
Abstract
Platinum (Pt) is the most common catalyst in Polymer Electrolyte Membrane Fuel Cells due to its ability to effectively promote the oxidation of hydrogen and reduction of oxygen. However, as a noble metal, the use of Pt should be minimized. Alloying Pt with low-cost transition metals is an effective strategy to improve catalytic activity and reduce Pt use. In this context, we report on a one-step synthesis of a Platinum/Copper (PtCu) catalyst, which can be used at both the anode and the cathode of a fuel cell. Catalysts with various Cu to Pt ratios were synthesised and in particular the PtCu catalyst at a Cu to Pt ratio of 0.5 demonstrated a high activity for hydrogen oxidation and oxygen reduction, i.e. 2.4 times superior to Pt alone. This enhanced catalytic activity was confirmed in a self-breathing PEMFC with a power output of 45.16 mW cm−2, which corresponds to a 1.4-fold increase compared to Pt alone. This is a significant improvement because 40% more power was obtained with 22% less Pt.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proton Exchange Membrane Fuel Cells (PEMFC) have gathered great attention in the last decades as alternatives to deliver electrical power to a range of applications [1,2,3,4,5,6]. However, several issues such as: (1) excessive dependence on precious metals, e.g. Platinum (Pt) as the main catalyst, (2) costly manufacturing mainly at small scale, and (3) reliance on auxiliary systems such as compressors/ air cylinders, hinder improvements in efficiencies and significant cost reduction to trigger a wide deployment of PEMFC beyond large scale systems and automotive applications [1].
Until now, Pt has remained the catalyst of choice at both the anode and the cathode in commercial systems [1, 7]. In addition to its scarcity, kinetic losses at both anode and cathode has been reported during charge transfer in Hydrogen Oxidation Reaction (HOR) and Oxygen Reduction Reaction (ORR) due to strong binding of O2, H2 and their reaction intermediates, i.e. HO, HOO and O, resulting in “blockage” of the active catalyst sites [8, 9]. Such inhibition effects become significant especially at low Pt loading (< 0.05 mg cm−2) as a result of reduced Pt amounts in PEMFC stacks [9].
Regarding the replacement of Pt, most research efforts have focussed on alternative catalysts for the ORR [10]. However, developing better catalysts for HOR at the anode is equally important as both reactions rely on noble metals [1]. The main reason for the extensive research efforts on the ORR is the slow kinetic of this reaction (1), i.e. ~ 5 orders of magnitude lower than at the anode (2) [1, 11, 12].
At the cathode:
At the anode:
Overall reaction:
Current state-of-the-art ORR catalysts are still Pt based, with some non-noble catalysts such as those based on iron (Fe)/nitrogen (N)/carbon (C) [13], Fe/nickel (Ni)/N-graphene (G) [14] and N/C composites [15], which have shown good electrocatalytic performance, e.g. 2.5 mA cm−2 (at half wave potential, \(E_{1/2} = 0.76\) VSHE, 0.05 M H2SO4, 1600 rpm) for FeNC [16]. Pd alloyed with other noble metals, e.g. Pt, Ru and Ir have also been reported [17]. For example, Pt0.66Pd0.33 delivered an electrocatalytic activity of 1.7-fold (at \(E_{1/2} = 0.84\) VSHE, 0.1 M HClO4, 1600 rpm) that of Pt only, and the electrocatalytic activity of trimetallic PtPdCu exceeded that of Pt by 4.7-fold (at, \(E_{1/2} = 0.94\) VSHE, 0.1 M HClO4, 1600 rpm) [18, 19].
Alloying Pt with other transition metals such as Nickel (Ni), Cobalt (Co), Chromium (Cr) and Fe, has allowed to reduce the amount of Pt, but also was found to lead to better electrocatalytic activities owing to the more active sites [1, 20]. For example, the following trend: Pt < Pt3Ti < Pt3V < Pt3Ni < Pt3Fe ≈ Pt3Co has been reported, where the electrocatalytic activity of Pt3Co is 3 time that of Pt (at 0.9 VRHE, 0.1 M HClO4, 60 °C) [21,22,23].
Upon alloying Pt with various transition metals, a shortening in the Pt–Pt bond distance is expected and this would result in a compressive lattice strain leading to a decrease in the chemisorption energy of adsorbates such as O⁎ and HO⁎ at catalytically active sites. Additional effects including: (1) an increase in surface roughness due to the dissolution of the alloyed transition metals (e.g. Co, Ni) during operation, and (2) a downshifting of the d-band centre of Pt, would result in a weaker adsorption of intermediates such as HO⁎, HOO⁎ and O⁎ and thus enhanced O2/H+ catalytic reactivity [11, 21, 24]. In particular, a weaker adsorption of HO⁎, O⁎ and HOO⁎ oxygen intermediate species at the catalytic sites would limit the formation of an inhibiting oxide layer [1, 25].
