
Abstract
The WRKY gene family is widely distributed in plants and plays essential roles in plant development and stress response. Despite extensive characterization of WRKY transcription factors in many plants, a genome-wide analysis of the WRKY gene family in Pisum sativum is still lacking. The central method of this study focused on the retrieval and characterization of WRKY gene sequences in the pea genome and further looking up for their modulation in response to sulfur and water stress. This involved a combination of bioinformatics tools and gene expression study to identify, characterize, and understand the properties of the WRKY gene family in pea. In this study, 86 PsWRKY genes were identified in the pea genome, categorized into five phylogenetic groups. They were dispersed across all seven chromosomes, with Chromosome 5 showing the highest enrichment. Synteny analysis revealed orthologs of 69 PsWRKY genes in Arabidopsis and Medicago. Transcriptome analysis identified 36 differentially expressed PsWRKY genes in response to sulfur (S) stress, water (W) stress, and their combination (W + S). Real-time PCR validation confirmed significant upregulation of PsWRKY23, PsWRKY58, PsWRKY64, and PsWRKY83 under water stress, PsWRKY58 and PsWRKY84 under sulfur stress, and PsWRKY03 under combined (W + S) stress. Overall, this study provides important insights on genetic information of the WRKY gene family in Pea and its possible role in water and sulfur stress, which will help further to study these candidate WRKY gene with a future objective of understanding their role in abiotic stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pea (Pisum sativum L) is a widely consumed legume, serving as a crucial staple in both the food and feed industries globally (Kreplak et al. 2019). As one of the top four most cultivated and harvested legumes in the world, along with soybeans, common beans and chickpeas (FAO, 2023), pea plays a significant role in feeding a rapidly growing global population. Improving the traits of pea through conventional breeding or genetic engineering is a pressing concern. With the advent of pea’s reference and pan-genome, new opportunities have emerged in the fields of functional genomics and genetic engineering (Burstin et al. 2015; Kreplak et al. 2019; Yang et al. 2022). The availability of pea’s genome sequence, readily accessible online (Pea Genome project, 2019), has facilitated gene family mining and characterization, thereby increasing the potential for genetic improvement.
Transcription factors play a crucial role in gene regulation, binding to the promoter region to modulate gene expression. Several key transcription factors, such as the ABRE-binding factor (ABF)/ABA-responsive-element-binding (AREB), bZIP, bHLH, DREB, ERF, HD-ZIP (Hu et al. 2022), MYB, MYC, MADS, NAC and WRKY (Burke et al. 2020), are involved in the regulation of genes in response to both biotic and abiotic stress. The WRKY gene family, which comprises the largest number of genes in higher plants, is characterized by a WRKYGQK conserved sequence in the N-terminal region and a distinctive zinc-finger-like motif (Eulgem et al. 2000). WRKYs bind to the cis-regulatory element known as the W-box. In addition to the WRKYGQK motif, variants such as WRKYGEK, WKKYGQK, and WRKYDHK have also been reported in various plant species (Nan et al. 2020). These transcription factors are involved in a wide range of developmental processes, including flowering time, seed size, fruit ripening, leaf senescence (Phukan et al. 2016), and metabolic processes. They also play an important role in regulating nutrient starvation responses and root development (Devaiah et al. 2007). In soybean, for instance, the expression of GmWRKY13 is induced by abiotic stress, particularly nutritional stress, leading to an increase in lateral root development (Zhou et al. 2008). The overexpression of Citrullus lanatus WRKY20 in Arabidopsis has been shown to enhance resistance to lower temperature and salt stress (Zhu et al. 2022). In Populus alba, WRKY75 regulates negatively in response to salt and osmotic stress (Zhao et al. 2019). The expression of soybean WRKY17, WRKY21, and WRKY54 is also known to be involved in regulating salt, cold, and drought stress (Zhou et al. 2008). The interference of auxin transport and ABA signaling pathways in Arabidopsis has been demonstrated by overexpressing the OsWRKY72 gene (Yu et al. 2010). AtWRKY50 and AtWRKY51 negatively regulate jasmonic acid signaling, while positively regulating salicylic acid signaling (Gao et al. 2011).
To date, WRKY gene family members have been identified in various legume species, such as medicago, soybean, chickpea, etc. (Song et al. 2018). To the best of our knowledge, the genome wide characterization of WRKY transcription factors in pea (P. sativum L) and their response to environmental stress has not been studied. In the present study, we have identified 86 WRKY TFs in the pea genome, named PsWRKY1 to PsWRKY86, and analyzed their motifs, gene structures, chromosomal locations, phylogenetic and synteny relationships. Gene expression under abiotic stress (water, sulfur deficiency, and a combination of both) was analyzed using transcriptome data and significantly modulated gene expression were further validated using Real-time PCR.
