
Abstract
The kinetics of the oxidation of imipramine and opipramol using peroxydisulfate salts in the presence of a large excess of dibenzoazepine derivative (TCA) in acidic sulfate media was studied using UV–vis spectroscopy. The reaction between imipramine and S2O8 2− proceeds via the formation of two intermediates: a free organic radical and a dimeric dication. Further reaction of the intermediate dimeric dication leads to a positively charged radical dimer as one final product. Simultaneously, two other substituent cleavage degradation processes occur, leading to two dimeric derivatives. The first product, the positively charged radical dimer, and the next main product, a radical dimer without one alkyl substituent, were identified by EPR measurements. The measured kinetic trace is not first order and revealed a sigmoid shape with a characteristic induction time. The rate constants were determined by numerical analysis based on ordinary differential equations (ODEs). The reaction between opipramol and S2O8 2− proceeds by a two consecutive reaction scheme. The kinetics of the first degradation step were studied independently of the slower degradation reactions. Linear dependences, with zero intercept, of the pseudo-first-order rate constants (k obs) on [TCA] were determined for the first degradation process of opipramol.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The pharmaceutical industry is a critical component of the group of chemical enterprises. Pharmaceutical companies provide more effective active pharmaceutical ingredients (APIs) by designing new compounds with increased potency, bioavailability and degradation resistance. However, the growing pharmaceutical industry and the resulting increase in pharmaceutical pollution are becoming a greater risk to the environment. Persistent, bioaccumulative and toxic (PBT) substances pose a particular challenge for chemical safety management. The ecotoxicities of several human pharmaceuticals have been reviewed recently [1–3]. Another important issue from the modern chemistry and the environmental perspectives is the development of methods for removing the small quantities of drugs that have entered the environment (e.g., via groundwater, which is a source of drinking water). [4] To protect the environment, it is important that these methods belong to the category of “green chemistry”. Therefore, this study is focused on promoting a “green chemistry” method of removing toxic pharmaceuticals during water purification and on eliminating methods based on chlorination. Among these “green chemistry” methods, photolytic and oxidative degradation are distinguished by the following compounds and processes: O3, H2O2/UV, Fenton/H2O2, O3/H2O2, and Fe-TAML/H2O2. An attempt to develop methods for the degradation of pharmaceuticals in the environment is described in a previous study on the photocatalytic decomposition of imipramine [5]. Imipramine is a medicine that is difficult to degrade in the environment, is easy to bioaccumulate and has a high ecotoxicity. This paper explores the degradation of antidepressants by the strong oxidant peroxydisulfate in aqueous solutions. Because green chemistry involves the design of chemical processes that reduce or eliminate the use and generation of hazardous substances, and safe processes that allow us to remove hazardous contaminants from the environment, we can use these safe processes to remove pharmaceuticals and pharmaceutical pollution. Chemical oxidation aims to selectively remove persistent components via the conversion to readily biodegradable intermediates that can be subsequently treated biologically. Compounds that are particularly susceptible to oxidation often contain heteroatoms with lone pairs of electrons (e.g., tricyclic antidepressants and TCA), which suggests that APIs are candidates for oxidative treatment. The potential use of peroxydisulfate as a “green oxidant” has been intensively studied in the UV/S2O8 2− system [5–7]. Characteristic features of this oxidant are the one-electron transfer after homolytic decomposition or the direct one-electron transfer between many organic compounds in noncatalytic and catalytic processes, the relatively low oxidation rate and the strong influence of the concentration of sulfate(VI) and the H+ ion concentrations on both the rate of oxidative degradation processes and on the rate of decomposition of S2O8 2− [8]. In the oxidation process, sulfate ions are generated as a final product [9]. The SO4 2− ion is practically inert and is not considered to be a pollutant [7, 10]. Therefore, we studied the kinetics and mechanisms of the spontaneous aqueous oxidative degradation by peroxydisulfate.