In contrast to ORR catalysts, significantly less work has been reported on HOR. Attempts have been made to increase the electroactive surface area by using carbon nanotubes and the activity enhanced by 1.7-fold over Pt/C (at 0.1 VRHE, 0.1 M H2SO4) [26]. Substituting Pt by Pd has also been extensively explored mostly by alloying with other noble metals, e.g. PdIr, PdRu [1, 17]. PdIr has been reported to show improvements in electrocatalytic activity by a factor 1.8 (at 0.07 VSCE, 0.5 M H2SO4, 1600 rpm) [17]. In spite of the tremendous potential shown by noble-non noble metal alloys for both ORR and HOR, significant issues still limit the use of these catalysts in real systems. These include: (1) complex synthesis steps often difficult to scale-up, (2) leaching of the non-noble metal in the acidic environment, which eventually results in activity degradation, and (3) limited validation of performances observed by RDE under PEMFC environment [11].
Herein, we report on a one step synthesis method to prepare highly active PtCu catalyst on Vulcan Carbon (VC) using the polyol process [27]. The selection of Cu was motivated by the fact that among the 3d transition metals, Cu has a maximum number of possible d electrons (3d9) to facilitate O–O bond activation [28, 29]. Also, it has been reported that the addition of Cu to Pt, shifts the d-band centre of Pt down with respect to the Fermi level, which in turns weakens H2 chemisorption and improves HOR activity [30]. In addition, it has been reported the Cu has the ability to promote faster electron transfer to O2 during ORR owing to the higher redox potential of Cu (E° = 0.34 V vs SHE) compared to other 3d transition metals [28]. Hence, alloying Cu with Pt is expected to not only enhance Pt catalytic properties but also lead to some synergetic bifunctional effects for HOR and ORR, as Cu itself is a good catalyst for O2 reduction [31]. This work thus reports on this new catalyst for both HOR and ORR, as well as on its performance in a self-breathing PEMFC configuration with bipolar plates 3D printed from plastic. Developing means to operate and test the PEMFC in self-breathing mode can further help in terms of overall efficiency by eliminating the need of secondary devices to provide air [1, 32]. Most of the catalysts developed so far in the literature have been characterised at RDE to determine their catalytic activity, without considering performance at the Membrane Electrode Assembly (MEA) level under fuel cell operating conditions [33].
Experimental section
Materials
H2PtCl6·xH2O (≥ 99.9% trace metals basis), CuCl2·2H2O, ethylene glycol (anhydrous 99.8%), 2-propanol (99.7%), perchloric acid (70%, 99.99%) were purchased from Sigma-Aldrich. Vulcan XC 72R, Nafion™ 212, Nafion ™ Dispersion (10 wt.%), Freudenberg H23C2, 40 wt.% Platinum on Vulcan Carbon (VC) (Fuel Cell grade) were purchased from Fuel Cell Store. For all the synthesis, Milli Q water was used.
Synthesis of PtCu/VC
PtCu/VC was prepared based on the polyol process [27, 34]. In brief, 50 mg of VC mixed with 33 ml of ethylene glycol, 17 ml of Milli Q water, 34 mg of H2PtCl6 × H2O and 18 mg of CuCl2·2H2O in a 100 ml single neck round bottom flask. The mixture was sonicated for 5 min for homogenisation and was continuously stirred overnight (15 h) at room temperature. The next day, the round flask (with total mixture) was heated to 120 °C for 2 h under reflux and continuously stirred for the reduction of Pt and Cu on the carbon substrate. After cooling, the obtained catalyst was washed three times with Milli Q water and separated by centrifugation at 10,000 rpm. The resulting material was dried under vacuum at 60 °C for 15 h. The same procedure was used to synthesize all the catalysts of varied amounts of Cu/Pt. The synthesized catalysts supported by VC are noted as 18%Pt 9%Cu as Cu/Pt (0.5), 18%Pt 15%Cu as Cu/Pt (0.8), 18%Pt 20%Cu as Cu/Pt (1.1), 18%Pt as Pt and commercial 40% Pt/VC as Pt*. The number in brackets corresponds to the mass ratio Cu/Pt. By ICP-OES, it was confirmed that Cu/Pt (0.5) contained 17.7 ± 0.2% of Pt and 8.8 ± 0.1% of Cu, which means that the material corresponded to the initial amounts of Pt and Cu used for the synthesis.
Preparation of the catalyst ink
1.1 mg of synthesized catalyst, 20 µl of Nafion 10% in water, 480 ml of Milli Q water and 120 ml of 2-propanol were mixed in a vial and sonicated for 5 min to form a homogenous mixture. A glassy carbon electrode was polished using an alumina suspension and a microcloth disk. 2 µl of the prepared catalyst ink was taken in a pipette and dropped onto the polished glassy carbon electrode surface (3 mm diameter) and allowed to dry at room temperature. The catalyst (metal on VC) loading for all the experiments on Rotating Disk Electrode was 50 µg cm−2.
For making the fuel cell, 24 mg of synthesized catalyst was added to a vial. 125 µl of Milli Q water, 100 µl Nafion 10% in water and 500 µl of 2-propanol were added. The final volume was adjusted by adding 1.5 ml of 2-propanol. The mixture was sonicated for 5 min and left to stir overnight at a room temperature.
Preparation of membrane electrode assembly
The required amount catalyst ink was dispersed on the microporous layer of a carbon paper (Freudenberg H23C2) using the doctor blade technique. This was then dried in an oven at 60 °C for 1 h. Nafion 212 was used without pre-treatment. The MEA was formed by placing the catalyst coated gas diffusion electrodes on either side of the membrane and by hot pressing at 0.18 MPa at 90 °C for 2 min. The catalyst (metals on VC) loading in all the MEAs was 0.8 mg cm−2, the ionomer (Nafion) in the MEA was 30 wt.% of the solid dispersed in catalyst ink. The active area of the MEA was 2 cm2.