Materials and Methods
Sequence Retrieval of the WRKY Gene Family in Pea
To mine the WRKY encoded sequence from pea genome, we used already reported WRKY genes from ten different plant species namely Arabidopsis thaliana, Medicago truncatula, Cicer arietinum, Cajanus cajan, Glycine max, Lotus japonicus, Vigna radiata, Cajanus cajan, Trifolium pretense, and Arachis ipaensis as a query sequence. All the query sequences were downloaded from the plant transcription factor database v5.0 (http://planttfdb.gao-lab.org/). To identify homologous WRKY sequences in pea genome, BLASTP program on URGI (https://urgi.versailles.inra.fr/Species/Pisum/Pea-Genome-project) were used (Tayeh et al. 2015). The presence of the WRKY domain in each protein sequences was checked using the Hidden Markov Model (HMM profile) and HMMER program (http://hmmer.org/). Furthermore, presence of the WRKY domain using ScanProsite (https://prosite.expasy.org/scanprosite/) and the NCBI-Conserved Domain Database (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) were done. All sequences having WRKYGQK domain were selected as WRKY gene family member and designated as PsWRKY.
In silico Characterization of PsWRKY Gene Family
The physicochemical properties of the PsWRKY, including the molecular weight (MW) and isoelectric point (pI), were determined using the online ExPASy-ProtParam tool (http://web.expasy.org/protparam/). The predictions of a signal sequence, which determines the subcellular location of PsWRKY were carried out using TargetP1.1 (http://www.cbs.dtu.dk/services/TargetP/). Organelle localization within cells was determined by WoLF PSORT (https://wolfpsort.hgc.jp/). The amino acid sequences of the WRKY proteins from A. thaliana (At), Brassica rapa (Br), M. truncatula (Mt), and N. tabacum (Nt), were retrieved from the plant transcription factor database v5.0 (Tian et al. 2020). These protein sequences were subjected to multiple sequence alignment using the Clustal X program. Phylogenetic tree was constructed via the Neighbor-Joining (NJ) method using the MEGA 7.0 software tool with 1000 bootstrap iterative (https://www.megasoftware.net/) (Kumar et al. 2016).
The conserved domains with PsWRKY proteins were anticipated using NCBI CD-Search, (www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) and Pfam search (http://pfam.xfam.org/). The architectures of motifs present in the PsWRKY protein family were discovered by MEME 5.0.1 (http://meme-suite.org/) (Bailey et al. 2015). To further characterize WRKY proteins based on the number of WRKY domains and the type of zinc-finger motif they possess, pea WRKY proteins were classified into three groups. Group I WRKY proteins have two WRKY domains along with a zinc-finger motif. Group II WRKY proteins feature a single WRKY domain and a CX4-5CX22-23HXH zinc-finger motif and can be further divided into subgroups. Group III WRKY proteins contain a single WRKY domain and a CX7CX23HXC zinc-finger motif.
Additionally, to study the evolution of WRKY proteins in peas compared to other legumes, WRKY proteins from nine different legume crops were downloaded from the Plant Transcription Factor database (https://planttfdb.gao-lab.org/index.php) and classified into three groups based on their WRKY domain and zinc-finger motif. A phylogeny of the selected legumes was constructed based on the NCBI taxonomy browser (https://www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi).
Chromosomal Location, Synteny and Gene Structure of PsWRKY Genes
The identified PsWRKY genes were depicted on their respective chromosomes using MG2C tool (http://mg2c.iask.in/mg2c_v2.1/) and pea genome annotation (Chao et al. 2015). The synteny analysis diagrams of M. truncatula, A. thaliana, and P. sativum were generated in a globe plot using the Circos program (http://circos.ca/) (Krzywinski et al. 2009). The exon–intron organization of PsWRKY was generated by Gene Structure Display Server 2.0 (GSDS 2.0) using transcript and genomic sequences information (Hu et al. 2015).
Transcriptome-Based Expression Analysis of PsWRKY Genes
Expression of PsWRKY gene family was analyzed under abiotic stress (sulfur and water stress) conditions using the transcriptome data from the NCBI SRA database (http://www.ncbi.nlm.nih.gov/bioproject/) having the accession number PRJNA517587. The raw RNA-seq reads were filtered to remove adaptor sequences, low-quality reads and k-mer contamination using the fastp tool, whereas the quality of trimmed reads after processing was assessed using the FastQC tool (Chen et al. 2018). The clean reads of each RNA-seq dataset were mapped to a reference Pea genome separately with the TopHat2 program (Trapnell et al. 2009). The mapped reads were assembled separately for each dataset into transcripts based on the reference Pea genome with StringTie (v1.3.1) software using the default Parameters (Pertea et al. 2016). All transcripts with strand information, number of fragments per kilobase of transcript per million mapped reads (FPKM) greater than 2 in multiple exons in at least one sample were retained for analysis. The quantification of the expression level of protein-coding transcripts and candidate WRKY genes was calculated based on the FPKM values using StringTie software. Heatmap was prepared using pheatmap module in R.
