
Abstract
The carboxymethylation of cinnamylalcohol with dimethyl carbonate was performed using low-cost catalysts obtained from desulfurization slag. Processing of steel slag performed by different techniques was resulted in a wide range of the catalysts with different morphological and structural properties. Catalytic evaluation of the slag catalysts illustrated diversity of the obtained results strongly dependent on the surface area, crystal morphology and basicity. Catalytic materials demonstrated high variability of the conversion (8–85%) exhibiting similar selectivity to the desired product – cinnamyl methyl carbonate (ca. 80%). A significant impact of ultrasonication on catalytic activity was observed. Comparison of the synthesized samples with commercial basic materials illustrated competitive ability of the slag catalysts. Based on the results of catalytic evaluation and product analysis the reaction network was proposed and verified by thermodynamic analysis. A kinetic model was developed to describe concentration dependencies in carboxymethylation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Global environmental issues, limited natural resources as well as a need to mitigate a potentially negative impact of the chemical industry on humanity require development of systematic green approach in synthesis of valuable chemical products. An example of the process resulting in production of widely used compounds is synthesis of carbonate esters applied as green solvents, lubricating oils or fuel additives [1]. Previously used hazardous synthesis methods with utilization of phosgene [2, 3] were replaced by selective and catalytic processes applying a nontoxic reagent – Dimethyl carbonate (DMC).
Carboxymethylation of cinnamylalcohol (CA) with DMC (Fig. 1) as a green reactant and solvent produces cinnamyl methyl carbonate (product I), a valuable carbonate ester, as the main and the desired compound with some amounts of the side products [4]. Carbonate ester finds its application as an allylic electrophile affording the alkylated products in organic synthesis, namely the Tsuji–Trost reaction and asymmetric allylic substitution [5,6,7]. At the same time cinnamyl methyl carbonate can be utilized for phenol group protection [8] and used in synthesis of biologically active compounds [7, 9, 10].

Carboxymethylation of cinnamylalcohol with dimethyl carbonate
The reaction occurs at the temperature range 90–180 °C, where lower temperatures are used with inorganic oxides as catalysts, while higher ones are applied in the case of zeolites [11]. Numerous catalysts were applied for carboxymethylation of different alcohols with DMC including inorganic carbonates (K2CO3 [12]), oxides (nanocrystalline MgO, Al2O3, TiO2 and ZnO [13]), hydroxides (NaOH [4]) and basic supported catalysts (CsF/α-Al2O3 [12], NaAlO2/TiO2 [14]), hydrotalcite [4], sodium aluminate [4], homogeneous catalysts (triethylamine [4], ionic liquid [12]) or even zeolites (NaX and NaY [12]). Despite a wide utilization of catalysts exhibiting basic properties, some of the studies [15, 16] illustrated a positive effect of acidic catalysts – Brønsted and Lewis acids [15] or sulfonated mesoporous polymers [16], on conversion and selectivity to carboxymethylation products in carboximethylation of the alcohols and phenols with DMC.
Selection of the most cost effective, selective and environmentally friendly catalyst is an important step for the further industrial scale-up. Application of homogeneous catalysts is complicated by catalyst separation from the reaction products and their reusability, while utilization of the solid inorganic compounds allows achieving a high alcohol conversion together with a high product selectivity. However, it was found that some of inorganic oxides as well as such effective catalysts as K2CO3 [17] and NaAlO2 [18] are partially soluble in DMC implying de facto a homogeneous catalytic phase, which complicates the catalyst separation and contaminates the reaction products. Therefore, there is a clear need to develop efficient and stable heterogeneous catalysts, which synthesis and utilization will be responding to the zero waste principles simplifying separation of the catalysts from the reaction mixture.
The cost of the catalyst is another important factor for its application and industrial scale-up. Successful results were achieved in the reaction of glycerol with DMC over low-cost basic solid catalysts. Several works demonstrated a successful production of glycerol carbonate by in transesterification of glycerol and DMC over slag-based catalysts [19,20,21].
In particular interesting are low-cost slag catalysts obtained from desulfurization slag. This waste material representing a mixture of several oxides (CaO, SiO2, FeO, MnO, MgO and Al2O3) found its application in the building construction, CO2 capture, wastewater treatment and as a fertilizer [22,23,24,25,26]. At the same time, industrial ferrous waste, namely desulfurization slag, possesses high basicity mainly due to presence of basic oxides and, therefore, can be used as a feedstock for synthesis of basic catalysts. Thus, zeolite A [27], ZSM-5 [28] and hydroxyapatite–zeolite composite [29] were synthesized based on steel slag by several research groups.
Application of different treatment methods, especially variation of the treating agents for preparation of slag-based catalysts illustrated diversity of the catalytic properties depending on the treatment conditions [30,31,32]. Thus, low-cost catalytic materials treated with sodium hydroxide and ethylenediaminetetraacetic acid displayed cracking properties in the analytical pyrolysis of wood biomass enhancing conversion of the anhydrosugars to low molecular weight oxygenates [30, 31]. Utilization of the synthesized slag-based materials resulted in lowering the yields of the oxygen-containing compounds in the side chain structures of phenolic products, which was a clear evidence of potentially enhanced properties of the bio-oil. Application of ultrasound irradiation (US) showed that it had a high impact on crystallization kinetics [32]. Preliminary evaluation of the synthesized catalysts in carboxymethylation of CA with DMC also illustrated a significant impact of ultrasonication on catalytic activity [32].
The main reactant – cinnamylalcohol, is a fragrance compound applied in a wide range of fine fragrances and decorative cosmetics [33], as an important ingredient of pharmaceuticals production, namely Taxol [34] and Chloromycetin [35]. The main way of CA production is based on hydrogenation of cinnamaldehyde, a natural compound obtained from the bark of the cinnamon tree. Another interesting property of CA is its structure containing a propylene side chain with a terminal OH and similar to p-coumaryl alcohol, which allows consideration of its transformations as upgrading of the lignin-derived compounds into high-value chemicals [12].
Dimethyl carbonate is a well-known environmentally friendly agent obtained by oxidative carbonylation of methanol [36]. The chemical nature of DMC, namely a presence of carbonyl and the methyl groups, turns it into a compound with two reactive groups, promoting methylation if the nucleophile (Nu−) attacks the methyl carbon of DMC, and carboxymethylation if the nucleophile attacks at the carbonyl carbon with a cleavage the of acyl-oxygen bond (Fig. S1a) [2, 37, 38].
Moreover, DMC illustrates a selective behavior to different nucleophiles acting according to the Pearson HSAB (hard and soft acids and bases) principle (Fig. S1), where hard nucleophiles preferably react with hard electrophiles and vice versa [36]. Being a hard nucleophile [4, 12], cinnamylalcohol applied in the reaction with DMC results in both methylation and carboxymethylation products confirming the role of aromatic ring substituent in methylation and carboxymethylation reactions [39]. More precisely, softer phenoxide anions promotes methylation and harder ones allow both reactions (Fig. S1b).
As mentioned above preliminary evaluation of the synthesized slag materials demonstrated their potential applicability in the carboxymethylation of cinnamylalcohol with dimethyl carbonate [32]. The current work aims at a mechanistic investigation of this base-catalyzed reaction, elucidation of reaction kinetics and thermodynamics as well as analysis of catalytic activity and product distribution. The results were presented and analyzed for a broad range of catalysts synthesized by different methods and characterized by a wide range of analytical techniques. The physico-chemical and catalytic properties were correlated with each other. Efficiency of the slag-based catalysts was elucidated by comparing them with commercial materials. Stability of the synthesized slag catalysts was investigated by different methods.
Experimental
Catalyst preparation
Synthesis of the slag-based catalysts starting from the desulfurization steel slag provided by SSAB (Finland) is described in [32]. Only the pertinent details are thus presented here.
Catalytic materials used in the current study were synthesized by different methods upon variation of the treating agent type (distilled water, 0.6 M NaOH, 0.6 M HCl solution, 0.6 M EDTA (ethylenediaminetetraacetic acid) – 0.6 M NaOH mixture, 0.1 M TEAH (tetraethylammonium hydroxide) – 0.6 M NaOH mixture), US power (50 W, 80 W and 100 W) and time (4 h and 8 h), post-synthesis temperature (25 °C and 150 °C) and time (15 h and 48 h). One of the slag-based catalyst was produced by a multi-step synthesis with 1 M H3PO4 (Sigma-Aldrich) and 3 M NaOH, which was performed based on the procedure described in the literature [29]. The catalyst is designated in the work as “H3PO4–NaOH”.