Characterization
The samples were dispersed in ethanol and sonicated which was then dropped casted onto a carbon coated copper grid. The High Resolution Transmission Electron Microscopy (HRTEM), Selected Area Diffraction (SAD) and Energy Dispersive X-ray Spectroscopy (EDS) were performed with a JEOL JEM-F200 cold field emission gun operated at 200 kV with an attached windowless 100 mm2 silicon drift X-ray detector.
X-Ray Diffraction (XRD) was performed by using a Philips X’pert Multipurpose XRD system operated at 40 mA and 45 kV with a monochromated Cu Kα radiation (λ = 1.541 Å). The operation 2θ range was between 15 and 90°.
The amount of Pt and Cu in the synthesized catalysts were determined by inductively coupled plasma-optical emission spectrometry (ICP-OES) by using an Optima7300DV (PerkinElmer). The samples were prepared by digestion in acid (3HCl + 1HNO3).
Electrochemical test including Cyclic Voltammetry (CV) and Linear Sweep Voltammetry (LSV) was performed at a room temperature (25 °C) by using a VMP3-Biologic potentiostat. The potentiostat was connected to Rotating Disk Electrode (Basi RDE 2) having a three electrodes electrochemical cell. An Ag/AgCl saturated with 3 M NaCl was used as the reference electrode and Pt as the counter electrode. The use of Pt as a counter electrode instead of graphite was guided by the low levels of Pt dissolution reported under the current experimental conditions and the very moderate impact of this dissolution on the electroactivity measured [35]. The use of Pt as the counter electrode can lead to some overestimations in electrocatalytic activities at RDE owing to potential Pt dissolution (at 0.6–0.8 V vs SHE) and redeposition on working electrodes [36]. The use of graphite as counter electrode instead of Pt has been recommended to overcome this [35]. However, graphite can undergo severe oxidation even at a low potential of 0.1 V vs SHE and this results in the formation of CO and CO2 which rapidly poison Pt electrocatalytic sites during ORR [35, 37].
Freshly prepared 0.1 M HClO4 was used as the electrolyte. The catalyst activity reported was determined by subtracting the background measurement and iR correction. Regarding the surface area of the electrode, a real surface area \(\left( {A_{real} } \right)\) was calculated which is in line with Mayrhofer et al. [38] and several other literatures [39,40,41,42]. It has been reported that in case of nano catalysts, the real surface area of the electrode is substantially different than the geometric area \(\left( {A_{real} } \right)\) due to the presence of surface roughness introduced by the catalyst, \(A_{real} > A_{geo}\) [38,39,40,41]. The roughness factor \(\left( {r_{f} } \right)\) of the catalyst is the ratio between \(A_{real}\) and \(A_{geo}\) and is calculated by the relation below [38, 43],
where \(A_{real}\) and \(A_{geo}\) are the real and geometric surface area of the electrode, respectively, \(Q_{H}\) is the charge obtained from the integrated area of hydrogen adsorption in the region 0.03–0.3 V of CV and \(q_{H}^{^\circ }\) is the charge density of bulk polycrystalline Pt equal to 210 µC cm−2.
Fuel cell testing was done by using a home-made self-breathing single PEMFC fabricated with a 3D printer (Form 2, Formlab) as shown in Fig. 1. The opening at the air cathode was 55% and the anode had parallel flow fields. The active area of a cell was 2 cm2. Stainless steel was used as current collector on both anode and cathode and nitrile rubber gaskets were used for sealing. The fuel cell test was done at ambient condition. Operating temperature, pressure, and humidity were 25 °C, 1 bar and 20% RH (RH: Relative humidity), respectively. The H2 flow was maintained at 5 ml min−1.

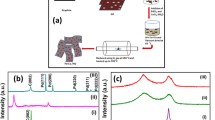
Photo of the components of the 3D printed self-breathing PEMFC used for testing the catalysts
Results and discussion
Synthesis of copper-platinum alloy on Vulcan carbon
The anticipated mechanism of Pt and Cu reduction on Vulcan carbon is summarized in Scheme S1, where the dehydration of ethylene glycol in interaction with H2PtCl6 and CuCl2 results in the formation of acetaldehyde, which finally oxidizes upon Pt4+and Cu2+ reduction to lead to diacetyl [44]. Ethylene glycol is widely used in the polyol process because it leads to a good control over the spatial dispersion and size distribution of nanoparticles [45]. During the polyol process, Pt and Cu nuclei are generated in the liquid phase and further nucleation and growth is expected to occur at defective sites of VC [46].
The XRD of the synthesized Pt on VC is shown in Fig. 2. The diffraction peaks at 39.4°, 67.4° and 81.6° were assigned to Pt (111), Pt (220) and Pt (311), respectively. The XRD of Cu/Pt (0.5) revealed that the peak position of Pt (111), Pt (220) and Pt (311) previously observed (Fig. 2) has shifted to higher diffraction angles and this indicates the formation of the cubic Pt7Cu kitagohaite phase [47,48,49]. In particular, the diffraction peaks at 40.1°, 46.6°, 68.0° and 81.9° were assigned to Pt7Cu (222), Pt7Cu (400), Pt7Cu (440) and Pt7Cu (622), respectively.