Plant Materials and Treatments
Pisum sativum L. (Cameor genotype) seed were used for the experiment. Seed were sterilized using 2% sodium hypochlorite then pre-germinated for 5 days in a control growth germinator at 20 °C in the dark. The germinated seed were then planted in 2-L pots filled with sterile perlite and sand at a temperature of 20 °C during the day and 15 °C at night with a 16-h photoperiod (250 μmol m2 s–1) under artificial lighting. All the growth condition for plant in control growth chamber were monitored using HortiMaX-Go greenhouse control system (https://ridder.com/ridder-hortimax-go/). The stress treatment procedure was in accordance with the methodology outlined by Zuber et al. (2013). We devised four separate treatments for our experiment, commencing from the seedling phase: a control group (adequately hydrated and supplied with all necessary nutrients), an S-stress group (suffering from sulfur deficiency due to limited hydration), a W group (subjected to water stress starting from the flowering stage for 9 days, followed by rehydration and sample collection), and a W + S group (enduring both sulfur deficiency and water stress). The stress conditions were sustained for a period of 9 days, after which immature seeds were harvested for RNA extraction and gene expression analysis. The plants were subsequently rehydrated with the necessary nutrients until maturity, as previously described by Henriet et al. (2019).
Mining Plant Stress-Related QTLs and WRKY Genes in Pea
We investigated plant stress-related QTLs for Pisum sativum using the Pulse Crop Database (https://www.pulsedb.org/). The search was focused on the Pisum sativum Cameor cultivar, specifically targeting QTLs associated with plant stress. Only QTLs with available genomic information were included in this study. Additionally, we identified WRKY genes located in the vicinity of these QTLs for further analysis.
RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
Total RNA was extracted using the SV Total RNA isolation system kit (Promega, Madison, WI, USA) as per manufacture protocol. RNA integrity was visualized on a 1.5% agarose gel. RNA was quantified using NanoDrop® Spectrophotometer ND-1000. cDNA synthesis was done using Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) according to the manufacturer’s protocol. Real-time PCR was done using SYBR Green chemistry with a LightCycler® 96 instrument (Roche, Mannheim, Germany). The PCR amplification parameters were 95 °C for 2 min, 40 cycles at 94 °C for 15 s then 60 °C for 30 s, and melting curve temperature ramping from 65 °C to 95 °C with fluorescence detection at every increment of 0.5 °C. Actin (Psat5g063760) was used as housekeeping gene for study. Three biological and two technical replicates were done in each experiment. For primer detail, see Supplementary Table 1. Quantification of fold change was done using 2−∆∆CT method (Livak and Schmittgen 2001). Each plate was run with respective positive control (genomic DNA as template), negative control (MQ water as template) and actin as housekeeping gene (internal control).
Results
Identification of PsWRKY Genes in Pisum sativum L.
Based on the amino acid sequence of WRKY genes from other plant system such as Arabidopsis, Medicago and Soybean, we mined a total 110 putative PsWRKY genes from pea genome. Out of all, 86 sequences showed the conserved WRKY motif (WRKYGQK). That’s confirmed using multiple sequence alignment (Supplementary Fig. 1). We named WRKY gene family from pea as PsWRKY1 to PsWRKY86 with their ID according to original pea genome. The length of PsWRKY proteins varies between 91 to 783 amino acids. The molecular weight and isoelectric points of these protein range from 10.02 to 90.49 kDa and 4.25 to 10.16, respectively (Table 1). The in silico subcellular localization results showed that most of the WRKY genes (94%) were localized in the nucleus, and only the WRKY1, WRKY5 and WRKY78 genes were localized in the cytosol.
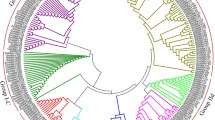
To further explore the phylogenetic relationship of the WRKY transcription factor family in Pea, a phylogenetic tree of Pea was constructed by using MEGA software as shown in Fig. 1a, which intuitively reflects the evolutionary status and grouping attribution of 86 members of the WRKY family. According to WRKY protein sequences and clustering analysis, the 86 identified members of the WRKY family were sorted into five groups namely I, II, III, IV and V. Group II and V were the largest with 20 genes in each group, followed by 17 in Group III, 16 in Group IV, and 13 in Group I the smallest group in phylogenetic distribution based on finding.

Phylogeny based classification and synteny analysis of PsWRKY genes from Pea. (a) Phylogeny-based classification of PsWRKY genes was determined using the neighbor-joining method with bootstrap value set to 1000 in the MEGA 6.0 software. (b) Synteny diagram of WRKY among A. thaliana, M. truncatula, and P. sativum genome. Colored line shows the different othrologous pair of PsWRKY with other genes from Medicago and Arabidopsis. The synteny circle, depicted by the interconnecting-colored lines between chromosomes, highlights the evolution relation between the chromosomes in the comparison with model plant genome databases
PsWRKY Genes Location on Chromosomes and Synteny
A total number of 86 identifiably distinct PsWRKY genes have been positioned across 7 separate chromosomes in the pea genome (Supplementary Fig. 2). The largest congregation of 20 PsWRKY genes is located on chromosome 5, while chromosome 3 accommodates 15 genes information can be also found in supplementary (Supplementary Table 2).
A comparative synteny analysis of WRKY genes among P. sativum, M. truncatula, and A. thaliana is depicted in Fig. 1b. The results of this analysis demonstrate a pronounced relationship among these plants in terms of duplication, evolution, functionality, and expression of WRKY genes. Synteny analysis shows the conservation of different WRKY genes compared to other plant species. We identified 69 pairs of orthologous genes, 32 of which were PsWRKY genes that showed orthology with both plant species (PsWRKY23-AT4G01720.1/Medtr2g023930.1 and PsWRKY46-AT4G31550.1/Medtr1g009613.1). 24 PsWRKY showed orthology only with Medicago, while 13 showed orthology only with Arabidopsis (for detail see Supplementary Table 2). When comparing chromosome numbers, we found that Pea chromosome 3 had a high number of orthologues with Medicago chromosome 7. This information is integral in comprehending the relationship and evolution of genes in terms of biological functionality.
