.1063/1.466847
Cao X, Dolg M, Stoll H (2003) Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. J Chem Phys 118:487–496. https://doi.org/10.1063/1.1521431
Cao X, Dolg M (2004) Segmented contraction scheme for small-core actinide pseudopotential basis sets. J Mol Struct (THEOCHEM) 673(1–3):203–209. https://doi.org/10.1016/j.theochem.2003.12.015
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105:2999–3093. https://doi.org/10.1021/cr9904009
Scalmani G, Frisch MJ (2010) Continuous surface charge polarizable continuum models of solvation. I General formalism. J Chem Phys 132:114110. https://doi.org/10.1063/1.3359469
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113(18):6378–6396. https://doi.org/10.1021/jp810292n
Jellinek HHG, Urwin JR (1954) Ultraviolet absorption spectra and dissociation constants of picolinic, isonicotinic acids and their amides. J Phys Chem 58(7):548–550. https://doi.org/10.1021/j150517a009
Acknowledgements
The authors thank Dr. A. Morgenstern for advice.
Funding
The calculations have been carried out using resources provided by the affiliations of the authors (Minnesota Supercomputer Institute and JRC Karlsruhe site).
Author information
Authors and Affiliations
Contributions
Both authors contributed to the study conception and design. The B3PW91/ZORA calculations were performed by Z. Varga, the B3PW91/PP ones by A. Kovács. The data analysis and the first draft of the manuscript were made by A. Kovács and both authors commented on subsequent versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics declarations
The ethical standards have been met.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 112 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kovács, A., Varga, Z. H2O coordination in macropa complexes of f elements (Ac, La, Lu): feasibility of the 11th coordination site. Struct Chem 32, 643–653 (2021). https://doi.org/10.1007/s11224-020-01717-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01717-3