XRD of the synthesized catalysts: Pt, Cu/Pt (0.5), Cu/Pt (0.8) and Cu/Pt (1.1)
Further characterization of Cu/Pt (0.5) by TEM revealed uniformly dispersed PtCu particles with an average size of ~ 3 nm well dispersed on Vulcan carbon (Fig. 3). Further lattice measurement by HRTEM shows that the particles observed have a d spacing of 0.224 nm (Fig. 3b–c), which corresponds to Pt7Cu (222) in agreement with the XRD analysis. In addition, STEM and elemental mapping of Cu/Pt (0.5) using EDS show the distribution of Pt, Cu on carbon substrate to be uniform with Pt and Cu co-located on the VC support. It can be concluded that uniform Pt7Cu nanoparticles have been synthesised on VC at Cu/Pt 0.5 mass ratio.

a, b HRTEM images, c SAD, d EDS MAP of Cu/Pt (0.5)
Electrocatalytic activity of PtCu for the HOR
Figure 4a shows the CV profile of synthesized Pt obtained in 0.1 M aqueous HClO4 under saturated Ar and H2 at the scan rate of 50 mV s−1. Under Ar, the cathodic and anodic CV features between 0.05 and 0.2 V correspond to the adsorption and desorption of underpotential deposited H (HUPD), respectively [50]. Similarly, the anodic feature in the 0.7–1.0 V range corresponds to the formation of Pt surface oxides (Eq. 4), and the cathodic one in between 0.6 and 1.0 V is due to the reduction of the oxide layer at the Pt surface (Eq. 5) [51].

HOR activity for: a Pt, b Cu/Pt (0.5), CV in 0.1 M aqueous HClO4 under saturated Ar and H2 at the scan rate of 50 mV s−1 @ 1600 rpm c LSV comparison of Pt and Cu/Pt (0.5) under saturated H2 at the scan rate of 10 mV s−1 @ 1600 rpm, catalyst loading was 50 µg cm−2 for all samples
Under saturated H2, the CV profile was slightly shifted upward due to the occurring HOR, and revealed an increase in intensity of the typical feature associated with the adsorption and desorption of HUPD at the Pt surface [50].
For Cu/Pt (0.5), these observations were more pronounced, and under saturated H2, the entire CV profile shifted upward with larger currents due to the occurring HOR and peaks in between 0.05 and 0.2 V corresponding to the adsorption and desorption of HUPD at the catalyst surface. This more intense shift in current density for Cu/Pt (0.5) in comparison to Pt clearly indicates that Cu/Pt (0.5) is a better HOR catalyst than Pt (Fig. 4b).
This improved performance of Cu/Pt (0.5) can be explained as alloying Pt with Cu would lower the d-band centre of Pt, and this should imply an optimal binding for adsorbed H2 [52,53,54]. Thus, HOR occurs at a lower potential at PtCu compared to Pt [53]. This was also confirmed from the LSV profile (Fig. 4c), where HOR started at a slightly lower potential (20 mV vs SHE) in case of Cu/Pt (0.5).
Electrocatalytic activity of PtCu alloy for the ORR
Synthesized Pt was also investigated as a potential catalyst for the ORR. Figure 5a shows the CV of the synthesized Pt under saturated Ar and O2 at the scan rate of 50 mV s−1, as a reference point. As in the case of HOR, under saturated Ar, the cathodic and anodic CV profile in the range 0.05–0.2 V is assigned to adsorption and desorption of HUPD, respectively. Similarly, the anodic features in between 0.7 and 1.0 V correspond to the formation of Pt surface oxides, and the cathodic feature in the voltage range 0.6–1.0 V is due to the reduction of these surface oxides.

ORR activity for: a Pt, b Cu/Pt (0.5), CV in 0.1 M aqueous HClO4 under saturated Ar and O2 at the scan rate of 50 mV s−1 @1600 rpm, c LSV comparison of Cu/Pt (0.5) and Pt under saturated O2 at the scan rate of 10 mV s−1 @ 1600 rpm, catalyst loading was 50 µg cm−2 for all samples
Under saturated O2, in addition to the typical features associated with the underlying adsorption and desorption of HUPD larger currents due to the reduction and oxidation of dissolved O2 at the Pt surface are observed [50].
In the case of Cu/Pt (0.5), under saturated O2, the features associated with the formation and reduction of oxides at the Pt surface were slightly more pronounced. As a result, the larger cathodic current corresponding to the reduction of Pt surface oxides, at Cu/Pt (0.5) in comparison to Pt suggest that Cu/Pt (0.5) is a better catalyst for ORR than Pt (Fig. 5b).
Higher activity of Cu/Pt (0.5) can be understood considering that when Cu is alloyed with Pt, the chemisorption energy of ORR intermediates (OH⁎, OOH⁎, and O⁎) is weakened, because of the downshifting in the d-band centre of Pt and this results in increased ORR [1, 55, 56]. This was further confirmed from LSV profiles (Fig. 5c), where the improvement in half wave potential (50 mV vs SHE) was observed in the case of Cu/Pt (0.5) compared to Pt only.
Influence of Cu/Pt ratios
It is thus apparent from the results above that alloying Cu with Pt leads to some significant improvements not only in terms of HOR but also ORR. To determine the optimum amount of Cu, different Cu/Pt ratios were investigated by increasing the amount of Cu in the alloy.