Coding of the synthesized catalysts was done using the following principle. All sampled are marked according to the treating agent, US conditions (power, time) and post-synthesis conditions—mode of stirring (rotation (rot) vs stirring (st)), time and temperature (only in the case of hydrothermal synthesis). For example, the catalyst labeled as H2O US (80 W, 4 h) rot 48 h, 150 °C was synthesized using distilled water as a treating agent with ultrasonication for 4 h at power 80 W with the post-synthesis in the rotation mode for 48 h at 150 °C.
Properties of the synthesized slag-based catalysts evaluated by a wide range of analytical techniques are presented in the work [32].
Evaluation of catalytic properties
Carboxymethylation of cinnamylalcohol (98%, Sigma Aldrich) with dimethyl carbonate (99%, ReagentPlus®) used both as a reactant and a solvent (20:1 molar ratio of DMC to CA) was carried out in 300 mL batch reactor (Parr Instruments). Initial concentration of CA was 0.59 mol/L. The autoclave was equipped with a mechanical agitator and a sampling line with a 5 μm filter to prevent the catalyst withdrawal from the reactor during sampling. The reactor was surrounded by an electrical heater and contained a water cooling system to keep the desired temperature and prevent overheating. One day prior to the experiment the catalyst was placed into an oven preheated to 110 °C for moisture removal. The reactor was filled with CA dissolved in 100 mL of the solvent and the dried catalyst (1.18 g), sealed and flushed with argon (AGA, 99.999%) for 10 min to remove air from the reaction media. Thereafter, the autoclave was pressurized to 10 bar with argon and kept for 5 min for the leakage test. The temperature was increased to 150 °C with the ramping rate 5 °C/min and the final total pressure was increased to 16 bar. The conditions of experiments were selected based on extensive testing of slag catalysts in the temperature range of 90–180 °C. The stirring speed during the reaction was 540 rpm to prevent external mass transfer limitations. Stirring was initiated after reaching the desired temperature. The samples were periodically withdrawn from the reactor and analysed by GC (gas chromatography) equipped with a DB-1 column (30 m, 250 µm, 0.50 µm). The peaks were identified via GC–MS (gas chromatography–mass spectrometry, Agilent Technologies 5973 GC/MSD) equipped with a DB-1 column (30 m, 250 µm, 0.50 µm) and compared with the corresponding data for the neat compounds.
The following materials were used as a comparison with the synthesized slag-based catalysts: potassium carbonate (K2CO3, ReagentPlus®, 99%), hydrotalcite (Aldrich), magnesium oxide (MgO, Fluka, ≥ 98%), sodium aluminate (NaAlO2, BDH Chemicals Ltd, technical grade) and a sodium form of Y zeolite (HY (Si/Al = 5.1), Zeolyst International). Hydrotalcite was calcined prior to experiment at 600 °C [40]. The sodium form of Y zeolite was obtained by ion exchange of HY with 0.1 M NaNO3 during 24 h with a further calcination at 400 °C [41]. All reference catalysts were dried before experiments at 100 °C for 7 h.
Recycling experiments were performed by the same procedure described for carboxymethylation of CA with DMC using several catalysts selected for the two catalytic runs. After each experiment the catalyst was washed with acetone, dried in air at 100 °C and calcined at 400 °C for 4 h.
NMR analysis of the reaction mixture
The sample preparation prior to measurements consisted of the following steps. Approximately 0.5 mL of the reaction mixture was air blown until the majority of the dimethyl carbonate had been removed. The residue was dissolved in 600 µL CDCl3 (Chloroform-d). The samples were analyzed using a Bruker AVANCE III HD spectrometer, operating at 600.16 MHz (1H) and 150.9 MHz (13C) equipped with a TCI CryoProbe™ optimized for proton detection, and a Bruker AVANCE III spectrometer operating at 500.2 MHz (1H) and 125.8 MHz (13C) equipped with a Prodigy BBO CryoProbe™ optimized for carbon detection.
Inductively coupled plasma optical emission spectroscopy (ICP-OES)
Leaching of inorganic components from some of the slag catalysts into the reaction media was analyzed after 24 h experiments by inductively coupled plasma optical emission spectrometry (ICP-OES). The liquid samples for analysis were taken after filtration of the catalyst from the reaction media. The measurements were carried out using Optima 5X00™ DV ICP-OES spectrometer (PerkinElmer Inc.).
Hot filtration test
The hot filtration test was performed for verification of catalyst stability to leaching into the reaction solution. The reaction was carried out for 4 h using the catalyst pretreated with ultrasound with further interruption, cooling and separation of the reaction mixture from the solid catalyst. The separation was performed by centrifugation with the further filtration (PVDF syringe filter, 0.45 u). The filtrate was loaded into the autoclave, heated to 150 °C and run for another 20 h.
Analysis of the spent catalysts
The surface area and the pore volume of the spent catalysts were analyzed by nitrogen physisorption using MicroActive 3Flex™ 3500 (Micromeritics®). BET (Brunauer–Emmett–Teller) method and BJH (Barrett–Joyner–Halenda) method were applied for determination of the specific surface area and pore volume, respectively. Scanning and transmission electron microscopies were applied for detection of changes in crystal morphology and the internal structure, respectively. Scanning microscopy was performed using LEO Gemini 1530 Scanning Electron Microscope with a Thermo Scientific UltraDry Silicon drift detector (SDD). Internal structure was investigated using JEM-1400 Plus with 120 kV acceleration voltage and resolution of 0.38 nm equipped with OSIS Quemesa 11 Mpix bottom mounted digital camera.
Results and discussion
Catalyst characterization results
Detailed characterization of the synthesized slag catalysts is presented in [32] and only the main results are presented below. Most types of treatment led to an increase of the surface area in comparison with the initial material (6 m2/g). The surface area of the synthesized catalysts was varied in the range 7 – 64 m2/g. The highest impact on development of the surface area and pore volume was made by a multi-step synthesis with 1 M H3PO4 and 3 M NaOH, which was amounted to 64 m2/g and 0.26 cm3/g, respectively. Such effect was achieved by partial leaching of calcium observed by Energy dispersive X-ray analysis (EDXA).
Treatment conditions, especially the treating agent type, had a significant influence on the chemical composition of the synthesized materials [32]. The slag treatment mostly affected the calcium and sulfur content. Amount of the catalytic poison, sulfur, in the desulfurization slag was determined by EDXA reaching 3%. Processing of the industrial slag by different techniques contributed to sulfur reduction (ca. 1%). Application of ultrasonication and variation of its parameters did not have influence on the sulfur content in comparison with the treatment without ultrasonication. The most important parameter affecting the sulfur content was the treating agent type. The highest impact on the sulfur leaching was observed for the treatment with the surfactant – TEAH (ca. 0.3%) and multi-step synthesis with H3PO4 and NaOH, where the trace amounts of sulfur were observed. The treatment with water contributed to the sulfur reduction up to 0.8%, while the alkaline- and EDTA-treatment led to 0.35% and 0.55% of the sulfur content, respectively. Almost the same trends were observed in the case of calcium. The initial slag with a high calcium content (ca. 48%) underwent significant changes during the catalysts synthesis contributing morphology and basicity of the final product. Partial leaching of calcium resulted in the surface area increase giving access of the reactants to the active sites.
Treatment of the industrial slag had an effect on basicity of the synthesized catalysts as well. The original material has a relatively high basicity (75.6 μmol CO2/gcat) due to the presence of high amounts of basic oxides, which underwent significant transformations during the treatment attributed to recrystallization of the certain phases. Thus, utilization of distilled water as a treating agent and application ultrasonication for intensification of allows significant increase of basicity up to 136 μmol CO2/gcat. Hydrothermal synthesis as well as prolonged sonication time (8 h) have a negative impact on the basic sites concentration in comparison with synthesis at ambient conditions.
Synthesis with an alkaline solution resulted in a lower amount of the basic sites in comparison with water treatment. Such effect can be explained by higher leaching ability of the NaOH solution. Application of the intensification step as well as an increase of US power results in stronger leaching of slag components and a significant decrease of basicity. The same trend of basicity dependence from the leaching ability of the solvent was observed for EDTA-treated materials. Exhibiting a high leaching impact on calcium, EDTA contributed to its leaching from the original material two fold resulting in a shapeless morphology of the final catalysts and the lowest basic sites concentration as well. Utilization of the surfactant in the catalyst synthesis allows a slight increase of basic sites concentration. However, application of ultrasonication on different stages of the multi-step synthesis with EDTA and TEAH had a negative influence on basicity.