PsWRKY Genes Structural Organization
To investigate the evolutionary progression of PsWRKY genes, an examination of their exon/intron boundaries was conducted. The gene size including coding and non-coding sequences ranges between around 0.5 and 12 kb (Fig. 2a). In our result, out of 86 identified PsWRKY genes, 44 have 3 exons, this is around 37% of all identified, while 10 individual PsWRKY contained total number of 4 exons, 15 PsWRKY have 5 exons, 7 PsWRKY have 2 exons and the 6 PsWRKY belong in group I and Group IV, PsWRKY6, PsWRKY11, PsWRKY16, PsWRKY23, PsWRKY54 and PsWRKY55 have maximum number of 6 exons out of total 86 individual PsWRKY genes, while the lowest exons were found on only two PsWRKY17 and PsWRKY52 genes that belong in group V. However, number of exons varies in between each of PsWRKY genes. To understand better clues about the evolution of the PsWRKY gene between their family members including specific features.

Organization of identified 86 PsWRKY in pea. (a) Indication of intron–exon structure with solid blue color boxes and solid yellow color boxes respectively. (b) Schematic distribution of conserved motifs in PsWRKY protein. By using MEME software tool (http://meme-suite.org), we identified 86 WRKY in pea protein in pea genome
Conserved Domains and Motif in PsWRKY
The motif is a defining structural feature located on a protein sequence and is used to determine its unknown function. This information is also crucial in tracing the evolutionary history of genes. Our study identified 10 unique conserved motifs in various PsWRKY proteins, labeled as motif 1 to motif 10 (Fig. 2b). A total of 86 protein sequences were found to possess the WRKYGQ motif, which serves as the signature of the WRKY gene family. The signature sequences of all 10 motifs for each PsWRKY can be found in Supplementary Table 1. Most PsWRKY proteins contain between 3 and 8 motifs, with Group I containing only two WRKY motifs, except for PsWRKY39 from Group V, which boasts the largest number of motifs at 9. The PsWRKY from Cluster Groups I and II share three distinct motif arrangements, consisting of 1, 2, and 3 motifs. Cluster Group V shares arrangements consisting of 4 to 8 motifs. WRKY transcription factors are characterized by a WRKYGQK heptapeptide at the N-terminal end and a zinc-finger motif (either CX4-5CX22-23HXH or CX7CX23HXC) at the C-terminal end. These proteins are categorized into three groups (I–III) based on the number of WRKY domains and the type of zinc-finger motif. Group I WRKY proteins have two WRKY domains and a zinc-finger motif. Group II WRKY proteins have a single WRKY domain paired with a CX4-5CX22-23HXH zinc-finger motif, and Group III WRKY proteins feature a single WRKY domain with a CX7CX23HXC zinc-finger motif.
To understand the evolution of WRKY proteins in pea compared to other legume crops, we downloaded WRKY transcription factors from the Plant Transcription Factor Database (https://planttfdb.gao-lab.org/index.php) for nine legumes (Cajanus cajan, Glycine max, Phaseolus vulgaris, Vigna angularis, Vigna radiata, Cicer arietinum, Medicago truncatula, Lotus japonicus, Arachis duranensis) and classified them into groups I, II, and III along with pea (Supplementary File 1). Additionally, we constructed a phylogenetic tree of these 10 legume crops based on taxonomy data, showing that pea is closely related to Medicago truncatula and Cicer arietinum (Supplementary Fig. 3a).
Analyzing the WRKY protein dynamics in the selected legumes revealed that Group II WRKY genes are the most evolved and widely distributed among all legume crops. Glycine max has the highest number of WRKY genes, while Vigna radiata has the fewest (Supplementary Fig. 3b).
WRKY Genes in the Vicinity of Plant Stress–Related QTLs in Pea
A total of 27 plant stress–related QTLs were identified in Pisum sativum using data from the Pulse Crop Database (https://www.pulsedb.org/). Among these, three QTLs were located on chromosome 3, while the remaining QTLs were on chromosome 5. Analysis of WRKY genes in the vicinity of these QTLs revealed six WRKY genes (PsWRKY26, PsWRKY28, PsWRKY49, PsWRKY51, PsWRKY52, PsWRKY53) near some of the QTLs (Supplementary File 2). Notably, the WRKY gene PsWRKY50 was found within a QTL associated with resistance to Pseudomonas syringae. For detailed information, refer to Supplementary File 2.