Experimental
Materials
Imipramine hydrochloride (Sigma-Aldrich), opipramol dihydrochloride (Sigma-Aldrich), Na2S2O8 and all other chemicals used in this study were analytical grade reagents. Ultrapure water was obtained from a Milli-Q system (Millipore/Waters, Milford, MA, USA) and was used to prepare all of the solutions. Sulfuric acid solutions of the desired concentration were prepared from reagent grade H2SO4 (65 %, Aldrich). The NaClO4 salt was added in the kinetic experiments to maintain a constant ionic strength. Aqueous imipramine and opipramol solutions were prepared immediately before measurements. The S2O8 2− solutions were freshly prepared in sulfuric acid just before mixing with other reactants. Both the concentration and stability studies of peroxydisulfate were performed iodometrically.
EPR measurements
EPR spectra were recorded with a Radiopan EPR SE/X 2541 M spectrometer in the X band (ca. 9.25 GHz) with a 100 kHz modulation. The microwave frequency was monitored with a frequency meter. The magnetic field was measured with an automatic NMR-type magnetometer. EPR spectra were recorded at room temperature using both (i) a stationary technique and (ii) a continuous flow technique, and the measurements were performed at ca. 1 s after the initial mixing of the reagents. A flat quartz cell was used. The solution concentrations were [TCA] = (5–25) × 10−4 M, [S2O8 2−] = (5–100) × 10−4 M, and [H2SO4] = 1.0 M.
High-performance chromatography and mass spectra analyses of the products
Chromatographic separations followed by measurements using a MS analyzer were performed on a C18 column (Waters 100 × 2.1 mm i.d.). The injection volume was 20 μl and the flow rate was 0.6 ml/min. Gradient mobile phase composition (A: water containing 0.2 % formic acid, B: acetonitrile containing 0.2 % formic acid) was adopted as follows: 0–1 min 2 % B, 12 min 75 % B, 18 min 90 % B. Measurements were performed on a HPLC system 1100 (Agilent Technologies, Waldbronn, Germany) equipped with a quaternary pump and a UV–VIS detector and coupled with the Agilent 6410 Triple Quad LC/MS mass spectrometer equipped with an ESI ion source, a capillary voltage of 4.5 kV, a drying gas temperature of 300 °C, a flow of 9.0 l/min and a nebulizer pressure of 40 psi. The acquisition method used was previously optimized during tuning sections for the parent compound (capillary, magnetic lenses and collimating octapole voltages) to achieve the maximum sensitivity. Mass spectra were collected in full-scan positive mode in the mass-to-charge ratio (m/z) range from 100 to 850.
NMR and IR analyses
1H and 13C NMR spectra were recorded in D2O solutions using a Bruker Avance III 400 spectrometer (13C) and a Bruker Avance III 700 spectrometer (1H) operating at typical frequencies. In the case of imipramine, the D2O solution was acidified with 10 mM TFA-d. IR spectra were measured using a Perkin-Elmer Spectrum 2000 Fourier Transform Infrared spectrometer using KBr discs (4,000–400 cm−1).
Opipramol
1H NMR data and assignments δ(ppm): H1 = 7.26 (t, 2H, J = 7.7 Hz); H2 = 7.09 (d, 2H, J = 7.7 Hz), H3 = 7.06 (d, 2H, J = 7.7 Hz); H4 = 6.99 (t, 2H, J = 7.7 Hz); H5 = 6.78 (s, 2H); H6 = 3.76 (t, 2H, J = 4.9 Hz); H7 = 3.75 (t, 2H, J = 6.3 Hz); H8 = 3.44 (s, 4H); H9 = 3.33 (s, 4H); H10 = 3.21 (t, 2H, J = 5.3 Hz); H11 = 3.17 (t, 2H, J = 7.7 Hz); H12 = 1.83 (q, 2H, J = 7.7 Hz). 13C NMR data and assignments δ(ppm): C1 = 149.3; C2 = 133.2; C3 = 132.1; C4 = 129.8; C5 = 129.4; C6 = 124.4; C7 = 120.4; C8 = 58.02; C9 = 55.11; C10 = 55.07; C11 = 48.72; C12 = 48.67; C13 = 46.52; C14 = 21.16.
IR data (cm−1): 452, 474, 487, 510, 586, 627, 718, 764, 791, 843, 877, 905, 942, 966, 1029, 1058, 1072, 1102, 1119, 1130, 1230, 1246, 1301, 1325, 1393, 1439, 1466, 1485, 1573, 1592, 2317, 2409, 3004, 3048, 3333.
