
Abstract
Background and Aims
Nitrogenous fertilizers provide a short-lived benefit to crops in agroecosystems, but stimulate nitrification and denitrification, processes that result in nitrate pollution, N2O production, and reduced soil fertility. Recent advances in plant microbiome science suggest that genetic variation in plants can modulate the composition and activity of rhizosphere N-cycling microorganisms. Here we attempted to determine whether genetic variation exists in Zea mays for the ability to influence the rhizosphere nitrifier and denitrifier microbiome under “real-world” conventional agricultural conditions.
Methods
To capture an extensive amount of genetic diversity within maize we grew and sampled the rhizosphere microbiome of a diversity panel of germplasm that included ex-PVP inbreds (Z. mays ssp. mays), ex-PVP hybrids (Z. mays ssp. mays), and teosinte (Z. mays ssp. mexicana and Z. mays ssp. parviglumis). From these samples, we characterized the microbiome, a suite of microbial genes involved in nitrification and denitrification and carried out N-cycling potential assays.
Results
Here we are showing that populations/genotypes of a single species can vary in their ecological interaction with denitrifers and nitrifers. Some hybrid and teosinte genotypes supported microbial communities with lower potential nitrification and potential denitrification activity in the rhizosphere, while inbred genotypes stimulated/did not inhibit these N-cycling activities. These potential differences translated to functional differences in N2O fluxes, with teosinte plots producing less GHG than maize plots.
Conclusion
Taken together, these results suggest that Zea genetic variation can lead to changes in N-cycling processes that result in N leaching and N2O production, and thereby are selectable targets for crop improvement. Understanding the underlying genetic variation contributing to belowground microbiome N-cycling into our conventional agricultural system could be useful for sustainability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
More than half the world’s population depends on crops grown with synthetic nitrogen (N) fertilizers (Bowles et al. 2018). Regrettably, most of these synthetic N fertilizer inputs escape the agroecosystem, degrade natural areas, and harm human health (Vitousek et al. 2013; Zhang et al. 2015). Stimulation of soil nitrogen cycling microorganisms (i.e. nitrifiers and denitrifiers) is a major contribution to “leaky” agricultural systems (Kuypers et al. 2018). To improve the sustainability of agricultural systems, we need to understand how ecological drivers, such as genetically controlled plant–microbe interactions play out under “real world” conditions to influence soil’s nitrogen cycling microorganisms and the movement of nitrogen, and how this can be managed to reduce nutrient losses (Moreau et al. 2015, 2019; Favela et al. 2023).
Genetic variation within crop species has been shown to play a significant role in plant-microbiome assembly and recruitment (Peiffer et al. 2013b; Bouffaud et al. 2016; Walters et al. 2018). Across large-scale and multi-year field trials, researchers find consistent sets of heritable core microbial taxa associated with specific plant genotypes (Walters et al. 2018; Xu et al. 2018). These community composition differences are functionally relevant, as microorganisms contain a diverse biochemical repertoire that allows plants to escape nutrient, drought, and pathogen stress (Philippot et al. 2013a, b; Compant et al. 2019; Trivedi et al. 2020). In the rhizosphere, plants exude chemical cocktails of metabolites, the production of which is directed by the plant genome. These exudate traits, in tandem with root phenology and physiology, act as an ecological filter that has direct fitness consequences for the surrounding soil microbial communities (Huang et al. 2019; Canarini et al. 2019; Favela et al. 2023; Baggs et al. 2023). Soil microorganisms that are phytochemically competent to the ecological filter of the rhizosphere are able to survive and persist near the plant root (Philippot et al. 2013a, b). It follows, then, that the ecological filtering for rhizosphere occupancy that arises from genotypic variation directing microbiome selection should have consequences for the biogeochemical cycling activities carried out by the rhizosphere microbiome.