The XRD of synthesized PtCu with Cu/Pt ratios of 0.8 and 1.1 are shown in Fig. 2. In case of Cu/Pt (0.8), the diffraction peaks at 40.1°, 46.6°, 68.0° and 81.9° are assigned to Pt6Cu2 (222), Pt6Cu2 (400), Pt6Cu2 (440) and Pt6Cu2 (622), respectively. In contrast, for Cu/Pt (1.1), the diffraction peaks at 40.2°, 46.7°, 68.8.0° and 82.5° are assigned to PtCu (111), PtCu (200), PtCu (220) and PtCu (311), respectively. It is thus apparent that increasing the amount of Cu in the ratio (Cu/Pt) from 0.5 to 1.1 changed the phase from Pt7Cu to Pt6Cu2 to PtCu. This phase evolution is mainly the result of changes in the concentration of Cu and Pt precursors at various Cu/Pt ratios, and the difference in the reduction rates of the Pt and Cu precursors (redox potential of [PtCl6]2−/Pt = + 0.742 V vs SHE, Cu2+/Cu = + 0.340 V vs SHE, while that of ethylene is glycol 2.24 V vs SHE) [57,58,59]. In this process, the galvanic replacement reaction between the pre-reduced metal (Pt) and the other metal ion, i.e. Cu2+, in the growth solution may also influence the final phases [60]. It is noteworthy that the various PtCu phases observed are in line with the Cu/Pt phase diagram (Fig. S1) [61]. In addition, from the analysis of the diffraction patterns of PtCu, it was found that the crystal size and microstrain of the later increased with the addition of Cu (Fig. S2a, b). This suggests the alloying of Pt with Cu [62].
The morphology of PtCu particles with different Cu/Pt ratios was further analysed by TEM (Fig. S4–S6). In all the materials, the PtCu particles were found to be well dispersed on VC and the size increased from 2 to 4 nm with the addition of Cu (Fig. S4, S5). This increase in particle size along with the increase in Cu concentration is explained by the easier reduction of the Pt vs Cu precursor, and the associated nucleation and growth, as increasing amounts of CuCl2 would result in an extended generation of Cu nuclei and thus the growth of larger particles [57]. Increased amounts of Cu in Cu/Pt (0.8) in comparison to Cu/Pt (0.5) is also noticeable by EDX analysis (Fig. S6).
Influence of Cu/Pt ratios on electrocatalytic activities
The electrocatalytic activity for the HOR of the synthesized catalysts with different Cu/Pt ratios is summarised in Fig. 6a and S7, and a comparison with the commercial catalyst is also made. The properties of this commercial catalyst are further discussed in the supplementary section (Figure S3–4 and S7–9). All the Cu/Pt catalysts performance for HOR was found to be superior, 2.4-folds over commercial catalyst (Fig. 6a and S7). The electrochemical surface area (ECSA) of all the catalysts is shown in Fig. S8a, and the ECSA of Pt/Cu (0.5) is marginally higher (89 m2 g−1) than Pt (85 m2 g−1).

Activity comparison of the synthesized catalysts at various Cu/Pt ratios, a HOR activity @ 0.2 V vs SHE, b ORR activity @ 0.85 V vs SHE. The activity value is from LSV in 0.1 M aqueous HClO4 under saturated H2 (a) and O2 (b) at the scan rate of 10 mV s−1 @1600 rpm, shown in Fig. S7d and S9d, catalyst loading was 50 µg cm−2 for all samples
For the ORR activity, the maximum activity was obtained with Cu/Pt (0.5) as shown in Fig. 6b and Fig. S9d. However, with increasing Cu amounts (> 0.5) the ORR activity was found to decrease. It is known that the desired ORR path should involve four-electrons (Eq. 1). However, the two-electron path is also possible during the dissociative chemisorption of O2 on Pt (Scheme 1). For the four-electron path (E° = 1.229 V vs SHE), O2 adsorption should be in the bridge configuration (Scheme 1a–c). This may explain the decrease in ORR activity observed with increasing Cu amounts because O2 bridges across neighbour Pt atoms would not be possible due to presence of Cu atoms (Scheme 1b–c) [1, 63]. The mass activity of all the catalysts at 0.9 V vs SHE is shown Fig. S8b, and the mass activity of Cu/Pt (0.8) was found to be superior (297 mA mg Pt−1) to that of Pt (151 mA mg Pt−1) at 0.9 V vs SHE.

This was also confirmed by calculating the number of electrons transferred per O2 molecule during ORR. The detailed calculation is discussed in the supporting information (Fig. S10). The number of electrons involved during ORR decreased from 3.9 to 3.3 after increasing Cu to Pt ratio from 0.5 to 0.8 (Fig. 7). This means that on Cu/Pt (0.5), a four electrons dissociative O2 chemisorption can be assumed to occur, while on Cu/Pt (0.8) it is possible that the reaction may also occur along the two electrons path [1, 63].