Processing of an industrial waste made a significant contribution in the textural transformations of the slag components. Changes in the crystal morphology of the synthesized slag catalysts in comparison with the neat slag demonstrated a clear dependence of the crystals type from the treatment conditions. Differences in the catalyst texture compared with the initial material occur due to dissolution of the original material with a subsequent recrystallization of the new phases under certain synthesis conditions. Investigation of the internal structure demonstrated formation of the pore and channel structure determined by TEM analysis.
Catalytic performance
Slag-based catalysts were evaluated and compared with the commercial materials exhibiting properties of solid bases in carboxymethylation of CA with DMC at 150 °C. Catalytic results, namely CA conversion after 24 h, are displayed in Table 1. Conversion curves as function of time, dependences of selectivity to desired product from CA conversion, product distribution and correlation between physico-chemical and catalytic properties of the synthesized materials are discussed below in different sections corresponding to respective treatment conditions.
Slag variation
First of all, carboxymethylation of CA with DMC was performed over desulfurization and granulated slags as well as without any catalyst. Conversion curves as a function of time are given in Fig. 2. A blank experiment showed ca. 10% of CA conversion with selectivity to the desired product of 69% after 24 h. The lowest activity was observed over the granulated slag exhibiting just 1% of conversion. Application of desulfurization slag as a catalyst allows conversion of 34% with a high selectivity to the desired product I (89% of selectivity at 30% of conversion) without its further transformations to the product III. Selectivity to p-methoxyprop-1-enylbenzene (product II) and other products was 1% and 10%, respectively.

Conversion of CA as a function of time over different slags at 150 °C
Water-treated catalysts
For slag catalysts synthesized using distilled water as a treating agent and ultrasound irradiation as an intensification tool, catalytic results are displayed in Table 1. Dependences of CA conversion from reaction time are presented in Fig. 3. A more detailed comparison of conversion dependencies is given in Fig. S2.

Conversion of CA as a function of time over the water-treated slag catalysts tested at 150 °C. Notation: H2O st 48 h (●); H2O rot 48 h, 150 °C (○); H2O US (50 W, 4 h) st 48 h (▼); H2O US (80 W, 4 h) st 48 h (∆); H2O US (100 W, 4 h) st 48 h (■); H2O US (50 W, 8 h) st 48 h (□); H2O US (50 W, 4 h) rot 48 h, 150 °C (♦); H2O US (100 W, 4 h) rot 48 h, 150 °C (◊)
Water-treated slag catalyst synthesized by stirring for 48 h exhibits the same conversion of CA as untreated desulfurization slag (34%) with a lower selectivity to cinnamyl methyl carbonate (84% vs 89% at 30% of conversion). Treatment at hydrothermal conditions (Table 1, entry 5) did not affect significantly the catalytic activity giving almost the same conversion and selectivity despite basicity and the surface area exceeding that of the catalyst synthesized at ambient conditions.
Comparison of conversion curves for these catalysts (Figs. 3 and S2b) illustrates a higher initial activity of the material treated at a higher temperature (Table 1, entry 5). Application of ultrasonication in production of the slag-based materials allows achievement of a higher conversion in comparison with the catalyst synthesized without US step (H2O st 48 h vs H2O US (50 W, 4 h) st 48 h). Despite significant differences in the catalytic behavior (Fig. 3) for all water-treated materials selectivity to the desired product was almost the same and varied in the range 82–85% (at 30% of CA conversion). An exception was the catalyst treated under prolonged US time (Table 1, entry 9) and the material obtained by hydrothermal synthesis after sonication at 50 W (Table 1, entry 10), which displayed the lowest conversion among the water-treated slag catalysts. A plausible explanation can be related to a low total basicity in the former and a low surface area in the latter case (Fig. S4) [32]. On the contrary, an increase of US power up to 100 W with a subsequent hydrothermal synthesis at the same conditions (Table 1, entry 11) resulted in a material with improved physico-chemical and catalytic properties.
The conversion curves (Fig. S2d) for the catalysts synthesized at high temperature (Table 1, entries 5, 10 and 11) display a gradual elevation of CA conversion over the catalyst synthesized without US as opposed to the material produced at 50 W of US, which exhibited low activity undergoing deactivation after 18 h. The catalysts synthesized via the hydrothermal treatment were inferior in activity to materials produced at ambient conditions. Most likely, this difference can be explained by predominance of the phases of Fe2O3 or TiO2 formed after hydrothermal synthesis, which are inactive in CA carboxymethylation [32].
An increase of the US power among catalysts synthesized via stirring at ambient conditions showed ambiguous results. Overall, these materials illustrate higher CA conversion than the one not exposed to US (Table 1, Fig. S2c). Conversion (37%) as well as selectivity to the product I (82% at 30% of CA conversion) were the lowest for the catalyst synthesized at 80 W, while the best result was achieved for the US power at 100 W exhibiting 76% of conversion and 85% of selectivity to the product I (at 30% of CA conversion). Catalytic activity of these three catalysts (Table 1, entries 6–8) cannot be solely attributed to the basicity and the surface area.
Dependences of selectivity to the product I on CA conversion are given in Fig. 4. For the most water-treated slag catalysts the character of the curve and selectivity levels are similar showing an increase of selectivity at the beginning of the reaction levelling off soon thereafter on displaying a maximum due to consecutive transformations. Elevation of selectivity to the desired product at the beginning of the reaction can be attributed to side transformations of CA and impurities presented in CA, such as hydrolysis of cinnamaldehyde to benzaldehyde. A decrease of the selectivity to product I at higher conversion levels is attributed to cinnamyl methyl carbonate consumption in the side reactions.

Dependence of selectivity to the desired product on CA conversion at 150 °C over water-treated slag catalysts. Notation: H2O st 48 h (●); H2O rot 48 h, 150 °C (○); H2O US (50 W, 4 h) st 48 h (▲); H2O US (80 W, 4 h) st 48 h (∆); H2O US (100 W, 4 h) st 48 h (■); H2O US (100 W, 4 h) rot 48 h, 150 °C (◊)
An exception was the material synthesized using ultrasonication with the post-synthesis at hydrothermal conditions avoiding formation of the side products at the beginning of the reaction. The catalyst synthesized at 100 W of US also exhibited the lowest formation of the side products.
More side products (products II, III and others) were observed with the material synthesized at 80 W (Fig. S3). All water-treated catalysts exhibited the same low selectivity to the product III and a moderate selectivity to the product II. The main difference between these catalysts was in formation of some undetected side products. As mentioned above, the material produced at 80 W of US displayed the highest selectivity to the side products, while the most active catalyst (Table 1, entry 8) showed the lowest amount of the side products.
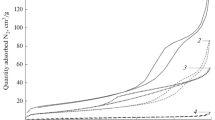
A straightforward correlation between the catalyst properties and behavior in the reaction can be difficult to establish because of the system complexity. An attempt of such correlation between CA conversion, the surface area and the total basicity of the water-treated slag catalysts is given in Fig. S4. The highest CA conversion was achieved for catalysts exhibiting both a high surface area and basicity. It should be noted that the amounts of weak and strong basic sites are similar for all water-treated slag catalysts [32]. The main difference is attributed to the medium basic sites probably influencing catalytic behavior.
Alkaline-treated catalysts
Dependence of CA conversion on reaction time over alkaline-treated slag catalysts is presented in Figs. 5 and S5.