Rhizosphere nitrifying and denitrifying microorganisms are key contributors to essential ecosystem functions. Processes such as nitrification and denitrification are controlled by microorganisms in the soil interacting with the abiotic environment and can result in a considerable loss of nitrogen from an ecosystem (Philippot et al. 2007; Davidson et al. 2012). An extensive amount of work in nitrifiers and denitrifiers has shown that edaphic soil history and agricultural management plays a significant role in structuring the functions of these communities (Qian et al. 1997; Hayatsu et al. 2021; Raglin et al. 2022). Furthermore, it is well known that the carbon/nitrogen inputs and uptake from plants can structure the functions of these critical N-cycling taxa (Haller and Stolp. 1985). In addition to this we know that the cultivated crop above these soils contributes to their nitrogen functions (Weier et al. 1993; Lucas et al. 2023). Furthermore, emerging research is beginning to show that genetic variation between species and within plant species can have a considerable role in driving both community composition and the activity of biogeochemically-relevant microorganisms (Subbarao et al. 2013; Pérez-Izquierdo et al. 2019). For example, it has been shown that historic selection on plant genotype can drive the assemblage of the nitrogen cycling rhizosphere microbiome, both by changing the taxonomic composition and representation of nitrogen-cycling functional genes (Bouffaud et al. 2016; Favela et al. 2021). Yet we lack evidence linking genotype-driven microbiome changes to altered nitrogen cycling processes within the complex and more microbially diverse conventional agricultural systems. For example, large research efforts have focused on showing plant inhibition of nitrification under highly-controlled culture conditions with only a few microbial isolates (Kaur-Bhambra et al. 2022; Otaka et al. 2023; Petroli et al. 2023). While these results are mechanistically interesting, they are no guarantee for how diverse soil communities will behave. Here, we attempted to address this gap in an agroecological field setting. Specifically, we sought to determine whether plant genetic variation involved in root phenotypes is an important explanatory variable for understanding changes in the soil microbiome, particularly microbial functional groups, and to connect variation in this extended phenotype back to changes in important nitrogen cycling processes.
To evaluate the influence of plant genetic variation in root phenotypes on nitrogen cycling microbial functional groups, we grew a diverse panel of Zea mays (including elite inbreds, their hybrids, and wild teosintes) and measured their contribution to differences in microbial community assembly and nitrogen cycling processes. By doing this in a single field, we were able to control for edaphic factors and estimate the plant genetic contribution driving soil microbiome function in an agronomically relevant field setting. Previously, our greenhouse research in elite inbred maize suggested that breeding of maize has resulted in changes to microbiome recruitment, with more modern cultivars recruiting fewer N-fixing taxa and more denitrifiers/nitrifiers; outcomes that we would predict would change nutrient retention, and thereby shape sustainability (Favela et al. 2021, 2022). Informed by this prior research, we included maize hybrids to estimate if microbiome functions would be regained through heterosis (Wagner et al. 2021). In addition to this, within Zea mays, we found microbiome recruitment and N-cycling functional groups differed the most between modern inbred maize and wild teosinte (Favela et al. 2022). Therefore, wild Zea was included as an outgroup to evaluate the influence of pre-domestication genetics and their ecological filtering traits on microbiome assembly and function (Brisson et al. 2019; Schmidt et al. 2020). These treatments give us an understanding of how much plant genetic variation is necessary to induce changes in the microbiome of soils and provide insight into how domestication and breeding for performance in high N environments altered microbiome functions. With this information, we hoped to better understand how genetic alterations in maize can have cascading effects in plant microbiome recruitment and nitrogen cycling activity in conventional agricultural settings. This work in maize serves as a model for optimizing sustainable N cycling in other modern crops such as sorghum, which is an emerging bioenergy feedstock crop. Understanding the potential for plant genetics to contribute to these functional processes in an agroecological field setting is critical for improving the sustainability of maize production.
Methods
Field design
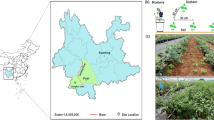
Field plots were located at the Crop Sciences Research and Education Center (CSREC)—South Farms at the University of Illinois, Urbana-Champaign, IL (40°03′30.4"N 88°13′50.4"W). We used a panel of Zea cultivars that encompassed modern ex-PVP inbreds, their hybrids, and the wild progenitor of maize, teosinte (27 genotypes in total: Description of genotypes in Table S1). Germplasm was obtained from USDA-ARS Germplasm Resources information Network (https://www.ars-grin.gov/). Teosinte was represented by the subspecies Zea mays spp. mexicana and Zea mays spp. parviglumis, with three genotypes per subspecies). Across our field, each cultivar was replicated four times. The replications were arranged in a randomized complete block design (Fig. S1-2). Each maize plot contained four rows with 15 plants per row. Maize inbreds and hybrids were machine planted on 4/30/2017 while teosinte cultivars were hand planted 5/19/2017. Due to seed limitations teosinte was planted in 2-row plots. Fields are managed by the CSREC in a Corn-Soy rotation, tilled yearly, and plant density of 32,000 plants/acre. Fertilization application was applied homogenously across all plots and match those on a typical conventional agricultural farm in Illinois (82 N kg/acre, 35 P kg/acre, 23 K kg/acre). CSREC staff carried out fertilization the week before planting (4/19–4/24). Urea-ammonium-nitrate (28%) solution was applied to fields at a rate of 60 gallons/acre.