Comparison of number of electrons transferred during ORR by the synthesized catalyst at various Cu/Pt ratios, the calculation is done by Koutecky-Levich (K-L) method and is discussed in the supplementary section and Fig. S10
Testing on self-breathing PEMC
To determine the effectiveness of the catalysts developed, these were implemented in a 3D printed self-breathing PEMFC, and their activity was compared to that of a commercial Pt based catalyst. Based on the activity result of RDE, Cu/Pt (0.5) was found to be the best catalyst for both HOR and ORR in comparison to Pt. However, Cu/Pt (0.8) revealed to be a better HOR catalyst, but its ORR activity was significantly lower. Accordingly, fuel cell testing was done along three scenarios: (1) Cu/Pt (0.5) as the anode catalyst, (2) Cu/Pt (0.8) as the anode catalyst, and (3) Cu/Pt (0.5) as both the anode and the cathode catalyst.
In the initial configuration with Cu/Pt (0.5) as the anode catalyst and commercial Pt as the cathode catalyst, a maximum power density of 37.5 mW cm−2 was observed (Fig. S11). With Cu/Pt (0.8) as the anode catalyst and the commercial Pt as the cathode catalyst, a similar maximum power density of 40.37 mW cm−2 was observed (Fig. S11). The result corresponds to the result observed at RDE (Fig. 6) where the activity of Cu/Pt (0.8) is marginally higher than Cu/Pt (0.5). Both these power densities were superior to the power density (32.46 mW cm−2) observed with the commercial Pt catalyst as both the anode and the cathode (Fig. S11), in agreement with the observation made by RDE measurements.
Given the similarities in power density observed for both Cu/Pt (0.5) and Cu/Pt (0.8), a cell was made with Cu/Pt (0.5) at both the anode and the cathode (Fig. 8a–b). In this case, a superior power density of 45.16 mW cm−2 was observed in comparison to the 32.46 mW cm−2 obtained with commercial Pt. In addition to variable loads testing, the self-breathing PEMFC with Cu/Pt (0.5) at both the anode and cathode was operated under a constant load of 90 mA cm−2 to assess its initial stability, and throughout the operation a stable output voltage was observed (Fig. 9). It is thus apparent that the catalyst developed with 22% less Pt is a far superior catalyst for self-breathing PEMFC operation.

Performance of Cu/Pt (0.5) and Pt* catalysts in self-breathing PEMFC, a Current density vs voltage, b Current density vs power density, catalyst loading at the active area of both anode and cathode was same, i.e. 0.8 mg cm−2 for both Cu/Pt (0.5) and Pt* catalysts

Operation of self-breathing PEMFC for assessing the initial stability of the Cu/Pt (0.5) catalyst under a constant load @ 90 mA cm−2
To determine any early degradation of the Cu/Pt (0.5) catalyst during operation and confirm the stability test, the cell was disassembled, and the catalyst was removed for characterisation. Analysis by XRD showed similar diffraction peaks to the as-synthesised catalyst (Fig. 10).

XRD of Nafion coated with Cu/Pt (0.5) after self-breathing PEMFC operation for 300 min
Similarly, TEM analysis of the Cu/Pt (0.5) catalyst after fuel cell operation revealed uniformly dispersed particles on VC (Fig. 11) with a mean particle size of ~ 3 nm and no observable particle agglomeration or leaching (Fig. 11), which indicates that the catalyst was stable for the duration of the fuel cell test. Furthermore, there was no change in the d spacing of 0.224 nm, which corresponds to Pt7Cu as determined by HRTEM (Fig. 11c–d), in agreement with the XRD analysis. Additional STEM analysis and elemental mapping (EDS) confirmed the uniformed distribution of Pt, Cu on the carbon substrate with Pt and Cu “co-located” on the VC support. Accordingly, no significant alteration of the catalyst was observed during this initial test. This was also confirmed by the EDX analysis (Fig. S12) where the ratio of Cu and Pt is similar to the one obtained before fuel cell testing (Fig. S6a). This demonstrates the superiority of PtCu catalysts as compared to state of art Pt/VC. Indeed, other transition metals alloyed with Pt, e.g. Ni and Co, have been reported to leach out from the Pt alloy structure during the initial hours (< 5 h) of operation [64,65,66].

TEM after self-breathing PEMFC operation, a, b HRTEM images c SAD, d EDS MAP of the Cu/Pt (0.5) catalyst
Conclusions
PtCu alloys on VC were successfully synthesized by using the polyol process with ethylene glycol as a reducing agent. The obtained particles were highly dispersed on VC, with a particle size varying from 2 to 4 nm with increasing Cu to Pt ratios from 0 to 1.1. The catalyst Cu/Pt (0.5) delivered superior HOR and ORR activity in comparison to Pt only. Further analysis revealed that Cu/Pt (0.5) facilitated a four-electron path which does not involve in the formation of H2O2 but facilitates formation of H2O. This also results in higher output voltage. This promising Cu/Pt (0.5) catalyst was further evaluated in a self-breathing PEMFC. Much improved performance over commercial Pt based catalyst was obtained with a power density of 45.16 instead of 32.46 mW cm−2, and a good initial stability of the catalyst as observed by TEM analysis. In addition, incorporation of Cu reduced Pt content which significantly reduces the cost. By enabling the developed catalysts for both the anode and cathode, this work opens new avenues for enabling self-breathing PEMFC for small power applications.