Dependence of CA conversion on reaction time for alkaline-treated materials evaluated at 150 °C. Notation: NaOH st 48 h (●); NaOH rot 48 h, 150 °C (○); NaOH US (50 W, 4 h) st 48 h (▼); NaOH US (80 W, 4 h) st 48 h (∆); NaOH US (100 W, 4 h) st 48 h (■); NaOH (50 W, 4 h) rot 48 h, 150 °C (□)
Alkaline-treated slag catalysts showed a visible difference in the catalytic behavior compared to the water-treated ones (Fig. S5a). Exhibiting almost the same conversion after 24 h, the slag catalysts treated with an alkaline solution (Table 1, entry 12) displayed a sharp increase of conversion only after 15 h of the reaction (Fig. S5a). At the same time, water- and alkaline-treated materials synthesized at the high temperature showed significant differences in catalytic activity and obtained conversion levels (Fig. S5a). Therefore, the treatment with an alkaline solution and application of the high synthesis temperature resulted in a less active catalyst (Table 1, entry 13) than when water was used as a treating agent (entry 5). Such difference can be explained not only by a different crystal morphology, namely needle shaped crystals for water treatment and round shape with platelets for alkaline one [32], exhibiting different reactivity, but also basicity and access to active sites. Thus, two-fold higher basicity of the water-treated material resulted in a corresponding increase of conversion. The size of the pores is also playing an important role regulating access to the active sites. In the case of alkaline-treated sample, the pore size was two times lower.
Application of ultrasound irradiation gave a positive effect on activity for the treatment at both ambient and hydrothermal conditions (Figs. 5, S5b and S5c). Elevation of US power resulted in an increase of the conversion level going through a maximum at 80 W of US (Table 1, entries 14–16; Fig. S5b). Alkaline-treated catalysts did not exhibit a clear correlation between catalytic activity and material properties (Fig. S6). Thus, catalyst synthesized at 80 W of US and exhibited the highest activity, has the lowest basicity among the alkaline-treated catalysts synthesized with US step. The surface area and the pore volume of the catalysts were decreased with the US power [32]. Absence of a correlation with basicity and surface area implies that other factors, such as crystal morphology, phase distribution, or even their combination, also play an important role.
In the case of synthesis at hydrothermal conditions, ultrasonication allows to achieve a higher conversion than without an US step (Fig. S5c). The main differences between these catalysts were in basicity of the final materials and crystal morphology. For US-treated catalyst the crystalline structure consists of a mixture of round shape and needle shape crystals, while inactive agglomerates were formed in the absence of ultrasound [32].
Alkaline-treated catalysts afforded higher selectivity to the desired product than the water-treated counterparts exhibiting similar selectivity dependencies on CA conversion (Fig. S7). Selectivity to desired product I was varied in the range 84–87% (at 30% of CA conversion). The highest selectivity was achieved over the catalyst synthesized at 80 W of US also exhibiting the highest conversion (Table 1, entry 15) among alkaline-treated materials. Side products formation over catalysts synthesized via alkaline treatment (Fig. S8) keeps the same ratio between formed by-products as for water-treated slag catalysts.
EDTA- and surfactant-treated catalysts
Such types of treating agents as EDTA, HCl and surfactant-EDTA mixture gave catalysts with a broad range of activity (Table 1) and similar selectivity.
Thus, EDTA treatment influenced in a negative way catalytic properties due to high leaching of the catalytically active components during catalyst synthesis. No improvement was obtained implying the US treatment, contrary to water- and alkaline-treated materials (Fig. S9a). A decrease of the CA conversion level with synthesis time increase (Table 1, entries 19 and 20) can be linked to the calcium content and catalyst basicity [32].
Application of a surfactant (TEAH) after the EDTA-treatment had a positive effect not only on the physico-chemical properties of the materials, but also on catalytic activity. Structure of the surfactant-treated materials underwent significant transformations during the treatment consisting of crystal morphology changes and formation of internal channels improving the reactant access to the active sites [32]. In contrast to the EDTA-treated materials, TEAH catalysts possessed high activity (Table 1, entries 21–24) with a slightly increased selectivity to cinnamyl methyl carbonate (85% vs 83% over EDTA-treated catalyst). Interestingly, that application of US and its placement in the overall synthesis procedure had an influence on the catalytic activity and the shape of conversion curves (Fig. S9b). Thus, the material synthesized without US showed a gradual increase of cinnamylalcohol conversion with a reaching the maximum reaction rate between 7 and 13 h of reaction defined as a slope of conversion curves. Explanations for such catalytic behavior can be attributed to phase transformations or leaching of metals in the reaction media.
Application of US for both procedures involving EDTA and TEAH treatment steps changes the catalytic behavior. The same conversion and selectivity levels as for the material synthesized without ultrasonication were achieved for the catalyst TEAH-US EDTA-US (Table 1, entry 24) exhibiting a steady conversion increase (Fig. S9b). Utilization of ultrasound irradiation only on one step of the catalyst synthesis has a negative influence on catalytic activity. Synthesis with an US step for the procedure involving EDTA treatment resulted in a catalyst of a low activity (Table 1, entry 22), which is just slightly higher than for the sample synthesized without the TEAH treatment (entry 21). A positive influence on conversion was achieved by application of a surfactant for the EDTA pre-treated material. Utilization of US in the TEAH-treatment step led to a higher conversion – 52% (entry 23), than for the previously discussed material (entry 22). However, the conversion curve is similar to the one for the TEAH-treated material without an US step (Fig. S9b). EDTA- and the surfactant-treated catalysts showed a clear dependence of CA conversion on surface area and basicity increasing with elevation of both parameters (Fig. S10).
Utilization of the acid-treated sample (HCl st 48 h) resulted in moderate activity (entry 25; Fig. S9c) with a relatively low selectivity to the desired product (83% at 30% of CA conversion) in comparison with other slag-based catalysts. High basicity (87.1 μmol CO2/gcat) and a relatively high surface area (47 m2/g) of this material led to a relatively high conversion level (47%). However, selectivity to desired product – cinnamyl methyl carbonate, was lower (83% at 30% of conversion) in comparison with other slag-based catalysts. Such catalytic behavior is similar to other slag materials with amorphous morphology, e.g., EDTA-treated catalysts.
Significantly different catalytic behavior was observed using the catalyst synthesized by a multi-step treatment with H3PO4 and NaOH (entry 26), which afforded the highest conversion (85%) and selectivity (89% at 30% conversion). Exhibiting the highest surface area (64 m2/g) and the pore volume (0.26 cm3/g) among all slag materials, this catalyst synthesized by combined H3PO4–NaOH treatment displays a relatively low basicity (29.4 μmol CO2/gcat). However, a unique internal structure consisting of well-developed pores with an average size of 13 nm facilitates an efficient access of the reactants to the active sites [32]. This material displayed a high reaction rate at the beginning and achieving a steady plateau after 8 h (Fig. S9c). Such dependence can be related to either thermodynamics addressed below, or even deactivation, namely coking by the reaction products, which blocks an access to the active sites and is manifested by a decrease of the surface area in the spent catalyst.
Dependence of selectivity to the desired product on conversion over EDTA- and TEAH-treated materials has a similar character as for the alkaline- and water-treated ones (Fig. S11). In the case of the synthesis with EDTA, selectivity to the product I decreases with conversion, while for TEAH catalysts side products were formed from the beginning. Application of US contributed to avoiding the side product formation at the beginning of the reaction. Distribution of the side products (Fig. S12) was also similar for water- and alkaline-treated catalysts.
Comparison of the most promising slag-based materials with the reference catalysts
The most promising low-cost slag materials were compared with the commercially available catalysts exhibiting base properties. Cinnamylalcohol conversion as well as selectivity to the product I at 30% and 60% of CA conversion are presented in Table 2.
Table 2 and Fig. 9 illustrate that these materials display diverse results. For example, MgO gave the lowest conversion among commercial and slag catalysts (Table 2, entry 29). A slightly better conversion was achieved using NaY zeolite (entry 31). This material showed the lowest selectivity to the desired product despite exhibiting a moderate basicity (38 μmol CO2/gcat) en par with the H3PO4–NaOH slag catalyst. In this case, the product II and other side products were formed similar to the results reported in [12]. The highest conversion and selectivity levels among commercial catalysts were achieved over K2CO3, hydrotalcite and NaAlO2.
Application of the potassium carbonate afforded 92% conversion and 81% selectivity to product I after 24 h (Table 2, entry 27) being somewhat different from [12], where 98% conversion and 82% selectivity at 165 °C in 3 h were reported. However, a clear disadvantage of this material is high solubility in the reaction media implying homogeneous rather that heterogeneous catalysis. Even a visual inspection of the catalyst amounts before and after the experiment showed a significant decrease of the initial catalyst weight undermining potential applicability and reusability of this material. The same effect was observed for NaAlO2 displaying moderate solubility into the reaction media explaining high reactivity at the beginning of the process with a decline in activity after 5 h (entry 30). Leaching of NaAlO2 into the solution was also proved by ICP analysis of the liquid sample after filtration of the catalyst (0.12 mg Al/L; 0.01 mg Na/L). The most suitable for carboxymethylation of CA with DMC among tested commercial materials is hydrotalcite exhibiting high conversion (86%) and selectivity to the desired product (82% at 30% of CA conversion). Comparison of hydrotalcite with NaAlO2 was also performed in [4] at 90 °C with a DMC to the substrate ratio lower (i.e., 1–10) than in the current work. A low activity (40% conversion) of calcined hydrotalcite together with a high selectivity to cinnamyl methyl carbonate (99%) was reported in [4], while experiments with sodium aluminate resulted in 76% conversion and 99% selectivity. A direct comparison between these results is not straightforward taking into account different reaction conditions.