10-[3-[4-(2-hydroxyethyl)-piperazinyl]propyl]-acridine
1H NMR data and assignments δ(ppm): H1 = 8.44 (d, 2H, J = 8.4 Hz); H2 = 8.41 (d, 2H, J = 8.4 Hz); H3 = 8.31(t, 2H, J = 8.1 Hz); H4 = 8.11 (s, 1H); H5 = 7.86 (t, 2H, J = 7.7 Hz); H6 = 3.79 (t, 2H, J = 5.6 Hz); H7 = 3.77 (t, 2H, J = 5.6 Hz); H8 = 3.46 (s, 4H); H9 = 3.37 (s, 4H); H10 = 3.23 (t, 2H, J = 6.3 Hz); H11 = 3.22 (t, 2H, J = 6.3 Hz); H12 = 1.88 (q, 2H, J = 7.7 Hz). 13C NMR data and assignments δ(ppm): C1 = 151.8; C2 = 140.9; C3 = 139.9; C4 = 132.3; C5 = 127.8; C6 = 126.7; C7 = 117.1; C8 = 58.02; C9 = 54.89; C10 = 53.28; C11 = 48.85; C12 = 48.66; C13 = 46.55; C14 = 22.74.
IR data (cm−1): 439, 470, 559, 592, 721, 859, 1063, 1171, 1203, 1266, 1304, 1387, 1457, 1547, 1614, 2308, 2448, 2549, 2655, 3003, 3422.
Imipramine
1H NMR data and assignments δ(ppm): H1 = 7.07 (t, 2H, J = 7.7 Hz); H2 = 7.06 (d, 2H, J = 7.7 Hz); H3 = 7.01 (d, 2H, J = 7.7 Hz); H4 = 6.88 (t, 2H, J = 7.7 Hz); H5 = 3.63 (s, 2H); H6 = 2.97 (s, 4H); H7 = 2.88 (s, 2H); H8 = 2.53 (s, 6H); H9 = 1.75 (s, 2H).
13C NMR data and assignments δ(ppm): C1 = 147.6; C2 = 134.6; C3 = 130.2; C4 = 126.9; C5 = 123.4; C6 = 119.8; C7 = 55.9; C8 = 46; C9 = 42.6; C10 = 31.3; C11 = 22.2.
Kinetic measurements
Spectra and preliminary kinetic measurements were studied using a Lambda 20 Perkin-Elmer spectrophotometer and a Schimadzu UV-1601 PC spectrophotometer equipped with an external Julabo F25 ultrathermostat. Measurements were performed in 1.0 M H2SO4 solution with constant ionic strength equal to 1.4 M that was maintained with a solution of NaClO4. In the oxidative degradation of opipramol, the concentration of an oxidant was used in excess and was altered over the range (1–5) × 10−2 M. The concentration of dibenzoazepine was fixed at 5 × 10−5 M. The other experimental conditions were as follows: [S2O8 2−] = (1–5) × 10−2 M, [H2SO4] = 1.0 M, I = 1.55 M (H+, Na+, HSO4 −, SO4 2−, ClO4 −, S2O8 2−), T = 303–323 K, λ = 350 nm. The rate was also analyzed at different H2SO4 concentrations: [H2SO4] = 0–1.0 M, pH = 7–0, I = 1.55 M (H+, Na+, HSO4 −, SO4 2−, ClO4 −, S2O8 2−), T = 313 K. In addition, one series of kinetic measurements was performed without maintaining constant ionic strength. Reactions were studied under pseudo-first order conditions in an excess of one of the reactants over the others. Kinetic data were analyzed by a Gauss–Newton nonlinear least-squares fit to the first-order dependence of the absorbance versus time for the fast first oxidation process. Absorbance versus time data were collected up to 95 % conversion degree. The reported rate constants are the mean values of at least three measurements. The relative standard errors of the pseudo-first order rate constants for a single kinetic trace were ca. 0.5–1 %, and the relative standard errors of the mean value were usually ca. 1–2 %.