Expression Analysis Profile of Pea WRKY Genes under Different Abiotic Stress
Out of the 86 identified PsWRKY genes, 36 gene transcripts were found to be expressed under abiotic stress according to the transcriptome data (Fig. 3). Based on transcriptome data analysis PsWRKY3, PsWRKY19, and PsWRKY81 were highly expressed in immature seeds in water stress under limiting sulfur conditions. PsWRKY11, PsWRKY8, PsWRKY47, and PsWRKY50 were downregulated under limiting sulfur conditions. To validate the transcriptome data, we selected 10 PsWRKY genes for real-time PCR experiment based on the differentially expressed genes found on analysis of transcriptome data (Fig. 4b). PsWRKY23, PsWRKY58, PsWRKY64, and PsWRKY83 exhibited significant upregulation under water stress conditions. Conversely, under sulfur stress, PsWRKY49 and PsWRKY81 were found to be downregulated, while PsWRKY58 and PsWRKY84 demonstrated upregulation in the same conditions. PsWRKY43 showed significant downregulation when subjected to water stress combined with sulfur deficiency, whereas PsWRKY3 exhibited significant upregulation as depicted in Fig. 4a.

Heat map-based representation expression profile of PsWRKY genes in abiotic stresses (Water stress, sulfur deficiency including combined of both) using the RNA-seq data in the immature seed of pea cultivar “Cameor”. (SRR8709721, SRR8709723, SRR8709724 represent RNA-Seq of Pisum sativum: immature seeds, water-stressed for 9 days, replicate 1, 2 and 3 respectively; SRR8709730, SRR8709731, SRR8709729 represent RNA-Seq of Pisum sativum immature seeds, water-stressed for 9 days under limiting sulfur conditions, replicate 1, 2 and 3 respectively; SRR8709722, SRR8709725, SRR8709726 represents RNA-Seq of Pisum sativum immature seeds, well-watered under limiting sulfur conditions, replicate 1, 2 and 3 respectively. SRR8709719, SRR87097120, SRR8709718 represent RNA-Seq of Pisum sativum: immature seeds, well-watered, replicate 1, 2 and 3 respectively

Expression profile of the selected PsWRKY genes based on transcriptome expression profile using qRT-PCR methodology. (a) qRT-PCR based validation of the selected PsWRKY genes expression of ten WRKY in immature pea seed of “Cameor” cultivar grown under water stress, sulfur stress and water combine with sulfur stress. (b) List of selected PsWRKY genes based DeSEQ analysis of transcriptome data. qRT-PCR experiment was conducted with three biological replicates. Student t test for carried out statistical analysis of data. * Indicate the p-value < 0.05
Discussion
P. sativum L is an important crop and third largest legume crop by production. The seeds are rich sources for protein. The growth and production of seeds depend upon various factors including adverse environmental conditions. Water and sulfur are essential abiotic factors that play a pivotal role in seed development. The composition of seed globulin is significantly modulated by sulfur deficiency, water stress, and their synergistic effect (Henriet et al. 2019). The transcriptional regulation of these biological processes involves various transcription factors (TFs), with the WRKY gene family being a prominent TF that is implicated in the transduction of signaling cascades and the modulation of plant growth, development, and stress responses to diverse abiotic stresses (Bakshi and Oelmüller 2014; Phukan et al. 2016). Therefore, the characterization of the WRKY gene family in pea is of paramount importance for a comprehensive understanding of the molecular mechanisms underlying seed development and stress responses. Thus far, numerous reports on WRKY genes have been extensively characterized in various plant species, including A. thaliana (Dong et al. 2003), M. truncatula (Hui Song 2014), Cajanus cajan (Singh et al. 2019), Cicer arietinum (Song et al. 2018), Lotus japonicas (Song et al. 2014), and Glycine max (Yin et al. 2013). However, best of knowledge till date PsWRKY gene family is not characterized in pea. Recently published pea genome sequence information provided an opportunity to carryout genome wide identification and characterization. Among legumes, WRKY genes have been extensively characterized in several species, such as M. truncatula (140), Cajanus Cajan (97), Cicer arietinum (94), and Glycine max (296). In this study, we identified 86 WRKY-encoded genes in the pea genome which is comparatively less in number than other legumes. In pea we found most of PsWRKY were contained WRKYGQK heptapeptide domain. However, this domain is reported to bind with w-box cis element with core sequence C/TTGACC/T which lead to activate downstream genes to regulate different physiological and stress response gene in Arabidopsis (Eulgem et al. 2000). Some of Q-box variant such as WRKYGKK was found in five PsWRKY, whereas WRKYGEK were found in four of PsWRKY genes. Intriguingly, we found four other variants of Q-BOX domain: WRKYGMK, WRKYGRK, WRKYGDK and WRKYGHK. Four individuals PsWRKY contain one of each of different variation in Q-box domain, out of this one was founded with a bit unique variation in G and E-box that contain WRKYEDK motifs. In other plant such variation is also reported (Ciolkowski et al. 2008) which suggested that variation of WRKYGQK domain could affect the specificity of DNA binding, and the WRKY genes lost the motif may bind to other specific sequences. These variation in domain were also reported in Medicago truncatula and other legume crop (Song et al. 2018). A legume-specific WRKY gene, GmWRP1 (Glyma14g37960), was reported in Glycine max in 2015 (Chi et al. 2015). However, in pea and the other nine legume species studied, no closely related homologs were found. In pea, the closest homolog based on BLASTP results was PsWRKY17, which has 59% identity to GmWRP1. Among the ten legume crops studied, the WRKY gene Phvul.008G251700.1 from Phaseolus vulgaris showed the highest similarity to GmWRP1, with 69% identity. In a phylogenetic study, WRKY transcription factors (TFs) in pea were classified into five major groups, highlighting their close relationships with those in Arabidopsis and Medicago (Hui Song 2014). This distribution reveals some similarities among the compared plants within the phylogenetic groups, though there are also notable differences within these groups. Our exon analysis indicates structural variation among the identified PsWRKYs, suggesting functional diversification among PsWRKYs and their roles in various developmental processes and stress responses. Phylogenetic tree also explains about the putative role of pea WRKY genes based on the functionally characterized WRKY gene from Medicago and Arabidopsis. For example, PsWRKY23 gene from group II was close to AtWRKY42, which is known as a positive regulator of leaf senescence in Arabidopsis (Niu et al. 2020). A concise report has indicated that number of WRKY transcription factors (TFs) play a significant role in the response to various stimulate in plants such as 29 WRKY in hormone signaling, 12 in ion transport, 31 in antioxidant response, and 44 WRKY in osmotic response pathway were explored (Price et al. 2022). These findings suggest strong base about the role of WRKY gene in different biological process in plant, and it may aid in comprehending the levels of evolution by investigating the pea genome, as well as by recognizing their evolutionary connection, it can provide insight into pea physiological processes.