Such active and effective catalytic material as hydrotalcite did not find a broad industrial application, which can be related to fast poisoning under air, impracticable reusability and time-consuming regeneration as reported in [42].
Some of the synthesized slag catalysts can compete with commercial materials in selectivity to the product I. A lower activity of the slag materials in comparison with potassium carbonate and hydrotalcite is compensated by high selectivity to cinnamyl methyl carbonate (up to 89%). Surface area, basicity, texture and crystal morphology influence catalytic behavior of the slag materials. Thus, the same catalytic behavior of H3PO4–NaOH and the alkaline-treated catalysts consisting of a sharp increase of activity at the beginning of the reaction with further deactivation by coking is a different from the water- and surfactant-treated materials showing a gradual elevation of CA conversion (Fig. 6). Dependence of selectivity to the desired product on conversion over the most active slag catalysts exhibited the same trends as the water- and alkaline-treated materials (Fig. 7).

Conversion of CA as a function of time over commercial materials and the most active slag-based catalysts at 150 °C

Selectivity to cinnamyl methyl carbonate as a function of CA conversion over commercial materials and the most active slag-based catalysts evaluated at 150 °C
Contrary to the alkaline- and TEAH-treated catalysts H3PO4–NaOH treated material exhibited constant selectivity up to 75% of CA conversion. The catalyst synthesized by a treatment with H3PO4 and NaOH was in this sense rather exceptional in terms of its selectivity vs conversion behavior. It has relatively a high surface area, low basicity and a unique crystal morphology in comparison with other slag materials. Further work is required to fully explore the origin of its performance.
The catalyst treated with water displayed less prominent cinnamyl methyl carbonate consumption, while for other catalysts there was a sharper decrease of selectivity to the desired product during the reaction. Selectivity dependence on conversion for the commercial materials is significantly different from the slag catalysts. Thus, hydrotalcite gave a sharp decrease of selectivity to the desired product attributed to its subsequent transformation into the side products, including coking and polymerization. An unusual parabolic curve with a maximum selectivity (91%) at 46% of conversion follows from Fig. 7 for sodium aluminate. Potassium carbonate exhibited a sharp increase of selectivity with conversion. Distribution of the side products is similar to previously discussed catalysts treated with water or an alkaline solution (Figs. S13a and S13b). An exception was potassium carbonate exhibiting the highest selectivity to the product III (Fig. S13b). Apparently, further studies are needed to elucidate the reaction pathways for these materials.
Kinetic curves for the most active slag catalysts and some commercial materials are presented in Figs. 8 and S14, S15, S16. Water- (Fig. 8a) and surfactant-treated materials (Fig. S15) exhibited similar behavior with a gradual consumption of CA. For the catalysts treated with H3PO4–NaOH (Fig. 8b) and alkaline solution (Fig. S14) there was a constant increase of CA conversion with a sharp rise after a certain value. The final steady plateau typically indicates either catalyst deactivation by coking or thermodynamic limitations, which will be addressed in Sect. Thermodynamic analysis of carboxymethylation reaction.

Kinetic curves of CA carboxymethylation with DMC at 150° over the catalysts: a H2O US (100 W, 4 h) st 48 h; b H3PO4 –NaOH treated slag
Commercial materials – K2CO3 and NaAlO2, displayed catalytic behavior different from slag catalysts illustrated in Fig. S16. Application of sodium aluminate resulted in a smoother character of kinetic curves, while utilization of potassium carbonate is characterized by a sharp increase of CA consumption levelling off after 7 h.
Reaction network
Analysis of the product mixture after the reaction time of 24 h was performed by NMR resulting in the reaction scheme comprising different side reactions as presented in Fig. 9 and Table S1. Carboxymethylation of cinnamylalcohol (I) with dimethyl carbonate generates cinnamyl methyl carbonate (II) as the main product. Synthesis of carbonate ester is accompanied by its further reaction with cinnamylalcohol (reaction 2) observed over different base catalysts [4]. The extent of this reaction is certainly limited as cinnamylalcohol has much higher chances to react with DMC present in excess rather than with cinnamyl methyl carbonate (II).

The reaction network
Methanol formed as a side product of the main process (reaction 1) reacts with cinnamylalcohol giving p-methoxypropenylbenzene (IV) and water (reaction 3) [12]. At the same time, this compound IV can be obtained by in situ decarboxylation of methyl cinnamyl carbonate (reaction 4) [10, 12] or by methylation of CA. The reaction pathway of p-methoxypropenylbenzene formation is directly influenced by the strength of the basic sites with less basic sites favoring methylation of CA [12]. Complex chemical composition of the slag catalysts may promote undesired side reactions such as, for example, double bond migration in cinnamylalcohol catalyzed by basic sites of alumina (reaction 5) [43, 44]. Dehydrogenation of cinnamylalcohol to cinnamaldehyde can occur on the acid–base surface of alumina (reaction 6) [44,45,46]. Hydrogen formed in the reaction 6 is consumed in double bond hydrogenation of cinnamylalcohol (reactions 8 and10) to 3-phenylpropanol (VIII) and carbonate ester to carbonic acid methylhydrocinnamyl ester (IX). Formation of carbonic acid methylhydrocinnamyl ester can also occur by the reaction of DMC with 3-phenylpropanol (reaction 9) [47, 48]. Cinnamaldehyde obtained in the reaction 6 undergoes hydrolysis over basic sites of slag catalysts (reaction 7) with formation of benzaldehyde (VII) [46, 49].
The base nature of the catalysts results in allyl rearrangement of cinnamylalcohol into vinylbenzyl alcohol (reaction 11), which is a backward reaction of the one reported in [50]. Obtained vinylbenzyl alcohol reacts with preferably DMC due to its excess and also with formed methanol according to reactions 12 and 13, respectively.
Thermodynamic analysis of carboxymethylation reaction
Evaluation of possible reaction pathways was performed by thermodynamic analysis, which was implemented based on the enthalpy (ΔH0r) and Gibbs free energy (ΔG0r) for each reaction (j) using the thermodynamic approach presented in [51]. Enthalpy and Gibbs free energy at standard conditions for each reaction (j) at different temperatures (Tmin = 373.15 K; Tmax = 473.15 K) were computed from the standard enthalpy (ΔH0f) and Gibbs free energy (ΔG0f) of formation from the elements estimated with the Joback approach [52,53,54]:
The equilibrium constant of each reaction was calculated through the Gibbs energy by:
The temperature dependence of the free Gibbs energy for different reactions was calculated by implementing the Gibbs–Helmholtz equation at P = 1 bar (ΔGΦr,j):
The calculation results are presented in Figs. 10 (reactions 1–4) and 11 (reactions 5–13), and Table S2. The stochiometric matrix was built based on the reaction scheme and reported in Table S3. As can be seen from the thermodynamic analysis, there are some thermodynamic limitations in cinnamyl methyl carbonate synthesis and its further reaction with CA, which are less prominent upon temperature elevation (Fig. 10; Table S2). It was suggested that p-methoxypropenylbenzene (product II) can be formed from either cinnamylalcohol (reaction 3) or carbonate ester (reaction 4). Thermodynamic analysis does not support former suggestion considering high Gibbs free energy values for reaction 3, while formation of product II from cinnamyl methyl carbonate is thermodynamically favored (Fig. 10). Nevertheless, the product II can be formed from CA at the beginning of the reaction even before heating. Migration of the double bond in cinnamylalcohol (reaction 5) is thermodynamically allowed, while dehydrogenation of cinnamylalcohol to cinnamaldehyde (reaction 5) is not feasible. Benzaldehyde formation is possible (reaction 7) originating from cinnamaldehyde present in CA as an impurity.