Sample collection
Plots were sampled three times. Plants were sampled in approximately in the V4 (Maize and teosinte: 6/5/17), V6 (Maize: 6/20/17, Maize and teosinte:7/20/17), and R2 (Maize:7/12/17, Teosinte: 8/11/17) growth stages across the growing season (Nielsen et al. 1993). Staggered planting and sampling of teosinte and maize was carried out to control for differential growth rates among these genotypes. Staggered sample collection was accounted for in statistical models for nitrification and denitrification by modeling growth stage as a fixed factor and sampling date as a random factor. We found minor influence (R2 < 1%) of both. At each sampling event, four individual plants per plot were sampled and combined into a composite sample. Individual plants were not resampled. To accommodate the size of the experiment, true “rhizosphere samples” were not collected. Instead, root zone soil was collected as a proxy of the rhizosphere. Root zones soils here consisting of a mixture of rhizosphere and bulk soil present in the immediate proximity of the plant. This method allowed us to sample enough soil for nitrogen cycling assays and molecular work without having to carry out time consuming rhizosphere soil extractions from the roots. Samples consisted of a soil core (10 cm depth) obtained from the root zone of the plant (2 cm away from base of stem). Four root zone soil cores were collected from each plot and combined into a composite sample. Composite samples were placed on ice until they were transported to the lab. Processing of soil cores before assays and molecular work consisted of removal of all root tissue present in sample and homogenization of soil cores. Homogenization was performed by hand, for a minute per samples. Sieving homogenization was avoided as to not simulate mineralization and aeration. Once in the lab, soils were stored at 4 °C awaiting potential nitrification and potential denitrification assays (within 5 h). Aliquots for DNA extraction were frozen immediately. The frozen DNA aliquots were placed into 15 mL centrifuge tubes and lyophilized before DNA extraction (0.5 g total) using the FastDNA for Soil DNA extraction kit (MPBio, Solon, OH).
Soil samples were collected at the end of the season (9/15/17) for soil chemistry analyses carried out by Waypoint Analytical (Champaign, IL, USA). Analysis included buffer pH, nitrate, phosphorus, potassium, sulfur, manganese, copper, organic matter, estimated nitrogen release as estimated, cation exchange capacity, pH, sodium, iron, and boron. These were selected as they are typical reliable indictors of soil nutrition and would give us insights into how the soil nutrient profile was by the end of the season. Buffer pH measures the resistance of the soil to change pH and is done by adding a 7.5 pH solution to a soil and measuring the relative change. Estimated nitrogen release is calculated from the total SOC and is the amount of N thought to be stored with carbon. Cation exchange capacity and pH measure the number of cations in the soil and H + ions in the soil. Finally, all other measurements constitute macro and micronutrients important to plant nutrition.
Potential nitrification assay
The potential nitrification assay was developed and modified from (Schinner et al. 1996). This assay was performed at substrate saturation and values presented should be interpreted as the maximum potential rate of the transformation of ammonium to nitrite, the first-rate limiting step of nitrification. In principle, this assay uses ammonium sulfate (0.19 M, pH 8.5) as the substrate for the first step of nitrification during a 5-h incubation. Nitrite products released during the incubation period were extracted with potassium chloride and concentration is determined colorimetrically at 520 nm. Sodium chlorate (1.5 M) was added to the assay to inhibit nitrite oxidation during the incubation period. Sample tubes were incubated at room temperature on an orbital shaker for 5 h and control tubes were stored at -20 °C for 5 h. After incubation and thawing, KCl was used to extract nitrite from both samples and controls. Potential nitrification rates were arithmetically adjusted by initial soil moisture, soil weight, % dry matter, and initial nitrite in the sample. Potential nitrification data analyses are presented as (log (ng N g d.w soil −1 h −1)) and percent change ((population mean – genotype mean) divided by population mean).