References
P. Sapkota, C. Boyer, R. Dutta, C. Cazorla, K.-F. Aguey-Zinsou, Sustain Energy Fuels. 4, 2 (2020)
T. Reshetenko, V. Laue, U. Krewer, K. Artyushkova, J. Power Sources 458, 228032 (2020)
A. Caglar, T.A. Hansu, O. Sahin, H. Kivrak, Energy Storage (2021). https://doi.org/10.1002/est2.301
E. Higuchi, K. Okamoto, K. Miyatake, H. Uchida, M. Watanabe, Res. Chem. Intermed. 32, 5 (2006)
P. Sapkota, H. Kim, J Ind Eng Chem. 15, 4 (2009)
A. Oshima, T. Miura, S. Asano, S. Ichizuri, J. Li, S. Ikeda, M. Iida, C. Matsuura, Y. Tabata, Y. Katsumura, M. Washio, Res. Chem. Intermed. 31, 7 (2005)
D. Łomot, Z. Karpiński, Res. Chem. Intermed. 41, 12 (2015)
M. Shao, A. Peles, K. Shoemaker, Nano Lett. 11, 9 (2011)
D. Banham, J. Zou, S. Mukerjee, Z. Liu, D. Yang, Y. Zhang, Y. Peng, A. Dong, J. Power Sources 490, 229515 (2021)
M.B. Sassin, Y. Garsany, B.D. Gould, K.E. Swider-Lyons, Anal. Chem. 89, 1 (2017)
D. Banham, S.Y. Ye, ACS Energy Lett. 2, 3 (2017)
H. Gharibi, N. Dalir, M. Jafari, M.J. Parnian, M. Zhiani, Appl. Surf. Sci. 572, 151367 (2022)
T. Marshall-Roth, N.J. Libretto, A.T. Wrobel, K.J. Anderton, M.L. Pegis, N.D. Ricke, T.V. Voorhis, J.T. Miller, Y. Surendranath, Nat. Commun. 11, 1 (2020)
R. Sirirak, B. Jarulertwathana, V. Laokawee, W. Susingrat, T. Sarakonsri, Res. Chem. Intermed. 43, 5 (2016)
G. Ren, S. Chen, J. Zhang, N. Zhang, C. Jiao, H. Qiu, C. Liu, H.-L. Wang, J. Mater. Chem. A. 9, 9 (2021)
A. Mehmood, B. Ali, M. Gong, M.G. Kim, J.Y. Kim, J.H. Bae, A. Kucernak, Y.M. Kang, K.W. Nam, J. Colloid Interface Sci. 596, 148 (2021)
F. Tzorbatzoglou, A. Brouzgou, S. Jing, Y. Wang, S. Song, P. Tsiakaras, Int. J. Hydrog. Energy. 43, 26 (2018)
Z.Q. Li, X.T. Deng, H.K. Zhou, W. Xuan, Z.Y. Xie, F. Liu, J Solid State Electrochem. 24, 1 (2020)
H. Wang, S. Yin, Y. Xu, X. Li, A.A. Alshehri, Y. Yamauchi, H. Xue, Y.V. Kaneti, L. Wang, J. Mater. Chem. A. 6, 18 (2018)
E. Higuchi, K. Adachi, S. Nohara, H. Inoue, Res. Chem. Intermed. 35, 8 (2009)
M. Shao, Q. Chang, J.P. Dodelet, R. Chenitz, Chem. Rev. 116, 6 (2016)
Z.P. Wu, D.T. Caracciolo, Y. Maswadeh, J. Wen, Z. Kong, S. Shan, J.A. Vargas, S. Yan, E. Hopkins, K. Park, A. Sharma, Y. Ren, V. Petkov, L. Wang, C.J. Zhong, Nat. Commun. 12, 1 (2021)
C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H.L. Xin, J.D. Snyder, D. Li, J.A. Herron, M. Mavrikakis, M. Chi, K.L. More, Y. Li, N.M. Markovic, G.A. Somorjai, P. Yang, V.R. Stamenkovic, Science 343, 6177 (2014)
J. Durst, M. Lopez-Haro, L. Dubau, M. Chatenet, Y. Soldo-Olivier, L. Guetaz, P. Bayle-Guillemaud, F. Maillard, J. Phys. Chem. Lett. 5, 3 (2014)
M. Shao, J.H. Odell, A. Peles, D. Su, Chem. Commun. 50, 17 (2014)
T. Kim, Y. Kwon, S. Kwon, J.G. Seo, ACS Omega 5, 41 (2020)
F. Fievet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J.Y. Piquemal, L. Sicard, G. Viau, Chem. Soc. Rev. 47, 14 (2018)
A. Bhagi-Damodaran, M.A. Michael, Q. Zhu, J. Reed, B.A. Sandoval, E.N. Mirts, S. Chakraborty, P. Moenne-Loccoz, Y. Zhang, Y. Lu, Nat. Chem. 9, 3 (2017)
L.T. Cui, L.R. Cui, Z.J. Li, J. Zhang, H.N. Wang, S.F. Lu, Y. Xiang, J. Mater. Chem. A. 7, 28 (2019)
S.M. Alia, B.S. Pivovar, Y. Yan, J. Am. Chem. Soc. 135, 36 (2013)
D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A.P. Paulikas, V.R. Stamenkovic, N.M. Markovic, Nat. Chem. 5, 4 (2013)