The Gibbs free energy as a function of temperature for the reactions 1–4 from Table S2

The Gibbs free energy as a function of temperature for the reactions 5–13 from Table S2
Reactions 8 and 10, namely hydrogenation of CA double bond to 3-phenylpropanol and the carbonate ester transformation into the carbonic acid methylhydrocinnamyl ester, respectively, are thermodynamically feasible being favored upon temperature elevation (Fig. 11). Reaction of DMC with 3-phenylpropanol (reaction 9) is thermodynamically allowed, but to a lesser extent than reaction 10. An interesting case is reaction 11. As follows from Fig. 11 and Table S2, reactions 12 and 13 are not prohibited by thermodynamics giving products which were found in reaction mixture by NMR analysis. However, the reaction leading to these products (reaction 11) is not thermodynamically favored, which can be explained by a fast reaction of vinylbenzyl alcohol formed in the reaction 11 with DMC and methanol by reactions 12 and 13, respectively.
Verification of thermodynamical limitations for the main reaction resulting in the desired product – cinnamyl methyl carbonate, was performed using a homogeneous catalyst – triethylamine (Alfa Aesar®, 99%). The main advantages of this catalyst are not only absence of catalyst deactivation by coking and internal diffusion limitations, but also high activity. A high conversion of CA (ca. 95%) over triethylamine was achieved in 2 h remaining for the subsequent 8 h in line with thermodynamic limitations of the CA carboxymethylation reaction.
Thus, along with coking of the catalyst surface, thermodynamic limitations are responsible for levelling off kinetic curves visible for some catalysts (Fig. 8b and c).
Analysis of the spent catalysts
The spent catalysts were analysed by nitrogen physisorption, scanning and transmission electron microscopies. The surface area and the pore volume of the spent catalysts are presented in Table S4 and compared with the corresponding values for the fresh materials taken from [32]. Desulfurization steel slag as well as majority of the synthesized catalysts displayed a decrease of the specific surface area and the pore volume indicating strong adsorption of organic compounds or deposition of coke blocking the pores.
The crystal morphology as well as internal structure of the treated slag materials did not exhibit significant transformations during catalysis (Figs. S17, S18). Opposite to the synthesized catalysts, analysis of the spent untreated slag by TEM illustrated formation of the pores and metal oxide particles (Fig. S19). The synthesized slag catalysts kept the texture (Figs. S20, S21) exhibiting only a slight increase of the pore size after the reaction (Table S5) being still with the measurement errors [32].
Analysis of the recycled catalysts confirmed stability of the textural characteristics and the crystal morphology of the slag materials after two recycles (Figs. S22, S23, S24). An exception was the surfactant-treated material for which formation of the metal oxide particles after the second recycle was noticed (Fig. S23d). The structural properties of the investigated catalysts were changed due to carbon deposition in the pores and on the catalytic surface influencing the surface area and the pore volume.
Stability of the catalysts in the reaction media
Leaching of the catalyst components into the reaction media was verified by a hot filtration test (HFT) and ICP-OES analysis of some catalysts. According to the ICP analysis of the most active slag catalysts there was no leaching into the solution. However, presence of some leached metals in the amounts lower than the detection limits cannot be excluded. The hot filtration test was performed for the catalyst H2O US (100 W, 4 h) st 48 h also confirming a heterogeneous nature of the reaction. The results obtained in the HFT test were compared with the experiment performed with the catalyst during 24 h. Due to a non-catalytic reaction verified by a blank experiment (Fig. 2) the results of HFT were also compared with a thermal reaction after catalyst removal from the reaction media. A comparison of HFT, a blank and a catalytic experiment of the water-treated catalyst is illustrated in Fig. S25.
As can be seen from Fig. S25, results for the catalytic experiment and the initial phase of the HFT performed also with the catalyst coincide within the measurement errors. When the catalyst in the HFT was removed from the reaction media after 4 h followed by continuation of this experiment without any catalyst, the catalytic behavior was essentially the same as in the case of a thermal reaction. Conversion curves after catalyst filtration and data for the blank experiment overlap confirming absence of any leaching of the slag components into the solution.
Recycling of the catalyst
Reusability of the slag materials was evaluated in three cycles (Fig. 12, Table S6), where the zero cycle is attributed to the fresh catalyst, and “1” and “2” correspond to activity of once and twice used and regenerated materials. Regeneration of the spent catalysts was performed by washing with acetone, drying in air at 110 °C and calcination at 400 °C for 4 h. As can be seen from Fig. 12, the slag catalysts exhibited a loss of activity after the first run with a further decrease in the second recycle. Due to stability of the textural and crystallographic characteristics after recycling the most plausible explanation of such catalytic behavior is on incomplete regeneration of the spent catalysts. Another reason of activity loss can be formation of some chemical compounds from the catalyst components and the reaction products. Such effect was observed in the work of Okoye et al. [20], where deactivation of the slag-based materials used in glycerol transesterification reaction with DMC was ascribed to transformations of the active component of the catalyst (CaO) into calcium diglyceroxide.

Changes in conversion during recycling tests with the slag catalysts at 150 °C: A H2O US (100 W, 4 h) st 48 h; B TEAH-US EDTA-US; C H3PO4–NaOH treated slag
A link between catalyst deactivation and an access to the active site can be clearly seen from analysis of the surface area of the regenerated and spent catalysts. As can be seen from Table S6 displaying textural properties and catalytic behaviour during recycling tests lower activity corresponds to decrease of the surface area. Comparison of the spent catalysts with the regenerated ones clearly indicates that further research is needed to develop a more efficient regeneration procedure. An exception is the surfactant-treated material, which showed significant activity loss from the first to the second recycle. The loss in catalytic activity can be attributed not only to coking, but also to morphological changes, namely formation of the metal oxide particles (Fig. S23d).
Kinetic modelling
The concentration dependencies demonstrating an induction period in the carboxymethylation of cinnamylalcohol with DMC provide a strong argument in favor of using a concept of two catalytically active sites. The same approach was previously applied [55] for methylation of aniline with DMC as well where catalyst inactivation was also included in the reaction mechanism. A simplified reaction scheme is presented in Fig. 13 illustrating formation of active sites of type B, their inactivation and a reversible reaction of cinnamylalcohol with DMC. For the sake of simplicity other products formed starting from cinnamylalcohol were lumped into one pseudo-component.

A simplified reaction network used in kinetic modelling
In general, the reaction can proceed with both catalytic species A and B, however, a preliminary data fitting has demonstrated that only reactions on sites of type B are sufficient for modelling the concentration dependencies at least from the numerical viewpoint.
In the kinetic treatment of the present work the concentration of catalytic species with time can be expressed
With
while the concentration of inactivated species \(c_{{cat_{Bd} }}\) is
The kinetic equation for inactivation of species of type B is given by
Concentrations of all catalytic species are related through the mass balance \(c_{{cat_{C} }} = c_{{cat_{A} }}^{0} - c_{{cat_{A} }} - c_{{cat_{B} }}\), where \(c_{{cat_{A} }}^{0}\) is the initial catalyst concentration.
Transformations of cinnamylalcohol were supposed to follow the first order kinetic to the product II as well as to side products because DMC was used in excess resulting in
In Eq. (9) K1->II is the equilibrium constant for the carboxymethylation of cinnamylalcohol with DMC in excess, which according to the experiment using trimethylamine as a catalyst is equal to 19.
The generation of side products from reactant I is expressed by the following equation
Taking into account the generation rates of reactants in a batch reactor, e.g.,
where \(\rho_{b}\) is the catalyst bulk density (11.8 g/L), kinetic modelling was done to compare the measured concentration of reactants and products with the theoretical predictions. The estimation of the kinetic parameters was performed using the ModEst parameter estimation software [56].
The objective function (Q) used in the modelling was set to minimize the degree of explanation between the calculated and experimental values of concentrations
The residuals given by the proposed kinetic model in this way are compared with the residuals of the simplest model, where the estimated values correspond to the average value of all data points [56]. The parameter estimation was done by numerically solving the differential equations for the concentrations of three components (compounds I and II and one lumped by-product) with the Levenberg–Marquardt method incorporated in the software.
Preliminary calculations demonstrated that for an adequate description of the induction period and the composition at longer times the deactivation terms cannot be neglected thus four parameters \(k_{A - > B}\);\(k_{b1 \to II}\);\(k_{bS}\). \(k_{{B_{d} }}\) had to be evaluated by the data fitting.