In the oxidative degradation of imipramine, the experimental conditions were as follows: [TCA] = 1 × 10−4 M, [S2O8 2−] = (1–5) × 10−2 M, [H2SO4] = 1.0 M, I = 1.55 M (H+, Na+, HSO4 −, SO4 2−, ClO4 −, S2O8 2−), T = 293–313 K, λ = 400–800 nm. The rate was also analyzed at different H2SO4 concentrations: [H2SO4] = 1.0–6.0 M, [H+] = 1.2–7.2 M, I ≠ constant, T = 293 K. In a few cases, the rate was measured in an excess of imipramine. For these measurements, the spectroscopic analyses and measurements were difficult to perform at concentrations of imipramine above 1 × 10−4 M because the reaction product began to precipitate. The kinetic traces are not first order and revealed a characteristic induction time for that process. The reaction was initiated by an introduction of 0.025 cm3 0.004 M dibenzoazepine solution in H2O to a quartz cuvette that contained 1.975 cm3 of S2O8 2− solution of the appropriate concentration.
Results and discussion
The reaction between dibenzoazepine derivatives and peroxydisulfate ion was studied in acidic perchlorate–sulfate ion media ([TCA] = 5 × 10−5 M, [S2O8 2−] = (1–5) × 10−2 M, [H2SO4] = 1.0 M, I = 1.55 M (H+, Na+, HSO4 −, SO4 2−, ClO4 −, S2O8 2−)). The stability of peroxydisulfate ion has been widely analyzed in different acidic and alkaline media [11]. Peroxydisulfate ion undergoes a hydrolysis reaction, which can be facilitated by H+ ions, Na+ ions, or other positively charged counter-ions. Its stability decreases with decreasing pH and with decreasing S2O8 2− concentration. The S2O8 2− ions are more stable in pure water and slowly decompose with rate constants ranging from 6.7 × 10−6 to 4.7 × 10−5 s−1 and half-lives of 29–4 h in the pH range from 1 to 0 ([S2O8 2−] = 1 × 10−2 M, [H2SO4] = 0.1–1 M, T = 323 K) [11].
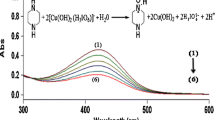
The reaction between opipramol and peroxydisulfate ion was studied spectrophotometrically in the 200–500 nm wavelength range. The characteristic spectral changes for the slow degradation of opipramol are consistent with the formation of a new band at 350 nm, and a simultaneous increase of the absorbance in the 400–500 nm range is attributed to the pale-green colored product (Fig. 1), which also revealed fluorescence in a characteristic emission band. The observed reaction of opipramol leads to acridine derivatives as a final product (Scheme 1). No EPR signal was recorded during the reaction, suggesting that the SO4 − and other radical reactive intermediates were not stable enough to be measured using the conventional EPR scanning method. The final product was detected by HPLC–ESI–MS. The chromatogram revealed the presence of only two fractions characteristic of opipramol with a retention time of 7.7 min and a product with a retention time shorter than that of opipramol and equal to 10.7 min. Electrospray ionization MS spectra (Fig. S1, Supporting Information) show the presence of molecular and characteristic fragmentation peaks for 10-[3-[4-(2-hydroxyethyl)-piperazinyl]propyl]-acridine (2): 350 (M+•), 171 (M–C13H9N), 143 (M–C15H13N); product 3: 366 (MH+, low abundance); product 4: 180 (MH+, low abundance); product 5: 196 (MH+, low abundance) m/z. The molecular and fragmentation peaks for opipramol (1) were as follows: 364 (MH+), 234 (M–C6H13N2O), 171 (M–C14H10N), 143 (M–C16H14N) m/z.

Spectral changes during opipramol degradation by peroxydisulfate ion. Inset the kinetic trace at 350 nm. Experimental conditions: [TCA] = 5 × 10−5 M, [S2O8 2−] = 5 × 10−2 M, [H2SO4] = 1.0 M, [H+] = 1.2 M, I = 1.55 M (H+, HSO4 −, SO4 2−, Na+, S2O8 2−, ClO4 −), T = 293 K, t = 40 min, Δt = 2 min

Products in the oxidative degradation reaction of dibenzoazepine derivatives
The samples of opipramol analyzed exhibit a very strong and broad absorption band at 3,333 cm−1, which is primarily due to O–H stretching. During oxidation, this absorption band increased in intensity and slightly shifted towards the higher frequency region, and it was covered by a new very strong and broad absorption band at 3,422 cm−1, which is primarily due to N–H stretching. The IR spectra of the final product do not exhibit a very strong band at 1,720 cm−1, which is characteristic of the carbonyl C=O stretching vibration. The lack of a carbonyl peak leads to the conclusion that the final product is the non-substituted acridine derivative rather than the acridinone derivative.