Y. Zhang, D.P. Wilkinson, F. Taghipour, Int. J. Energy Res. 45, 6 (2021)
D. Wu, X. Shen, Y. Pan, L. Yao, Z. Peng, ChemNanoMat. 6, 1 (2019)
S. Ott, A. Orfanidi, H. Schmies, B. Anke, H.N. Nong, J. Hubner, U. Gernert, M. Gliech, M. Lerch, P. Strasser, Nat. Mater. 19, 1 (2020)
S.G. Ji, H. Kim, C. Park, W. Kim, C.H. Choi, ACS Catal. 10, 18 (2020)
R. Chen, C. Yang, W. Cai, H.-Y. Wang, J. Miao, L. Zhang, S. Chen, B. Liu, ACS Energy Lett. 2, 5 (2017)
J. Lee, J.H. Bang, ACS Energy Lett. 5, 8 (2020)
K.J.J. Mayrhofer, D. Strmcnik, B.B. Blizanac, V. Stamenkovic, M. Arenz, N.M. Markovic, Electrochim. Acta 53, 7 (2008)
D. Voiry, M. Chhowalla, Y. Gogotsi, N.A. Kotov, Y. Li, R.M. Penner, R.E. Schaak, P.S. Weiss, ACS Nano 12, 10 (2018)
D. Li, C. Batchelor-McAuley, R.G. Compton, Appl. Mater. Today 18, 100404 (2020)
W. Zheng, M. Liu, L.Y.S. Lee, ACS Energy Lett. 5, 10 (2020)
D.A. Stevens, J.R. Dahn, J. Electrochem. Soc. 150, 6 (2003)
J.M.D. Rodríguez, J.A.H. Melián, J.P. Peña, J. Chem. Educ. 77, 1195 (2000)
Y. Holade, N. Sahin, K. Servat, T. Napporn, K. Kokoh, Catalysts 5, 1 (2015)
L. Gan, H.-D. Du, B.-H. Li, F.-Y. Kang, New Carbon Mater. 25, 1 (2010)
L. Dennany, P. Sherrell, J. Chen, P.C. Innis, G.G. Wallace, A.I. Minett, Chem. Phys. 12, 16 (2010)
W. Wang, Z. Wang, J. Wang, C.J. Zhong, C.J. Liu, Adv. Sci. 4, 4 (2017)
Y. Karatas, H. Acidereli, M. Gulcan, F. Sen, Sci. Rep. 10, 1 (2020)
A.R. Cabral, R. Skála, A. Vymazalová, A. Kallistová, B. Lehmann, J. Jedwab, T. Sidorinová, Mineral. Mag. 78, 3 (2018)
S. Tahmasebi, A.A. McMath, J. van Drunen, G. Jerkiewicz, Electrocatalysis 8, 4 (2017)
S. Prass, J. St-Pierre, M. Klingele, K.A. Friedrich, N. Zamel, Electrocatalysis 12, 1 (2020)
J. Hu, K.A. Kuttiyiel, K. Sasaki, C. Zhang, R.R. Adzic, J. Electrochem. Soc. 165, 15 (2018)
X. Liu, C. Liang, W. Yang, C. Yang, J. Lin, X. Li, RSC Adv. 11, 21 (2021)
W. Kang, R. Li, D. Wei, S. Xu, S. Wei, H. Li, RSC Adv. 5, 114 (2015)
H. Guo, X. Liu, C. Bai, Y. Chen, L. Wang, M. Zheng, Q. Dong, D.L. Peng, Chemsuschem 8, 3 (2015)
M. Oezaslan, F. Hasché, P. Strasser, J. Electrochem. Soc. 159, 4 (2012)
Y. Kang, B. Jiang, Z.A. Alothman, A.Y. Badjah, M. Naushad, M. Habila, S. Wabaidur, J. Henzie, H. Li, Y. Yamauchi, Chemistry 25, 1 (2019)
Y. Li, Y. Jiang, M. Chen, H. Liao, R. Huang, Z. Zhou, N. Tian, S. Chen, S. Sun, Chem. Commun. 48, 76 (2012)
F. Bonet, C. Guéry, D. Guyomard, R.H. Urbina, K. Tekaia-Elhsissen, J.M. Tarascon, Int. J. Inorg. Chem. 1, 76 (1999)
Y. Chen, Z. Lai, X. Zhang, Z. Fan, Q. He, C. Tan, H. Zhang, Nat. Rev. Chem. 4, 5 (2020)
S. Mehmood, U.E. Klotz, G. Pottlacher, Metall. Mater. Trans. A 43, 13 (2012)
J.D. Giallonardo, G. Avramovic-Cingara, G. Palumbo, U. Erb, J. Mater. Sci. 48, 19 (2013)
S.A.S. Machado, A.A. Tanaka, E.R. Gonzalez, Electrochim. Acta 36, 8 (1991)
O.T. Holton, J.W. Stevenson, Platin. Met. Rev. 57, 4 (2013)
H.R. Colón-Mercado, B.N. Popov, J. Power Sources 155, 2 (2006)
H.R. Colon-Mercado, H. Kim, B.N. Popov, Electrochem. Commun. 6, 8 (2004)
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sapkota, P., Boyer, C., Lim, S. et al. High performing platinum—copper catalyst for self—breathing polymer electrolyte membrane fuel cell. Res Chem Intermed 48, 3019–3037 (2022). https://doi.org/10.1007/s11164-022-04734-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-022-04734-5