Synteny analysis was used to understand the gene duplication, evolutionary progress and expansion of gene family (Cannon et al. 2004). Plant genome enlargement is predominantly influenced by occurrence of tandem and segmental duplication (Panchy et al. 2016). Evolution and duplication in gene can occur within the genome of same plant species, it’s also found higher with closely related plant species. Here in our finding, we explored within two closely related plant species among Arabidopsis and Medicago. In our result, it’s showed the WRKY gene conservation between Arabidopsis, Medicago and pea plant genome. Comparative mapping with Arabidopsis thaliana and Medicago tranculata established the paralogous and orthologues evolution relation with pea. However, Arabdopsis chromosome number 4 showed their higher orthologues with pea PsWRKY genes. Our results suggest that the pea genome is dynamic, and has undergone evolution, in part due to the influence of mobile elements in shuffling, duplication of chromosomes. Total number of 86 PsWRKY genes were distributed in total number of 7 PsWRKY chromosome (Table 1and Supplementary Fig. 1). In our study, we found that sixteen PsWRKY indicated the synteny with A. thaliana, and fourteen with M. truncatula. Higher PsWRKY gene orthologs are found in pea in comparison to Arabidopsis. These data demonstrate that pea plant WRKY gene expansion appeared to have occurred unequally during evolution and findings demonstrate the complex and dynamic evolution of the pea plant WRKY gene family.
Drought tolerance and sulfur deficiency are one of the major challenges in plant development and growth, leading to poor crop yield and quality. Because adverse environmental conditions and climate change such as water scarcity and soil nutrient occur, they are negatively impacting on plant growth and their yield productivity (Brendel 2021). In a biological process of plant development, under water and sulfur stress, the role of WRKY gene is important and reported in many other plants, such as in TaWRKY33 in wheat and OsWRKY11 in rice increased the level of drought tolerance (Wu et al. 2009; He et al. 2016). In soybean GmWRKY13, and AtWRKY57 in Arabidopsis improve drought tolerance (Zhou et al. 2008; Jiang et al. 2012). In our study, we observed a significant upregulation of PsWRKY23, PsWRKY58, PsWRKY64 and PsWRKY84 in response to water stress. Additionally, we found that PsWRKY23 is closely related to AtWRKY42 (AT4g04450.1) based on a phylogenetic tree analysis. AtWRKY42 is known to play a role in phosphate translocation and acquisition, which is influenced by a modulatory process that helps maintain phosphate homeostasis in Arabidopsis (Su et al. 2015). In response to sulfur stress, PsWRKY58 was significantly upregulated in pea (Fig. 4), and some reports suggest that manipulating transporters can improve sulfur utilization efficiency in Arabidopsis (Hawkesford 2000). Additionally, the sulfur deficiency-induced gene (sdi1) is important for increasing sulfate utilization under limited sulfur conditions in plants (Howarth et al. 2009). Despite sulfur’s significant role in plant improvement, no study has been conducted on impact of sulfur stress on WRKY genes in pea. During water and sulfur stress, we observed the downregulation of PsWRKY43 and the upregulation of PsWRKY03. In contrast, transcriptome data analysis by Henriet et al. (2019) highlighted only upregulated WRKY genes, specifically PsWRKY9, PsWRKY10, and PsWRKY82. These discrepancies in upregulated genes may be due to slight variations in the datasets used in our study compared to theirs. Furthermore, our results underscore the importance of qPCR validation for transcriptome data. Therefore, the characterization and expression analysis of PsWRKY gene in response to water and sulfur stress provide us that some key candidate genes could help in the development of strategies to improve and carry out the research in understanding role of WRKY gene in the sulfur and water stress in context to seed biology.
