
Abstract
The susceptibility of electric and energetic properties of two sets of molecules to perturbation in a uniform electric field was investigated. The molecules of one set were deca-1,3,5,7-pentaene terminated with two functional groups, R1 and R2; those of the second set were 4-R1-4′-R2-p-diphenylbenzenes, with R1 and R2 being the same as in the polyene set. The polyene and aromatic molecules had similar lengths of the system of conjugated bonds between the two most extreme carbon atoms. Dipole moments were used as determinants of the overall charge transfer within the molecules. The electric field was directed along the main (longest) axes of the molecules from the negative pole to the positive. Comparison of the effect of the charge relocation in both sets of conjugated molecules revealed that the charge transfer imposed by the electric field was more efficient in the polyenes than in the aromatic compounds; however, for the molecule pairs with the same R1 and R2, reversal of the dipole moment direction falls at the same field strength. Parabolic dependence of the molecules’ energy as a function of field strength can also be interpreted in terms of the response of electron density to an electric field.
Similar content being viewed by others
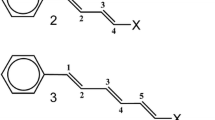

Avoid common mistakes on your manuscript.
Introduction
There are a number of papers dealing with the impact of a homogeneous electric field on the structural and electronic properties of molecules. A series of papers was devoted to atomic polarizabilities, that is polarization of the atomic charge distribution in molecules [1,2,3,4,5]. Among other effects, one can mention the tuning of aromaticity of para substituted benzene derivatives [6] and polycalicenes [7] imposed by the field. The most interesting result for us in the first paper was finding the dipole moment direction reversal in p-aminophenol, p-nitrobenzonitrile, and p-nitrophenol at a sufficiently high field strength and at its proper direction toward the main molecular axes.
There are also reports about the effects of electric fields (both in the inorganic and biological milieu) on chemical reactions, as the field can significantly alter the potential energy curves near the transition state region governing chemical reactions [8] and consequently influence course and rate constants of the reactions [9,10,11,12]. Most effects can be comprehended as the field-induced stabilization of ionic structures [16]. Electric fields also impact on the biological properties of cells and their components [12,13,14,15]. Therefore, application of a properly oriented electric field may be considered as a particular sort of catalysis [8,9,10]. These effects of the electric field were investigated both theoretically and experimentally. Comprehensive and informative reviews can be found in Ref. [16,17,18].
How large are the electric field strengths to cause the measurable catalytic effects? A study of the field effect to model reaction kinetics in the formic acid dimer [19] showed that a field strength of 3 × 109 V/m (0.3 V/Å) is found to be sufficient to double the proton transfer in the molecule [19]. The fields sufficiently strong to determine the selectivity between two competing reactions (hydroxylation vs epoxidation) are approximately 0.0125 atomic units (a.u.) to − 0.0125 a.u., that is 0.6 to − 0.6 V/Å [10]. In the biological milieu, e.g., the microenvironment of DNA, the local fields are estimated to be of the order 10−2–1 V/Å [19]. The lower limit is near to the estimated strength of the intramolecular field of the order 10−2 V/Å [20]. Fields of 0.01 V/Å accelerate photosynthetic reactions by an order of magnitude [8]. For a fully saturated phosphatidylcholine membrane model, the strength of the electric field can reach a value up to 0.1 V/Å [15]. In condensed phase systems (e.g., aqueous NaCl electrolytes), the internal (fluctuating) electric fields throughout the space between nuclei are much stronger (1 V/Å) [21].
We were interested in the impact of a steady electric field on the charge distribution in some molecules. The overall representation of the charge distribution is offered by the molecule dipole moment. Our first aim was to present and compare two ways of calculation of the field strength (E0) sufficient to reset to zero the overall dipole moment of a molecule. The fields stronger than that can reversely polarize molecules. It is interesting to compare the E0 field to fields which are produced within the chemical and biological environment.
The second aim was connected to the recent results of the comparison of the effect of substituents in cyclohexa-1,3-diene derivatives and in benzene [22]. It appeared that the donating ability of the NH2 group was around 1.4 times more strongly affected by a series of substituents in the para position to the NH2 group when the transmitting moiety was olefinic than when it was aromatic. We were interested in the comparison of the electric field effects on the properties of larger aromatic and olefinic structures, characterized by a similar length of conjugation.
The studied systems belong to the class of the so-called push-pull systems [23]. These molecules are relevant in the literature because they present a very high value of the calculated nonlinear optical properties. It has been found that the conventional density functional schemes generate overly large dipole moments in such systems as compared to the simple HF, Möller-Plesset, and coupled-cluster calculations. The quality of results depends on the choice of the XC functional [23, 24].
Calculation
The values of the electric field strength used in our calculations were taken from the interval − 0.01 to 0.01 a.u. (− 0.5 to 0.5 V/Å) for most cases. In two cases, two points at E = 0.014 a.u. had to be added to generate at least three points corresponding to molecules with both directions of dipole moments (Figs. 2, 3, 4, and 5).
The calculations were conducted using density functional theory (DFT) with Becke’s hybrid exchange functional [25] and Lee-Yang-Parr correlation functional (B3LYP) [26]. The 6-31+G(d) basis set with polarized and diffuse functions was employed in all calculations [27, 28]. Then, taking into consideration the Referee’s recommendation, the calculations were repeated with the CAM-B3LYP, a long-range corrected functional that fixes the incorrect electric field dependence modeled by the exchange functional of the traditional DFT methods as B3LYP [29, 30]. It was found that this long-range collected density functional removes a large part of the overestimation of dipole moments and hyperpolarizabilities (experimentally observed) and does better in modeling polarizability [31].
The plots of dipole moments and energies vs strength of electric field, calculated with the CAM-B3LYP, are included only for the polyene and diphenylbenzene with R1=NH2 and R2=NO2.
The calculations were performed using the Gaussian 09 program [32]. Molecular structures were optimized without any symmetry constraints, with and without external fields with the default convergence thresholds for the maximum displacements and residual forces on nuclei. The electric field was parallel to the main molecular axis and to the X-axis of the coordinate system and the E vector was directed toward the positive charge (as shown in Fig. 1).