Potential denitrification rates by acetylene-inhibition assay
Potential denitrification enzyme assays (here by referred to DEA) were carried out using a modified version of previously described assays (Schinner et al. 1996; Peralta et al. 2016). Field-moist root-zone soil samples were incubated under anaerobic conditions in the presence of 10% acetylene or 100% helium for 3 h at 25 °C. The assay was performed on 25 g of root-zone soil in 125-ml glass Wheaton bottles. Incubations were carried out at substrate saturation of carbon (dextrose 500 mg/L) and nitrogen (nitrate 720 mg/L). Chloramphenicol (10 mg/L) was added to the incubation to act as a bacteriostatic agent to prevent further microbial growth and protein synthesis. The incubation bottles were purged of oxygen with either helium or acetylene. Helium samples were used to estimate the amount of incomplete denitrification produced during the assay (referred to potential (N2O) DEA). Acetylene purged samples were used to measure complete + incomplete denitrification (referred to as potential (N2O + N2) DEA). Acetylene is a commonly known inhibitor of nitrous oxide reduction. Potential complete DEA (N2) was calculated by subtracting (N2O) DEA from (N2O + N2) DEA. These results are present in the supplemental materials (Table S8.7–9). Initial and final gas samples were collected at the start and end of the incubation period. Initial and final nitrous oxide in gas samples were quantified using a GC-2014 Gas Chromatograph (Shimadzu, Kyoto, Japan) with an electron capture detector (GC-ECD). Potential denitrification rates were arithmetically adjusted by initial soil moisture, soil weight, % dry matter, sample volume, and headspace. Potential denitrification data is presented as (log (ng N g d.w soil −1 h −1)) and percent change ((population mean – genotype mean) divided by population mean).
Carbon substrate utilization
Carbon substrate utilization assay was carried out using Biolog EcoPlates (Biolog Inc., Hayward, CA, USA). Biolog EcoPlates are a simple method to characterize the metabolic functions of microbial populations. Plates contain 31 different carbon substrates that can be used as the sole source of carbon. Each substrate is bound to a tetrazolium dye that changes colors once carbon compound is degraded. Assays were carried out with soils collected from the 7/20/17 sampling timepoint (represented by the R2 timepoint). Root zone soils (0.5 g) were diluted (1:4) in PBS, vortexed and centrifuged. Soil mixture supernatant (600 µL) was further diluted (1:25) in PBS. The diluted soil mixture was then added to the microplates and incubated for 5 days at room temperature. Absorbance at 590 nm was measured every 24 h using an Epoch microplate spectrophotometer (Santa Clara, CA, USA). Microbial metabolism was calculated as suggested in (Classen et al. 2003). This comparison focused on inbred B73, hybrid check1, and PI566677 teosinte (4 replicates per genotype). Data used in analysis consisted of carbon substrate usage (average well development) across 4 technical replicates at end of 5-day incubation. The SIMPER procedure in the ‘vegan’ R package was used to determine differences in substrate utilization across treatments (Oksanen 2017).