The kinetic data are presented in Table 1. The dependence of the absorbance with time is first order with respect to [TCA] (Fig. 1). The oxidation process is known in the literature to proceed primarily through a sequence of one-electron transfer reactions, whose kinetics is characterized by a zeroth order relationship, and a zeroth order increase in product concentration over time [12, 13]. We cannot conclude that the mechanism of this process may not be a radical mechanism that proceeds via the SO −•4 radical. The observed pseudo-first order rate constant is linearly dependent on [S2O8 2−] (Fig. 2); thus, the following simple second order rate equation was found to be valid:
where the second order rate constant for the forward reaction, k 1, according to Eq. 3, was obtained from the slope (Fig. 2). The linear regression data for this reaction are presented in Table 2. The observed rate constant did not change with decreasing concentrations of acid over the 1–0.1 M range (Table 1), but it increased at H2SO4 concentrations lower than 0.1 M. Therefore, the non-protonated form of dibenzoazepine derivatives with piperazine substituent undergoes a more effective oxidative degradation reaction and was a more reactive species than its protonated forms (pKa1 = 8.38, pKa2 = 3.81 1,4-dimethylpiperazine) [14]. A second, significantly slower process is consistent with the decrease of absorbance in the entire visible spectral region, when the intermediate converts to a pale yellow final product, which, as mentioned above, is acridine. The kinetics of the appearance of acridine is not within the scope of this work.

Plots of k obs versus [S2O8 2−] for the electron transfer reaction between opipramol and peroxydisulfate ion at different temperatures. [TCA] = 5 × 10−5 M, [H2SO4] = 1.0 M, [H+] = 1.2 M, I = 1.55 M (H+, HSO4 −, SO4 2−, Na+, S2O8 2−, ClO4 −), T = 303 K (filled square), 313 K (filled circle), 323 K (filled triangle)
The oxidation of dibenzoazepine derivatives has been proposed in the literature to lead to an intensively colored dimer, which is a product of a four-electron oxidation process [15]. Electrospray ionization MS spectra (Scheme 1, Fig. S2, Supporting Information) show the presence of molecular and characteristic fragmentation peaks for dimer 7: 557 (M–H), 279 (M–C19H23N2 monomer) m/z; dimer 8: 474 (MH+), 388 (M–C5H12N), 86 (M–C28H24N2) m/z; dimer 9: 388 (M+•) m/z; methyl derivative of dimer: 488 (MH+, low abundance) m/z; hydroxy derivative of imipramine: 297 (MH+, low abundance), 279 (M–OH), 86 (M–C14H13NO) m/z. The molecular and fragmentation peaks for imipramine (6) were as follows: 281 (MH+), 236 (M–C2H6N), 208 (M–C4H10N), 86 (M–C14H12N), 58 (M–C16H16N) m/z. A cleavage of the alkyl substituent, which leads to the formation of dimers 8 and 9, occurs at the stage of oxidation of the compound but is not due to the spontaneous degradation process because an analogous process does not occur in the starting solution of imipramine. When precipitation occurred in the solution, the 1H NMR spectra for the reaction mixture in the D2O and 10 mM TFA-d solution revealed only the signals from the piperazine substituent at 3.63–1.75 ppm, while the signals from the hydrogen atoms in the aromatic rings at 7.07–6.88 ppm disappeared. No changes occurred without acidification of the D2O solution because of the higher stability of the radical products in the acidic media.