Situation of polyenic and aromatic molecules toward the X-axis
This convention is the same as applied in Gaussian input. In the four aromatic structures with phenyl rings, the X-axis of the coordinate system passed through the carbon atoms in the para position. Position of the polyene structures with respect to the X-axis is shown in Fig. 1. The optimized geometries are deposited in the Supporting Information. The axis was oriented so that the dipole moments of all molecules without any applied electric field were positive—that is, the dipole moment vectors originate at the negative pole and point to the positive pole (Fig. 1). Therefore, all fields in this paper have the same direction as the X-axis along which they are aligned. The distances between two terminal C atoms, calculated for the two structures without substituents, where the terminal bonds were C-H instead of C-R1 and C-R2, were 11.47 Å for diphenylbenzene and 11.12 Å (in deca-1,3,5,7,9-pentaene).
Results
First, the dipole moments of the polyene and aromatic compounds were calculated in the absence of an external electric field (Table 1). Given the dipole moments as measures of polarization of the molecules, we can see that for the more polarized molecules (R1=NO2 and R2=NH2, as well as R1=NO2 and R2=OH), the difference between the dipole moments of a polyene and its aromatic counterpart is larger than for the two weakly polarized molecules (R1=NO2 and R2=NH2, as well as R1=NO2 and R2=CN).
Then, the dipole moments of all molecules were calculated and plotted against the strength of the electric field. Results obtained by using the B3LYP functional are shown in Figs. 2, 3, 4, and 5. It appeared that all plots were good linear functions. In all cases, the slope of the straight line for a polyene was significantly larger than that for the corresponding diphenylbenzene with the same substituents. Equations of linear functions describing plots for aromatic molecules (4-R1-4′-R2-p-diphenylbenzene) are shown at the left, and those for polyenes (R1-1,3,5,7-decapentaene-R2) are shown at the right of the pictures. These functions allowed determination of the values of the electric field (E0) for which dipole moments are zeroed. At this field strength, reversal of the dipole moment direction takes place. At E = E0, initial polarization of a molecule has the same absolute value (and the reverse direction) as the polarization induced by the field. The values of the ratios of the slopes (of polyenes vs aromatics with the same R1 and R2) are within the range 1.3–1.4. These significant data (values of E0 and the ratios of the slopes) are shown in Table 2. For the first four molecules with the higher dipole moments (7–15 Debye), the E0 is about 0.006–0.007 a.u., i.e., 0.3–0.4 V/Å. For the last four molecules possessing a relatively low dipole moment (up to 1.8 Debye), the E0 field is lower, of the order of 0.001 a.u., i.e., 0.05 V/Å.