Microbial community amplicon sequencing
For this experiment, we characterized the microbiome and diagnostic functional genes related to transformations that occur in the nitrogen cycle: nitrification, and denitrification. Amplicon sequencing was performed on bacterial and archaeal 16S rRNA genes, fungal ITS2, bacterial amoA, archeal amoA, nirS, nirK, and nosZ genes. The Fluidigm Access Array IFC system was used to prepare sequencing amplicons. This method allows for the simultaneous amplification of target functional genes using multiple primer sets (Fluidigm, San Francisco, CA). DNA sequencing was performed for bacterial, archaeal, and fungal amplicons using an Illumina NovaSeq Sp flowcell with 2 × 250 bp reads (Illumina, San Diego, CA). Primer information is provided in supplemental Table S2. Fluidigm amplification and Illumina sequencing were conducted at the Roy J. Carver Biotechnology Center, University of Illinois (Urbana, IL, USA). Fast Length Adjustment of Short reads (FLASH) (Mag and Salzberg 2011) software was used to merge paired-end sequences from bacterial and archaeal 16S rRNA genes. Due to the amplicon size for some functional genes, only forward read sequences were used. Once FLASH merging was performed, files were filtered by quality using the FASTX-Toolkit (Hannon 2014). Reads that did not have a minimum quality score of 30 across 90% of the bases were removed. Using the FASTX-Toolkit, nirK sequences were trimmed to the amplicon size of 165-bp (as the nirK amplicon size (165 bp) was smaller than the read length of 250 bp).Once quality preprocessing was performed, FASTQ reads were converted to FASTA format. Using USEARCH-UPARSE version 8.1 (Edgar 2010), sequences were binned into discrete OTUs based on 97% similarity and singleton DNA sequences were removed. Quantitative Insights into Microbial Ecology (QIIME) was used to generate OTU tables for downstream statistical analysis and to assign taxonomic information, this is done with a combination of the UCLUST algorithm and SILVA 138.1 database (DeSantis et al. 2006; Caporaso et al. 2010; Edgar 2010). Once taxonomy was assigned, chloroplast and mitochondrial OTUs were removed from the dataset. Rarefaction was performed to correct for differential sequencing depth across samples. Singleton OTUs were filtered prior to statistical analysis. Functional gene sequences were also assigned using QIIME (Caporaso et al. 2010) with the BLAST algorithm (Altschul et al. 1997) and custom gene-specific databases generated from reference sequences obtained from the FunGene repository (Fish et al. 2013). All OTU tables used in statistical analyses were generated in QIIME.
The number of raw reads generated from sequencing run, reads present after quality filter, and the rarefaction level are reported in supplemental Table S3. Rarefaction level was determined by calculating the rarefaction curve asymptote. Amplicon sequence data for 16S rRNA genes, fungal ITS2 region, and N-cycling functional genes is available for download on the NCBI SRA database at accession number: PRJNA789877. (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA789877/). Code for sequence processing and statistical analysis is available in GitHub (https://github.com/favela3/Maize.N-cycle.Function).
Quantifying nitrogen cycling functional groups
Quantitative PCR (qPCR) was carried out to determine the abundance of functional genes in each of the root zone microbial communities. Specific target amplification (STA), explained in Ishii et al. (2014), was carried out on samples and standards to increase template DNA for amplification. STA and qPCR master mix recipes from (Edwards et al. 2018) were used for all samples. STA product and qPCR master mix were loaded into the Dynamic Array™ Microfluidics Fluidigm Gene Expression chip, where amplification and quantification of functional genes were carried out simultaneously (Fluidigm, San Francisco, CA). All samples and standards were analyzed in 12 technical replicates. Fluidigm Real-Time PCR Analysis software version 4.1.3 was used to calculate gene threshold cycles (CT). CT values were converted to gene copy number using gene length and standard curves. All Fluidigm qPCR was conducted at the Roy J. Carver Biotechnology Center (Urbana, IL, USA). The final copy number of each functional gene amplicon was standardized by the ng of template DNA in the qPCR reaction.
In situ N 2 O flux measurements
Net soil-atmosphere N2O fluxes were measured weekly from 6/20/17 to 8/23/17, samples were collected for a total of 6 weeks. As gas flux measurements are laborious and time consuming, sampling was targeted during peak primary productivity (plant growth) and focused on the plant treatments that were hypothesized to have the largest effect on the microbiome function based on previous studies and known plant nutrient demands (Gentry and Below 1993). Specifically, the comparison focused on the B73 inbred, check1 hybrid, and PI566677 teosinte. Flux measurements were measured using static flux chambers as described in USDA-ARS GRACEnet Project protocol (Parkin and Venterea 2010). Chambers were installed in the field during the first sampling timepoint and remained in place throughout the maize growing season. Chambers consisted of two-pieces: PVC pipe with a 30 cm diameter (base installed 20 cm into soil), and sampling lids (10 cm in height). Gas sampling events occurred in the mornings between 10 am-noon; during this time 15 mL of gas were collected from chambers every 10 min for 30 min. Samples were stored in evacuated aluminum crimp-top glass vials with a chlorobutyl stopper and sealed with clear silicone to prevent sample leakage. Gas samples were later quantified using a GC-2014 Gas Chromatograph with an electron capture detector (GC-ECD) (Shimadzu, Kyoto, Japan). Standard curves were used to quantify the amount of N2O in gas sample. N2O samples were corrected using ambient temperature and moisture conditions of the collection day. Temperature and moisture data was collected in the field and validated using Illinois Climate Network (https://www.isws.illinois.edu/warm/weather/). Four sampling timepoints were used to determine the rate of N2O flux (mg m−2 min−1). Linear interpolation was used to estimate cumulative N2O production (mg L−1) over the growing season (Parkin 2008).