The EPR results provide clear evidence for the formation of intermediate imipramine radical(s). The EPR spectra of the imipramine—S2O8 2− reaction mixtures were recorded for both 1:1 and 1:2 molar ratios. The studies with the equivalent molar quantity of the oxidant using the stationary technique slowly form radical(s). In this case, the intensities of the EPR signal are very weak. The radical species could be detected ca. 5 min after mixing the reaction solutions. Then, the radical concentration gradually increases, reaching its maximum after 25–30 min and subsequently decaying after ca. 100–120 min. The EPR spectra of the imipramine radical(s) exhibit a single nearly isotropic line with g = 2.003 and peak-to-peak width, ΔBpp, of ~1.2 mT. The EPR parameters are very similar to that observed for the imipramine—cerium(IV) system [16]. The studies with a two-fold molar excess of S2O8 2− yielded different results. As in the equivalent molar case, imipramine radicals are formed during a few minutes, and the same initial EPR parameters are observed (within experimental error). However, the EPR signal not only has approximately five-times higher intensity but also changes over time. The intensity increases to a maximum of approximately two-times more (after ca. 25 min.), and both the g factor and the line-width decrease. These changes are most likely related to further radical oxidation and the formation of more stable radical(s). The representative EPR spectrum is presented in Fig. 3 and can be attributed to the existence of a more stable radical generated from imipramine. The EPR spectrum of the imipramine radical exhibits a single nearly isotropic line with g = 1.999 and peak-to-peak width, ΔBpp, of ~0.75 mT. Fig. 3 shows the spectrum of the reaction system after ca. 21 min from the initiation, which is the time when the EPR parameters have not yet changed.

EPR spectrum recorded for 2.5 × 10−3 M imipramine and 5 × 10−3 M Na2S2O8 in 1 M H2SO4. Other conditions: time of reaction a ca. 10 min, b ca. 21 min microwave frequency 9.24810 GHz, room temperature
The reaction with imipramine and its analogues were observed to have a highly sophisticated course that leads to the formation of at least two different products. Dimer 7 absorbs intensively in the visible region (Scheme 1) at 609 nm (λlit, nm (εlit, M−1 cm−1): 625(29,300)). [17] The next product (8) is characterized by the electron transition band at 648 nm (λlit, nm (εlit, M−1 cm−1): 670(3,3000)) [17]. The solution turns blue, which enables the kinetics of the process to be monitored by observing the change in absorbance in the visible range. The kinetic traces of the reaction at the wavelength λ = 630 nm are shown in Fig. 4. The initial stage of the process (up to 200 s) shows a characteristic induction time of the reaction. The increase in absorbance with time, corresponding to the increase in the concentration of the final dimer(s), exhibits a sigmoidal course. Nevertheless, a further decrease in absorbance (not shown), resulting from the degradation of the product(s) and the earlier oxidation stage, was very slow, unlike the reaction with cerium(IV) [16]. Fig. 4 included the first stage of the degradation reactions of the dibenzoazepine derivatives. With increasing concentrations of the oxidant, the rate of dimer(s) production increases. The induction time of this reaction is reduced with increasing concentrations of the S2O8 2− ion from ca. 750 s for [S2O8 2−] = 0.01 M to ca. 200 s for [S2O8 2−] = 0.05 M at T = 293 K (Fig. 3). The kinetic studies were conducted in solutions of sulfuric acid at different concentrations in the 0.1–6.0 M range. The reaction rate does not change with changes in concentration of sulfuric acid in the 0.1–1 M range and decreases in more concentrated solutions of acid up to 5 M. No reaction occurs at concentrations above 5 M H2SO4. Increasing the concentration of H2SO4 causes significant prolongation of the induction time (Fig. 4) due to the hydrolysis of the S2O8 2− ion and the limited generation of the sulfate radical \( {\text{SO}}_{ 4}^{ \bullet - } \) [6].