Conclusions
This study aimed to conduct a comprehensive identification and characterization of the 86 PsWRKY genes from the P. sativum genome assembly. We elucidated the expression profiles of PsWRKYs and their distinct expression patterns in response to sulfur, water, and their combined stresses, revealing the presence of a complex molecular regulatory network in P. sativum. Our findings provide novel insights into the evolutionary and genomic divergence of the WRKY gene family, identifying key water and sulfur stress response genes, including PsWRKY23, PsWRKY58, PsWRKY83, and PsWRKY49. The present study paves the pathway toward improving crop trait against abiotic stress.
References
Bailey TL, Johnson J, Grant CE, Noble WS (2015) The MEME suite. Nucleic Acids Res 43:W39–W49. https://doi.org/10.1093/NAR/GKV416
Bakshi M, Oelmüller R (2014) WRKY transcription factors. Plant Signal Behav 9:e27700. https://doi.org/10.4161/PSB.27700
Brendel O (2021) The relationship between plant growth and water consumption: a history from the classical four elements to modern stable isotopes. Ann For Sci 78:1–16. https://doi.org/10.1007/s13595-021-01063-2
Burke R, Schwarze J, Sherwood OL et al (2020) Stressed to death: the role of transcription factors in plant programmed cell death induced by abiotic and biotic stimuli. Front Plant Sci 11:565787
Burstin J, Salloignon P, Chabert-Martinello M et al (2015) Genetic diversity and trait genomic prediction in a pea diversity panel. BMC Genomics 16:1–17. https://doi.org/10.1186/s12864-015-1266-1
Cannon SB, Mitra A, Baumgarten A et al (2004) The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol 4:1–21. https://doi.org/10.1186/1471-2229-4-10
Chao JT, Kong YZ, Wang Q, Sun YH et al (2015) MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Yi Chuan Hereditas 37(1):91–97
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. In: Bioinformatics. Oxford Academic, pp i884–i890
Chi Y, Yang Y, Li G et al (2015) Identification and characterization of a novel group of legume-specific, golgi apparatus localized WRKY and Exo70 proteins from soybean. J Exp Bot 66(11):3055–3070. https://doi.org/10.1093/jxb/erv104
Ciolkowski I, Wanke D, Birkenbihl RP, Somssich IE (2008) Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol Biol 68:81–92. https://doi.org/10.1007/s11103-008-9353-1
Devaiah BN, Karthikeyan AS, Raghothama KG (2007) WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol 143:1789–1801. https://doi.org/10.1104/pp.106.093971
Dong J, Chen C, Chen Z (2003) Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol Biol 51:21–37. https://doi.org/10.1023/A:1020780022549
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206. https://doi.org/10.1016/S1360-1385(00)01600-9
FAO’s Statistical Yearbook 2023 (2023) Food and Agriculture Organization of the United Nations (FAO). https://www.fao.org/faostat/en/#home
Gao QM, Venugopal S, Navarre D, Kachroo A (2011) Low oleic acid-derived repression of jasmonic acid-inducible defense responses requires the WRKY50 and WRKY51 proteins. Plant Physiol 155:464–476. https://doi.org/10.1104/pp.110.166876
Hawkesford MJ (2000) Plant responses to sulphur deficiency and the genetic manipulation of sulphate transporters to improve S-utilization efficiency. J Exp Bot 51:131–138. https://doi.org/10.1093/jexbot/51.342.131
He GH, Xu JY, Wang YX et al (2016) Drought-responsive WRKY transcription factor genes TaWRKY1 and TaWRKY33 from wheat confer drought and/or heat resistance in Arabidopsis. BMC Plant Biol 16:1–16. https://doi.org/10.1186/s12870-016-0806-4
Henriet C, Aimé D, Térézol M et al (2019) Water stress combined with sulfur deficiency in pea affects yield components but mitigates the effect of deficiency on seed globulin composition. J Exp Bot 70:4287–4303. https://doi.org/10.1093/jxb/erz114
Howarth JR, Parmar S, Barraclough PB, Hawkesford MJ (2009) A sulphur deficiency-induced gene, sdi1, involved in the utilization of stored sulphate pools under sulphur-limiting conditions has potential as a diagnostic indicator of sulphur nutritional status. Plant Biotechnol J 7:200–209. https://doi.org/10.1111/j.1467-7652.2008.00391.x
Hu B, Jin J, Guo AY et al (2015) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31:1296–1297. https://doi.org/10.1093/bioinformatics/btu817
Hu Y, Chen X, Shen X (2022) Regulatory network established by transcription factors transmits drought stress signals in plant. Stress Biol 2:26. https://doi.org/10.1007/s44154-022-00048-z
Hui Song ZN (2014) Genome-wide identification and analysis of WRKY transcription factors in Medicago truncatula. Yi Chuan 36:152–168
Jiang Y, Liang G, Yu D (2012) Activated expression of WRKY57 confers drought tolerance in arabidopsis. Mol Plant 5:1375–1388. https://doi.org/10.1093/mp/sss080
Kreplak J, Madoui MA, Cápal P et al (2019) A reference genome for pea provides insight into legume genome evolution. Nat Genet 51:1411–1422. https://doi.org/10.1038/s41588-019-0480-1