Statistical analysis
Statistical analysis was performed in R, with the packages ‘Vegan’, ‘ASReml’, and ‘WGCNA’. ‘Vegan’ was used to perform multivariate statistical comparisons for microbiome data among experimental treatments (Oksanen et al. 2007; Langfelder and Horvath 2008; Gilmour et al. 2017). ‘ASReml’ was used to perform univariate comparisons among genotypes and cultivar classes (inbred, hybrid, and teosinte) for potential nitrification, potential denitrification, and copies of nitrogen cycling genes as assessed by qPCR. Weighted Gene Correlation Network Analysis (WGCNA) was carried out to compare multivariate microbiome data to univariate nitrogen cycling function data. Model factors used in statistical analyses were growth stage, sampling date, the location of the block, the row of block position, range of block position, the genotype within the block, and the interaction between genotype and time (combined growth stage and sample date). A typical model of analysis is displayed below:
In PerMANOVA models, block factor was constrained in permutations. For the PerMANOVA models, sampling date and growth stage are combined as a time factor. In the ASReml mixed effect models for potential nitrification and denitrification, plant genotype and the genotype × growth stage interaction were treated as fixed factors, while all other factors (block, range, row, sampling date) were treated as random factors. Furthermore, a simple ASReml mixed effect model was generated to compare soil chemistry at the end of the growing season, plant type was treated as a fixed factor, while all other factors (range, row, block) were treated as random factors.
Results
Nitrogen cycling microbial functional groups
From our analysis of nitrogen cycling functional genes, we observed 210 archaeal amoA OTUs, 98 bacterial amoA OTUs, 21,022 nirK OTUs, 2607 nirS OTUs, and 7294 nosZ OTUs (DNA sequencing quality is described in Table S3). In response to genotype, the overall microbiome and 4 of 5 nitrogen cycling genes showed statistically significant changes in community membership (Fig. 1 and Table S4), 0 of 5 nitrogen cycling genes changed in abundance (Table S4.1). Conversely, plant classification (i.e., inbred, hybrid, teosinte) affected the composition of 4 of 5 nitrogen cycling genes, and the abundance of 1 of 5 nitrogen cycling genes (Table S4.2). Additionally, genotype classification interactions with time had a significant effect on the composition N-cycling microbial communities and functional gene abundance (Table S4.1–2; Fig. S5-6).

PerMANOVA results for the overall microbiome (16S rRNA) and different nitrogen cycling functional genes included in this study (nitrification: AamoA – archaeal amoA [ammonia monooxygenase], BamoA – bacterial amoA; denitrification: nirS and nirK – nitrite reductase, nosZ – nitrous oxide reductase). The y-axis shows R.2, percent variance explained by the treatment factor, and the x-axis shows the functional genes tested. (* – P < 0.05, ** – P < 0.01, *** – P < 0.001)
Nitrification genes and potential function
Bacterial and Archaeal nitrifiers (indicated by gene sequences for bacterial and archaeal ammonia monooxygenase – amoA) responded differently to plant genotype and plant classification. Plant genotype explained a small but significant amount of variation for archaeal amoA (R2 = 0.08, p < 0.001, Figs. 1, Tables S5.1–3), there was not a significant change in community composition of bacterial ammonia oxidizers in response to genotype (perMANOVA p = 0.16, Table S4, S5.4–6). Regarding abundance, neither archaeal nor bacterial ammonia oxidizers were significantly influenced by plant genotype (archaeal amoA p = 0.61, bacterial amoA p = 0.99, Table S6.1–7). Plant classification showed the same patterns as genotypes (Fig. 2d-e, S5, Table S4, S5, S6). Potential nitrification rate (log (ng N g d.w soil −1 h −1)) of root zone soils was influenced by both growth stage, genotype, and plant classification (Fig. 3a, S3a, Table S7.1–2). Specifically, teosinte genotypes had lower potential nitrification rate by 9% compared to population mean, on average, stimulated potential nitrification rates by 4% (means difference of 13% between inbred and teosinte, p < 0.05, Fig. 2d, S9.1). It should also be noted that some amount of variation in potential nitrification rates could be attributed to plant genotype (p < 0.05, Fig. 3, Table S7, S9). Furthermore, it appears that teosinte in the parviglumis compared to mexicana have greater abilities to lower potential nitrification. Furthermore, we regressed both nitrification and denitrification functional gene abundance, composition, and diversity against potential function across all time points and found that only bacterial amoA Shannon diversity was important in predicting potential nitrification function (p < 0.05), while abundance was not (ANOVA p > 0.05, Fig. 2a-b).