Plots of absorbance versus time for the reaction between imipramine and peroxydisulfate ion. Experimental conditions: [TCA] = 5 × 10−5 M, a [S2O8 2−] = 1 × 10−2 M, [H2SO4] = 1.0 M; b [S2O8 2−] = 2 × 10−2 M, [H2SO4] = 1.0 M; c [S2O8 2−] = 5 × 10−2 M, [H2SO4] = 1.0 M; d [S2O8 2−] = 5 × 10−2 M, [H2SO4] = 4.0 M, T = 298 K
Based on the electrospray MS spectra and the results of the kinetic measurements obtained during the oxidation reaction with the S2O8 2− ions, the following scheme of the reaction is proposed [11, 18, 19]:
The first step in the proposed reaction scheme is a spontaneous cleavage of the peroxide group in the S2O8 2− ion that results in the highly reactive radical \( {\text{SO}}_{ 4}^{ \bullet - } \). Then, the \( {\text{SO}}_{ 4}^{ \bullet - } \) radical reacts directly with imipramine. The \( {\text{SO}}_{ 4}^{ \bullet - } \) radical is less reactive than a hydroxyl radical in the hydrogen transfer reactions and in the addition to the double bond [20, 21]. Nevertheless, the \( {\text{SO}}_{ 4}^{ \bullet - } \) radical reacts with amines with a rate constant equal to 105–109 M−1 s−1 [22–24]. This process leads to the appearance of a dibenzoazepine radical, which dimerizes and is oxidized to a blue–colored dimeric radical dication, (TCA•+)2, in the two consecutive reactions between the S2O8 2− ion and both (TCA)2 and (TCA) •+2 . It is not very clear what detailed mechanism is concerning the production of the second product 8. While product 7 can react with the \( {\text{SO}}_{ 4}^{ \bullet - } \) radical with abstraction of the substituent and can be converted to the product 8.
The proposed set of reactions leads to the following ordinary differential equations:
Regarding imipramine and its derivatives, there are six differential equations and six concentrations. The TCA•+ concentration is expressed as a function of other concentrations using the mass balance equations:
The knowledge of the contribution of the particular reaction stages to the overall oxidation mechanism is determined by the detailed analysis of the entire absorbance decrease observed during the reaction. The set of differential equations describing a specific reaction model were solved for an initial set of parameters using the Mathematica® program (Wolfram) and the built-in function NDSolve—a numerical differential equation solver. The optimal values of the reaction constants k 3, k 4, k 5, and k 6 were obtained by minimizing the sum of the squared absorbance deviations: \( \sum\nolimits_{i} {\left( {A_{\text{model}} (t_{i} ) - A_{\exp } (t_{i} )} \right)^{2} } \) using the Simplex method [25]. A model was calculated from: \( A_{\text{model}} = l\varepsilon [(TCAr)_{2} ] + A_{bg} \), where l is the path length and A bg is the background absorption, which was assumed to be equal to the experimental value of absorption at t = 0.
It was not possible to obtain a satisfactory fit of the absorbance using Eqs. 7–13. Therefore, reaction 14, where (TCA•+)2 is transformed into a non-absorbing final product (accounting for the experimentally observed decrease in absorbance with time) was added:
The ordinary differential equation for reaction 14 is as follows:
Thus, Eq. 12 is changed to:
and Eq. 13 to:
Fig. 5 illustrates the quality of the fit performed for the chosen reaction model A. Kinetic traces were analyzed at 608 and 648 nm, which enables a more simplified description of the reaction scheme.

Examples of modeling using certain selected reaction models of the kinetic traces recorded at 648 and 608 nm for the oxidation of TCA by S2O8 2−. Experimental conditions: [TCA] = 1 × 10−4 M, [S2O8 2−] = 0.05 M, [H2SO4] = 1.0 M, [H+] = 1.2 M, I = 1.55 M (H+, HSO4 −, SO4 2−, Na+, S2O8 2−, ClO4 −), T = 298 K. Dots—experimental trace; dashed line—model A, line—model B
The assumption that the value of [(TCA•+)2] increase up to the saturation point improved the fit (see model B in Fig. 5). Introducing [(TCA•+)2]sat as an independent variable leads to the corresponding change of Eqs. 15–16:
The set of values of rate constants calculated for the two chosen reaction models are shown in Table 3. The value “opt” refers to the minimal value of SSR. The values of “min” and “max” refer to those which yield ca. 1 % lower and higher SSR value, respectively. An attempt to make calculations using a numerical modeling of higher-order ordinary differential equations gave satisfactory fit to experimental data. Nevertheless, the value of the second order rate constants k 5 can be concluded to not exhibit satisfactory approximated values over a wide range of the sum of squared residuals (SSR) in model A. However, in model B, its value was estimated with better precision and it was not dependent on the limit of SSR change.