Krzywinski M, Schein J, Birol I et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. https://doi.org/10.1101/gr.092759.109
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/MOLBEV/MSW054
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Nan H, Li W, Lin YL, Gao LZ (2020) Genome-wide analysis of WRKY genes and their response to salt stress in the wild progenitor of Asian cultivated rice. Oryza Rufipogon Front Genet 11:478232. https://doi.org/10.3389/fgene.2020.00359
Niu F, Cui X, Zhao P et al (2020) WRKY42 transcription factor positively regulates leaf senescence through modulating SA and ROS synthesis in Arabidopsis thaliana. Plant J 104:171–184. https://doi.org/10.1111/tpj.14914
Panchy N, Lehti-Shiu M, Shiu SH (2016) Evolution of gene duplication in plants. Plant Physiol 171:2294–2316. https://doi.org/10.1104/pp.16.00523
Pea Genome project (2019) The Pea Genome Consortium INRA Dijon managed the Consortium. INRA-GENOSCOPE cooperation
Pertea M, Kim D, Pertea GM et al (2016) Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11:1650–1667. https://doi.org/10.1038/nprot.2016.095
Phukan UJ, Jeena GS, Shukla RK (2016) WRKY transcription factors: molecular regulation and stress responses in plants. Front Plant Sci 7:192140
Price L, Han Y, Angessa T, Li C (2022) Molecular pathways of WRKY genes in regulating plant salinity tolerance. Int J Mol Sci 23:10947
Singh A, Singh PK, Sharma AK et al (2019) Understanding the role of the WRKY gene family under stress conditions in pigeonpea (Cajanus cajan l.). Plants 8:214. https://doi.org/10.3390/plants8070214
Song H, Wang P, Nan Z, Wang X (2014) The WRKY transcription factor genes in Lotus japonicus. Int J Genomics 2014:1–15. https://doi.org/10.1155/2014/420128
Song H, Sun W, Yang G, Sun J (2018) WRKY transcription factors in legumes. BMC Plant Biol 18:1–13. https://doi.org/10.1186/s12870-018-1467-2
Su T, Xu Q, Zhang FC et al (2015) WRKY42 Modulates phosphate homeostasis through regulating phosphate translocation and acquisition in Arabidopsis. Plant Physiol 167:1579–1591. https://doi.org/10.1104/pp.114.253799
Tayeh N, Aluome C, Falque M et al (2015) Development of two major resources for pea genomics: the GenoPea 13.2K SNP array and a high-density, high-resolution consensus genetic map. Plant J 84:1257–1273. https://doi.org/10.1111/tpj.13070
Tian F, Yang DC, Meng YQ et al (2020) PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res 48:D1104–D1113. https://doi.org/10.1093/nar/gkz1020
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. https://doi.org/10.1093/bioinformatics/btp120
Wu X, Shiroto Y, Kishitani S et al (2009) Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep 28:21–30. https://doi.org/10.1007/s00299-008-0614-x
Yang T, Liu R, Luo Y et al (2022) Improved pea reference genome and pan-genome highlight genomic features and evolutionary characteristics. Nat Genet 54:1553–1563. https://doi.org/10.1038/s41588-022-01172-2
Yin G, Xu H, Xiao S et al (2013) The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol 13:1–19. https://doi.org/10.1186/1471-2229-13-148
Yu S, Ligang C, Liping Z, Diqiu Y (2010) Overexpression of OsWRKY72 gene interferes in the abscisic acid signal and auxin transport pathway of Arabidopsis. J Biosci 35:459–471. https://doi.org/10.1007/s12038-010-0051-1
Zhao K, Zhang D, Lv K et al (2019) Functional characterization of poplar WRKY75 in salt and osmotic tolerance. Plant Sci 289:110259. https://doi.org/10.1016/j.plantsci.2019.110259
Zhou QY, Tian AG, Zou HF et al (2008) Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol J 6:486–503. https://doi.org/10.1111/j.1467-7652.2008.00336.x
Zhu L, Li S, Ouyang M et al (2022) Overexpression of watermelon ClWRKY20 in transgenic Arabidopsis improves salt and low-temperature tolerance. Sci Hortic (Amsterdam) 295:110848. https://doi.org/10.1016/j.scienta.2021.110848
Zuber H, Poignavent G, Le Signor C et al (2013) Legume adaptation to sulfur deficiency revealed by comparing nutrient allocation and seed traits in Medicago truncatula. Plant J 76:982–996. https://doi.org/10.1111/tpj.12350
Funding
This study was supported by research grant (HORhn-802.14) funded by the Ministry of Agriculture and Rural Development, Poland. PK acknowledges the Institute of Plant Genetics, Polish Academy of Sciences, Poland for PhD fellowship. We thank Vanessa Vernoud from INRAE, France, for providing “cameor” pea cultivar seed.
Author information
Authors and Affiliations
Contributions
Conceptualization, PA and PK; formal analysis, PK, PA, KK, AA, ST, HS; Transcriptome analysis, PA and PK; Experiment and real time PCR, MG, PK; original draft preparation, PK; Writing—Review and editing PA, WS, AA, PK, HS. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, P., Alok, A., Kaur, K. et al. Genome-Wide Identification of WRKY Transcription Factors in Pea (Pisum sativum L.) and their Response to Sulfur and Water Stress. Plant Mol Biol Rep (2024). https://doi.org/10.1007/s11105-024-01498-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11105-024-01498-7