The results of calculations given in Figs. 14 and S26 show a very good correspondence between calculations and experimental data for two catalysts H3PO4–NaOH and NaOH US (80 W, 4 h)st 48 h with the degree of explanation of 99.93% and 99.11% respectively.

Kinetic profiles for the consumption of cinnamylalcohol (blue) and formation of product I (red) and side products (black) as a function of reaction time for slag derived catalyst H3PO4–NaOH
The results presented in these figures clearly demonstrate that the model is able to capture the key kinetic features of the reaction such as the S-shaped behavior and a limiting value of conversion not related to the thermodynamic equilibrium.
As anticipated due a large number of parameters (4) compared to the available data on concentration profiles the estimated values of the parameters are not statistically reliable and therefore the statistical analysis was conducted using the Monte Carlo Markov Chain (MCMC) method [57, 58]. In this method, the samples are drawn randomly to approximate the probability distribution of parameters, which reliability is thus evaluated by treating all the uncertainties in the data and the modelling as statistical distributions [59]. Fig. S27 displays the statistical analysis with the most probable values of constants located at maxima [60]. As can be seen from Fig. S27 the Monte Carlo Markov Chain (MCMC) method allows identification of the constants with some certainty, which cannot be achieved otherwise.
Conclusions
Carboxymethylation of cinnamylalcohol with dimethyl carbonate was investigated using low-cost catalysts synthesized from desulfurization steel slag. The initial industrial residual material exhibiting poor textural and structural properties was treated by different methods including treatment at ambient and hydrothermal conditions with a variation of the treating agent type (distilled water, NaOH, HCl, EDTA and TEAH), temperature and time. Ultrasonication was successfully applied as an additional intensification tool for the synthesis of slag catalytic materials. The synthesized slag catalysts with physico-chemical properties depending on the treatment conditions illustrated a direct relationship between the catalytic results and the materials characteristics. A clear trend was observed between the catalytic activity, the catalyst surface area and basicity. Another important parameter influencing catalytic behaviour was the crystal morphology, which can be varied by treatment conditions. Application of ultrasonication was instrumental in transforming inactive crystalline phases into the active ones otherwise the same synthesis parameters. Analysis of the catalytic results showed similar selectivity to the desired product cinnamyl methyl carbonate varying in the range 80 – 90% at the same CA conversion, while activity could be different. More specifically conversion varied in a broad range between 8 and 85%. The most efficient slag catalyst was prepared by a multi-step treatment with H3PO4–NaOH mixture and exhibited well-developed textural and structural properties along with a moderate basicity. Carboxymethylation of CA with DMC over this material afforded a high conversion level (85%) with high selectivity to the desired product almost independent on conversion (e.g., 89% at 30 and 60% of conversion).
The most active slag catalysts exhibiting CA conversion higher than 65% were compared with the commercial basic materials. Slightly inferior in activity, slag-based catalysts can compete with the commercial ones due to stability of the former to leaching into the reaction media, while the commercial catalysts demonstrated non negligible solubility or even rather significant at the reaction conditions. Potential changes in the crystal morphology and texture of the slag-based materials during catalysis were verified by SEM and TEM analysis, while stability to leaching was proved by ICP and hot filtration tests.
Reusability of the slag catalysts was tested in two additional experimental runs using the spent catalysts, displaying a decrease in activity and selectivity. Analysis of the spent catalytic materials after each run confirmed stability of the textural and morphological properties, although there was a clear decrease of the surface area indicating carbon deposition in the pores. The regeneration procedure comprising washing with acetone, drying and calcination at 400 °C was only partly successful.
Based on GC and NMR analysis the reaction network was proposed and evaluated from the viewpoint of thermodynamic feasibility. Kinetic modelling was performed explaining the essential features of observed kinetic regularities including the induction period and thermodynamic limitations.
Overall, a possibility of using catalysts prepared from the industrial slag in comparison with commercially available materials of basic character was investigated confirming their applicability in the base-catalyzed carboxymethylation of cinnamylalcohol with dimethyl carbonate. The slag-based catalysts displayed high activity with conversion up to 85% and selectivity to the desired product reaching ca. 90%. Catalytic application of the synthesized slag materials in the current work is not only contributing to the waste minimization strategy, but also following the concepts of green chemistry by using renewable and nontoxic reagents in the production of valuable chemicals. Further in-depth research, however, are required for efficient regeneration of the spent catalysts.
References
Kenar JA, Knothe G, Dunn RO, Ryan TW III, Matheaus A (2005) Physical properties of oleochemical carbonates. J Amer Oil Chem Soc 82:201–205
Ono Y (1997) Dimethyl carbonate for environmentally benign reactions. Catal Today 35:15–25
Tan HZ, Wang ZQ, Xu ZN, Sun J, Xu YP, Chen QS, Chen Y, Guo GC (2018) Review on the synthesis of dimethyl carbonate. Catal Today 316:2–12
Ramesh S, Indukuri K, Riant O, Debecker D (2018) Synthesis of carbonate esters by carboxymethylation using NaAlO2 as a highly active heterogeneous catalyst. Org Proc Res Dev 12:1846–1851
Serra-Muns A, Pleixats R (2010) Tsuji-Trost allylations with palladium recovery by phosphines/Pd (0)-triolefinic macrocyclic catalysts. J Organometal Chem 695:1231–1236
Cheng Q, Tu HF, Zheng C, Qu JP, Helmchen G, You SL (2019) Iridium-catalyzed asymmetric allylic substitution reactions. Chem Rev 119:1855–1969
Lee Y, Shabbir S, Lee S, Ahn H, Rhee H (2015) Catalytic allylic arylation of cinnamyl carbonates over palladium nanoparticles supported on a thermoresponsive polymer in water. Green Chem 17:3579–3583
Jones AD, Zelle RE, Silverman IR (2014) U.S. Patent No. 8822.498 B2. Lexington, MA (US). Concert Pharmaceuticals Inc.