Plot of dipole moment (in Debye) against the electric field strength (in a.u.) for 4-NO2-4′-NH2-p-diphenylbenzene (+++) and 1-NO2-10-NH2-1,3,5,7-decapentaene (ooo)

Plot of dipole moment (in Debye) against the electric field strength (in a.u.) for 4-NO2-4′-OH-p-diphenylbenzene (+++) and 1-NO2-10-OH-1,3,5,7-decapentaene (ooo) calculated at B3LYP/6-31+G(d)

Plot of dipole moment (in Debye) against the electric field strength (in a.u.) for 4-OH-4′-NH2-p-diphenylbenzene (+++) and 1-OH-10-NH2-1,3,5,7-decapentaene (ooo) calculated at B3LYP/6-31+G(d)

Plot of dipole moment (in Debye) against the electric field strength (in a.u.) for 4-NO2-4′-CN-p-diphenylbenzene (+++) and 1-NO2-10-CN-1,3,5,7-decapentaene (ooo)
Taking into account that the B3LYP and other (“classical”) DFT functionals generated too high dipole moments for the π-conjugated systems [23], calculations were also made using a hybrid exchange-correlation functional including the Coulomb attenuating factor (CAM-B3LYP). Tables 1, 2, and 3 contain data obtained with both functionals for all four systems. The plots (for a diphenylbenzene and polyene) of dipole moments against E, for data calculated at the CAM-B3LYP, are shown in Fig. 6 for one, the most interesting system, (R1=NO2 and R2=NH2).

Plot of dipole moment (in Debye) against the electric field strength (in a.u.) for 4-NO2-4′-NH2-p-diphenylbenzene (+++) and 1-NO2-10-NH2-1,3,5,7-decapentaene (ooo) calculated at CAM-B3LYP/6-31+G(d)
The plots constructed by using data generated at CAM-B3LYP were similar. One of the systems with R1=NH2 and R2=NO2 (for a diphenylbenzene and polyene) is shown in Fig. 8.
The energy of molecules as a function of the external electric field strength
Next, let us see how the energies of the molecules responded to perturbation in their charge redistribution. The plots of the energies calculated every 0.005 a.u. at the B3LYP/6-31+G(d) level are shown in Fig. 7. Equations of the parabolas are given: for diphenylbenzenes at the top and for polyenes near the horizontal axis. They allowed determination of the Emax values given in Table 3.

Energies in a.u. of 4-R1-4′-R2-p-diphenylbenzene (+ + +) and of 1-R1-10-R2-1,3,5,7-decapentaene (o o o) plotted against field strengths (a.u.) calculated at the B3LYP/6-31+G(d) level
The plots constructed by using data generated at CAM-B3LYP were very similar. One of the systems with R1=NH2 and R2=NO2 (for a diphenylbenzene and polyene) is shown in Fig. 8.