Conclusions
The imipramine oxidation reaction proceeds through many steps and forms various products. The appearance of the main products proceeds via a free radical mechanism, in which the rate limiting step is a homolytic cleavage of the peroxide bond in the S2O8 2− ion [26]. The reaction proceeds with a certain induction time, characteristic for the certain concentrations of the reactants set, according to kinetics that are characterized by the first order equation with respect to the concentration of the S2O8 2− ion and according to the zeroth order kinetics with respect to the concentration of imipramine. In the course of the imipramine—S2O8 2− reaction, several radical species with different lifetimes and stabilities are likely formed due to the gradual oxidation of imipramine, particularly in the case of excess oxidant. Unlike imipramine, opipramol degradation proceeds according to first order kinetics with respect to the concentration of opipramol and the S2O8 2− ion. Analyzing the effectiveness of the S2O8 2− ion in the degradation of antidepressants, we note that peroxydisulfate may be an effective “green oxidant” in the oxidative degradation processes of pharmaceuticals and pharmaceutical pollution. Although the spontaneous reactions with this ion are not fast, these reactions have a sufficiently high oxidizing potential, which enables drugs to be converted into less toxic and less active derivatives. An analogous reaction does not occur in DMSO and other organic solvents, but the S2O8 2− ion reacts in an aquatic environment and may be applied in methods of water purification for the removal of toxic pharmaceuticals.
References
Fent K, Weston AA, Caminada D (2006) Aquat Toxicol 76:122–159
Jjemba PK (2006) Ecotoxicol Environ Saf 63:113–130
Crane M, Watts C, Boucard T (2006) Sci Total Environ 367:23–41
Khetan SK, Collins TJ (2007) Chem Rev 107:2319–2364
Calza P, Sakkas VA, Villioti A, Massolino C, Boti V, Pelizzetti E, Albanis T (2008) Appl Catal B 84:379–388
Lau TK, Chu W, Graham NJD (2007) Environ Sci Technol 41:613–619
Balazs GB, Cooper JF, Shell TE (1999) J Appl Electrochem 29:285–292
Minisci F, Citterio A, Giordano C (1983) Acc Chem Res 16:27–32
Maurino V, Calza P, Minero C, Pelizzetti E, Vincenti M (1997) Chemosphere 35:2675–2688
Weiner ER (2000) Applications of environmental chemistry. A practical guide for environmental professionals. Lewis Publishers, CRC Press LLC, Boca Raton, p 238
House DA (1962) Chem Rev 62:185–203
Henry NP, Allen TL (1968) J Am Chem Soc 90:1127–1131
Hindmash K, House DA, van Eldik R (1998) Inorg Chim Acta 278:32–42
Khalili F, Henni A, East ALL (2009) J Chem Eng Data 54:2914–2917
Bishop E, Hussein W (1984) Analyst 109:73–80
Wiśniewska J, Wrzeszcz G, Kurzawa M, van Eldik R (2012) Dalton Trans 41:1259–1267
Horria AM (1992) Anal Lett 25:63–71
Kolthoff IM, Miller IK (1951) J Am Chem Soc 73:3055–3059
Huie RE, Clifton CL (1993) Chem Phys Lett 205:163–167
Clifton CL, Huie RE (1989) Int J Chem Kinet 21:611–619
Clifton CL, Huie RE (1989) Int J Chem Kinet 21:677–687
Bobrowski K (1980) J Phys Chem 84:3524–3529
Steenken S (1989) Chem Rev 89:503–520
Koltzenburg G, Bastian E, Steenken S (1988) Angew Chem Int Ed Engl 27:1066–1067
Himmelblau DM (1972) Applied nonlinear programming. McGraw-Hill, New York, p 451
Rodríguez-López JN, Lowe DJ, Hernández-Ruiz J, Hiner ANP, García-Cánovas F, Thorneley RNF (2001) J Am Chem Soc 123:11838–11847
Acknowledgments
The authors thank Mr. Grzegorz Spólnik, Department of Organic Chemistry, Polish Academy of Science, Warsaw, for his help with the MS measurements.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wiśniewska, J., Wrzeszcz, G., Koter, S. et al. The oxidative degradation and C–C coupling reaction of dibenzoazepine derivatives by peroxydisulfate ion and sulfate radical in aqueous media. Reac Kinet Mech Cat 107, 1–17 (2012). https://doi.org/10.1007/s11144-012-0463-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-012-0463-z