Gao N, Guo XW, Zheng SC, Yang WK, Zhao XM (2012) Iridium-catalyzed enantioselective allylation of sodium 2-aminobenzenethiolate: an access to chiral benzo-fused N, S-heterocycles. Tetrahedron 68:9413–9418
Tundo P, Musolino M, Aricò F (2018) The reactions of dimethyl carbonate and its derivatives. Green Chem 20:28–85
Jérôme F, Luque R (2017) Bio-based solvents. Wiley-VCH, Germany
Stanley JNG, Selva M, Masters AF, Maschmeyer T, Perosa A (2013) Reactions of p-coumaryl alcohol model compounds with dimethyl carbonate. Towards the upgrading of lignin building blocks. Green Chem 15:3195–3204
Kantam ML, Pal U, Sreedhar B, Choudary BM (2007) An efficient synthesis of organic carbonates using nanocrystalline magnesium oxide. Adv Synth Catal 349:1671–1675
Ramesh S, Devred F, Debecker DP (2019) NaAlO2 supported on titanium dioxide as solid base catalyst for the carboxymethylation of allyl alcohol with DMC. Appl Catal A: Gen 581:31–36
Jin S, Hunt AJ, Clark JH, McElroy CR (2016) Acid-catalysed carboxymethylation, methylation and dehydration of alcohols and phenols with dimethyl carbonate under mild conditions. Green Chem 18:5839–5844
Kanakikodi KS, Churipard SR, Halgeri AB, Maradur SP (2020) Solid acid catalyzed carboxymethylation of bio-derived alcohols: an efficient process for the synthesis of alkyl methyl carbonates. Sci Rep 10:13103
Selva M, Benedet V, Fabris M (2012) Selective catalytic etherification of glycerol formal and solketal with dialkyl carbonates and K2CO3. Green Chem 14:188–200
Rittiron P, Niamnuy C, Donphai W, Chareonpanich M, Seubsai A (2019) Production of glycerol carbonate from glycerol over templated sodium-aluminate catalysts prepared using a spray-drying method. ACS Omega 4:9001–9009
Zheng L, Xia S, Lu X, Hou Z (2015) Transesterification of glycerol with dimethyl carbonate over calcined Ca-Al hydrocalumite. Chin J Catal 36:1759–1765
Okoye PU, Abdullah AZ, Hameed BH (2017) Stabilized ladle furnace steel slag for glycerol carbonate synthesis via glycerol transesterification reaction with dimethyl carbonate. Energy Convers Manag 133:477–485
Liu G, Yang J, Xu X (2020) Synthesis of hydrotalcite-type mixed oxide catalysts from waste steel slag for transesterification of glycerol and dimethyl carbonate. Sci Rep 10:10273
Yildirim IZ, Prezzi M (2011) Chemical, mineralogical and morphological properties of steel slag. Adv Civ Eng 201:1–13
Dhoble YN, Ahmed S (2018) Review on the innovative uses of steel slag for waste minimization. J Mater Cycles Waste Manag 20:1373–1382
EUROSLAG Statistics (2018) https://www.euroslag.com/products/statistics/statistics-2018/. Accessed 9 July 2021
Fisher LV, Barron AR (2019) The recycling and reuse of steelmaking slags — A review. Resour Conserv Recycl 146:244–255
Kuwahara Y, Hanaki A, Yamashita H (2020) A direct conversion of blast furnace slag to mesoporous silica–calcium oxide composite and its application in CO2 capture. Green Chem 22:3759–3768
Sugano Y, Sahara R, Murakami T, Narushima T, Iguchi Y, Ouchi C (2005) Hydrothermal synthesis of zeolite A using blast furnace slag. ISIJ Int 45:937–945
Anuwattana R, Balkus KJ Jr, Asavapisit S, Khummongkol P (2008) Conventional and microwave hydrothermal synthesis of zeolite ZSM-5 from the cupola slag. Micropor Mesopor Mater 111:260–266
Kuwahara Y, Ohmichi T, Kamegawa T, Mori K, Yamashita H (2009) A novel synthetic route to hydroxyapatite–zeolite composite material from steel slag: investigation of synthesis mechanism and evaluation of physicochemical properties. J Mater Chem 19:7263–7272
Kholkina E, Kumar N, Ohra-aho T, Lehtonen J, Lindfors C, Perula M, Peltonen J, Salonen J, Murzin DY (2019) Synthesis and characterization of novel catalytic materials using industrial slag: influence of alkaline pretreatment, synthesis time and temperature. Top Catal 62:738–751
Kholkina E, Kumar N, Ohra-Aho T, Lehtonen J, Lindfors C, Peurla M, Peltonen J, Salonen J, Murzin DY (2020) Transformation of industrial steel slug with different structure-modifying agents for synthesis of catalysts. Catal Today 355:768–780
Kholkina E, Kumar N, Eränen K, Peurla M, Palonen H, Salonen J, Lehtonen J, Murzin DY (2021) Ultrasound irradiation as an effective tool in synthesis of the slag-based catalysts for carboxymethylation. Ultrasonics Sonochem 73:105503
Letizia CS, Cocchiara J, Lalko J, Lapczynski A, Api AM (2005) Fragrance material review on cinnamylalcohol. Food ChemToxicol 43:837–866
Bonini C, Righi G (1994) Enantio-and stereo-selective route to the taxol side chain via asymmetric epoxidation of trans-cinnamylalcohol and subsequent expoxide ring opening. J Chem Soc Chem Comm 24:2767–2768
Yamanaka R, Nakamura K, Murakami M, Murakami A (2015) Selective synthesis of cinnamylalcohol by cyanobacterial photobiocatalysts. Tetrahedron Let 56:1089–1091
Aricò F, Tundo P (2010) Dimethyl carbonate: a modern green reagent and solvent. Russ Chem Rev 79:479–489
Tundo P, Selva M (2002) The chemistry of dimethyl carbonate. Acc Chem Res 35:706–716
F Aricò, P Tundo (2016) Methylethers from alcohols and dimethyl carbonate. Encyclopedia of Inorganic and Bioinorganic Chemistry, Wiley-VCH
Tundo P, Rossi L, Loris A (2005) Dimethyl carbonate as an ambident electrophile. J Org Chem 70:2219–2224
Storgårds F, Mäki-Arvela P, Kumar N, Peltonen J, Salonen J, Perula M, Eränen K, Russo V, Murzin DY, Grénman H (2018) Catalytic conversion of hexanol to 2-butyl-octanol through the Guerbet reaction. Top Catal 61:1888–1900
Käldström M, Kumar N, Heikkilä T, Tiitta M, Salmi T, Murzin DY (2011) Transformation of levoglucosan over H-MCM-22 zeolite and H-MCM-41 mesoporous molecular sieve catalysts. Biomass Bioenergy 35:1967–1976
Abelló S, Vijaya-Shankar D, Pérez-Ramírez J (2008) Stability, reutilization, and scalability of activated hydrotalcites in aldol condensation. Appl Catal A: Gen 342:119–125
Halász I, Gáti G (1982) Double bond migration of olefins on deuterated and chlorinated Al2O3 catalysts. React Kinet Catal Lett 19:389–391
Carre S, Gnep NS, Revel R, Magnoux P (2008) Characterization of the acid–base properties of transition aluminas by model reaction. Appl Catal A: Gen 348:71–78
Martin D, Duprez D (1997) Evaluation of the acid-base surface properties of several oxides and supported metal catalysts by means of model reactions. J Molec Catal A: Chem 118:113–128
Keresszegi C, Bürgi T, Mallat T, Baiker A (2002) On the role of oxygen in the liquid-phase aerobic oxidation of alcohols on palladium. J Catal 211:244–251
Ochoa-Terán A, Guerrero L, Rivero IA (2014) A novel one-pot and one-step microwave-assisted cyclization-methylation reaction of amino alcohols and acetylated derivatives with dimethyl carbonate and TBAC. Sci World J 14:1–10
Zhang B, Ding G, Zheng H, Zhu Y (2014) Transesterification of dimethyl carbonate with tetrahydrofurfuryl alcohol on the K2CO3/ZrO2 catalyst—Function of the surface carboxylate species. Appl Catal B: Env 152:226–232
Yadav GD, Fernandes GP (2013) Selective synthesis of natural benzaldehyde by hydrolysis of cinnamaldehyde using novel hydrotalcite catalyst. Catal Today 207:162–169
Kasashima Y, Uzawa A, Hashimoto K, Nishida T, Murakami K, Mino T, Sakamoto M, Fujita T (2010) Synthesis of cinnamyl ethers from α-vinylbenzyl alcohol using iodine as catalyst. J Oleo Sci 59:549–555
Zemansky MW, Abbott MM, Van Ness HC (1975) Basic engineering thermodynamics. McGraw-Hill, New York
Poling BE, Prausnitz JM, O'Connell JP (2004) The properties of gases and liquids, 5th edn, McGraw-Hill: New York
Joback KG (1984) A unified approach to physical property estimation using multivariate statistical techniques. S.M. Thesis, Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA
Joback KG, Reid RC (1987) Estimation of pure-component properties from group-contributions. Chem Eng Comm 57:233–243
Cabrerro-Antonio JR, Adam R, Wärnå J, Murzin DYu, Beller B (2018) Reductive N-methylation of amines: mechanistic insights through kinetic modelling. Chem Eng J 351:1129–1136
Haario H (2001) Modest 6.0-A user’s guide, ProfMath, Helsinki
Görlitz L, Gao Z, Schmitt W (2011) Statistical analysis of chemical transformation kinetics using Markov-Chain Monte Carlo methods. Environ Sci Technol 45:4429–4437
Murzin DYu, Wärnå J, Haario H, Salmi T (2021) Parameter estimation in kinetic models of complex heterogeneous catalytic reactions using Bayesian statistics. Reac Kinet Mech Catal. https://doi.org/10.1007/s11144-021-01974-1
Haario H, Saksman E, Tamminen J (2001) An adaptive Metropolis algorithm. Bernoulli 7:223–242
Matera S, Schneider WF, Heyden A, Savara A (2019) Progress in accurate chemical kinetic modeling, simulations, and parameter estimation for heterogeneous catalysis. ACS Catal 9:6624–6647
Acknowledgements
Authors acknowledge the financial support of the Finnish Funding Agency for Innovation (Tekes, 2016–2017) and Business Finland (L4Value project, 2017–2020). E. Kholkina is grateful to Åbo Akademi University for the partial financial support (Doctoral research grant 2020).
Funding
Open access funding provided by Abo Akademi University (ABO).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kholkina, E., Kumar, N., Eränen, K. et al. Carboxymethylation of cinnamylalcohol with dimethyl carbonate over the slag-based catalysts. Reac Kinet Mech Cat 133, 601–630 (2021). https://doi.org/10.1007/s11144-021-02021-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-021-02021-9