Energies in a.u. of 4-NO2-4′-NH2-p-diphenylbenzene (+ + +) and of 1-NO2-10-NH2-1,3,5,7-decapentaene (o o o) plotted against field strengths (a.u.) calculated at the CAM-CAM-B3LYP/6-31+G(d) level
The plots of energies of molecules in the presence of electric fields of varying strengths were found to be parabolic functions of the latter with coefficients of determination exceeding 0.99 in every case. Based on the equation of the parabola, one can find the field strength corresponding to the maximum energy of a molecule (Emax). These values are given in Table 3. For the molecule of both groups, the Emax values are almost the same and agree very well with those determined from the plots in Figs. 2, 3, 4, and 5, shown in Table 2. Thus, it appears that maximum energy falls for the molecule characterized by a dipole moment very close to zero. Then, going to negative E values, the molecule is more polarized in the same direction, as at E = 0 (Table 1) and its energy lowers. Minima of energy fall at the lowest considered E values, e.g., E = − 0.005 a.u. (for R1=NO2, R2=NH2) or (− 0.01 a.u., for R1=NO2, R2=CN). These negative fields enhance the values of the dipole moments of molecules with the same direction as those of the unperturbed molecules.
So, the energy of a molecule in external fields of strengths going from zero to the maximum negative value monotonically decreases. One could expect that the energy would increase at the subsequent positive value of the electric field strength, resulting in diminishing the dipole moment and shifting the negative charge from the negative substituent (e.g., NO2) to the positive (e.g., NH2). However, at the E = E0 value corresponding to μ equal zero, the energy reaches the maximum and at the more positive values begins to decline. For the points at right from the maximum, both the orientations of the dipole moment and of the external field are once again antiparallel and the increasing field brings about a lowering of energy.
Conclusion
The reaction to a steady electric field of aromatic and polyene molecules, with very similar lengths of conjugation due to unsaturated carbon-carbon bonds, was compared. It was found that the response of polyene molecules, as measured by the slopes of linear plots of dipole moments vs electric field strengths, was larger than that of aromatic molecules. This finding points to a greater resistance of the aromatic units than that of the polyene chains to repolarization. However, the strength of electric fields reversing the dipole moment of dipolar molecules is, as far as it can accurately be determined, the same for an aromatic molecule or polyene molecule possessing the same substituents. For molecules with a higher initial dipole moment, E0 is of the order of 0.03–0.04 V/Å; for less polar molecules, E0 is of the order of 0.01 V/Å. These values are of the order of the fields that are common in the environment of molecules, including those of biological importance as well as in the tunneling microscope (STM) and in intense laser radiation fields [19].
References
Laidig KE, Bader RFW (1990) Properties of atoms in molecules: atomic polarizabilities. J Chem Phys 93:7213–7224
Gough KM, Yacowar MM, Cleve RH, Dwyer JR (1996) Analysis of molecular polarizabilities and polarizability derivatives in H2, N2, F2, CO2, and HF with the theory of atoms in molecules. Can J Chem 74:1139–1144
Krawczuk A, Pérez D, Stadnicka K, Macchi P (2014) PolaBer: a program to calculate and visualize distributed atomic polarizabilities based on electron density partitioning. J Appl Crystallogr 47:1452–1458
Dos Santos LHR, Krawczuk A, Macchi P (2015) Distributed atomic polarizabilities of amino acids and their hydrogen-bonded aggregates. J Phys Chem A 110:3285–3298
Macchi P, Krawczuk A (2015) The polarizability of organometallic bonds. Comp Theor Chem 1053:165–172
Dominikowska J, Palusiak M (2018) Tuning aromaticity of para-substituted benzene derivatives with an external electric field. ChemPhysChem 19:590–595
Dominikowska J, Krygowski TM, Ozimiński WP, Palusiak M (2015) Aromaticity induced by electric field: the case of polycalicenes. J Organomet Chem 80:9091–9101
Sowlati-Hashjin S, Matta CF (2013) The chemical bond in external electric fields: energies, geometries, and vibrational stark shifts of diatomic molecules. J Chem Phys 139:144101-1–144101-14
Wang Z, Shaik S (2018) Oriented-external electric fields create absolute enantioselectivity in Diels-Alder reactions; importance of the molecular dipole moment. J Am Chem Soc 140:13350–13359
Shaik S, de Visser SP, Kumar D (2004) External electric field will control the selectivity of enzymatic-like bond activations. J Am Chem Soc 126:11746–11749
Fuller J Wilson TR, Eberhart ME, Alexandrova AN (2019) Charge density in enzyme active site as a descriptor of electrostatic reorganization J Chem Inf Model. In press. https://doi.org/10.1021/acs.jcim.8b00958
Franzen S, Goldstein RF, Bover SG (1990) Electric field modulation of electron transfer reaction rates in isotropic systems: long-distance charge recombination in photosynthetic reaction centers (1990). J Phys Chem 94(5135):5149
Popovic ZD, Kovacs GJ, Vincett PS, Alegria G, Dutton PL (1986) Electric field dependence of recombination kinetics in reaction centers of photosynthetic bacteria. Chem Phys 110:227–237
Gopher A, Blatt Y, Schönfeld M, Okamura MY, Feher G, Montal M (1985) The effect of applied electric field on the charge recombination kinetic in reaction centers reconstituted in planar lipid bilayers. Biophys J 48:311–319
De Biase PM, Doctorovich F, Murgida DH, Estrin DA (2007) Electric field effects on the reactivity of heme model systems. Chem Phys Lett 434:121–126
Shaik S, Ramanan R, Danovich D, Mandal D (2018) Structure and reactivity/selectivity control by oriented-external electric field. Chem Soc Rev 47:5125–514515
Ciampi S, Darwish N, Aitken HM, Dièz-Pérez I, Coote ML (2008) Harnessing electrostatic catalysis in single molecule, electrochemical and chemical systems: a rapidly growing experimental tool-box. Chem Soc Rev 47:5146–5164
Che F, Gray JT, Ha S, Krase N, Scott SL, McEwen J-S (2018) Elucidating the roles of electric fields in catalysis: a perspective. ACS Catal 8:5153–5174
Arabi AA, Matta CF (2011) Effects of external electric fields on double proton transfer kinetics in the formic acid dimer. Phys Chem Chem Phys 13:13738–13748
Piela L (2013) Ideas of quantum chemistry, 2nd edn, par. 12.4.3. Elsevier, Philadelphia
Sellner B, Valiev M, Kathmann SM (2013) Charge and electric field fluctuations in aqueous NaCl electrolytes. J Phys Chem B 117:10869–10882
Szatyłowicz H, Siodla T, Krygowski TM (2017) Olefinic vs aromatic way of substituent effects: the case of 3- and 4-substituted cyclohexa-1,30-dieneamine derivatives. J Phys Org Chem 30:e3694
Champagne B, Perpète EA, Jacquemin D, van Gisbergen SJA, Baerends EJ, Soubra-Ghaoul C, Robins KA, Kirtman B (2000) Assessment of conventional density functional schemes for computing the dipole moment and (hyper) polarizabilities of push-pull π-conjugated systems. J Phys Chem A 104:4758–4763
Becke AD (1993) Density-functional thermochemistry. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W (1998). Phys Rev B 37:785
Francl MM, Pietro WJ, Hehre WJ, Binkl S, Gordon MS, DeFrees DJ, Pople AA (1982) Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys 77(8):3654
Gill PMW, Johnsom BG, Pople JA (1992) The performance of the Becke-Lee-Yang-Parr (B-LYP) density functional theory with various basis sets. Chem Phys Lett 197:499–505
van Gisbergen SJA, Schipper PRT, Gritsenko OV, Baerends EJ, Snijder JG, Champagne B, Kirtman B (1999) Electric field dependence of the exchange-correlation potential in molecular chains. Phys Rev Lett 83:694–697
Yanai T, Tew DP, Handy NC (2004) A new hybrid-exchange-correlation functional using the Coulomb attenuating method (CAM-B3LYP). Chem Phys Lett 343(343):51–57
Tawada Y, Tsuneda T, Yanagisawa S, Yanai T, Hirao K (2004) A long-range corrected time-dependent density functional theory. J Chem Phys 120(18):8425:8433
Limacher PA, Mikkelsen KV (2009) On the accurate calculation of polarizabilities and second hyperpolarizabilities of polyacetylene oligomer chains using the CAM-B3LYP density functional. J Chem Phys 130:194114
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashy R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford
Funding
The computing grant G44-18 from the Interdisciplinary Center for Mathematical and Computer Modeling (ICM) of Warsaw University and the financial support from the Polish Ministry of Science and Higher Education are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sadlej-Sosnowska, N. The response of electronic and energetic properties of conjugated vs aromatic molecules to an external uniform electric field. Struct Chem 30, 1407–1413 (2019). https://doi.org/10.1007/s11224-019-01345-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-019-01345-6