The relationship between functional bacterial amoA diversity, abundance, and potential nitrification function (A-B) and NMDS ordinations based on Bray–Curtis dissimilarity (C-F) display functional gene composition. A. Shannon diversity of bacterial amoA regressed against potential nitrification function. Note size of point is gene abundance. B. Bacterial amoA qPCR abundance regressed against potential nitrification function. Note size of point is gene diversity. (C-F) Shows that inbred, hybrid, and teosinte maize lines host different microbial taxa in the root zone under the same environmental conditions within for timepoint 1 (young plants V2-V4). Each point represents a genotypic mean (within mean n = 4) of the microbial community. C. Bacterial amoA, D. displays archaeal amoA, E. displays nirK, F. displays nosZ (note: nirS was not presented here as p > 0.10. Statistic presented in the ordination is the PerMANOVA of the classification model on genotypic means. For Temporal patterns in N-cycling gene communities please refer to supplemental materials (Fig S11)

Seasonal variation in potential nitrification and denitrification rate compared among germplasm group across the season. LS Means and Standard Error were calculated using ASReml-R. A. Potential nitrification rate (log (ng N g d.w soil −1 h −1)) across the three sampling time points averaged over plant classification. B. Potential (N2O) DEA rate(log (ng N g d.w soil −1 h −1)) across the growing season averaged over plant classification, no differences in among plant classifications was observed, but potential denitrification rates increased slightly across the season. C. Potential (N2O + N2) DEA (N2O + N2) (log (ng N g d.w soil −1 h −1)) across the growing season averaged over plant classification, no differences in among plant classifications was observed, but potential denitrification rates increased slightly across the season. Lines in figures (A-C) were added easily track changes in potential rates across the growing season, and do not represent collected data in the intermediate time points. Plant classification influences rhizosphere N-cycling activities. D. Average genotypic effect across all timepoints of hybrid, inbred, and teosintes genotypes on the potential nitrification (log (n ng N g d.w soil −1 h −1)) determined across the population. Statistical analysis for both figures can be found in supplemental tables S7-9. E. Average genotypic effect across all timepoints of hybrid, inbred, and teosintes genotypes on the log of potential (N2O) DEA (N2O) (log (ng N g d.w soil −1 h −1)) determined across the population. Statistical analysis for both figures is included in supplemental tables S7-9. F. Average genotypic effect across all timepoints of hybrid, inbred, and teosintes genotypes on the log of potential (N2O + N2) DEA (log (ng N g d.w soil −1 h −1)) determined across the population. Percent change calculation described in Methods. Statistical tests associated with figures are presented in supplemental materials
Denitrification genes and potential function
All the denitrification gene composition surveyed were significantly different among genotypes and plant classification (Fig. 1, S6, Tables S4, S5.7–15). Communities of denitrifiers possessing the cytochrome cd1-type nitrite reductase (encoded by nirS) or the copper containing nitrite reductase (encoded by nirK) both varied significantly among plant genotypes (nirS: R2 = 0.09, Fig. 1, Table S5.8; nirK: R2 = 0.09, p = 0.003, Fig. 1, 3e: Genotype means, Table S5.11) In addition, nosZ, the gene that encodes typical nitrous oxide reductase and crucial for the consumption of N2O, was found to be affected by plant genotype (R2 = 0.09, p = 0.011, Fig. 1, 2f: Genotype means Table S5.14). Quantitative PCR of denitrification genes showed no difference in the abundance of genes in the root zone across plant genotype and largely for plant classification (Fig. S5-6, Table S4). One exception to this was nosZ, which was observed to be altered by plant classification (p < 0.05, Table S5.13, Fig. 3e, S4e). Similar to the observations for bacterial amoA, the denitrification genes also showed a strong and significant interaction between plant classification and time (Fig. S5-6). The teosinte root zone microbiome contained similar denitrification gene abundance for the first sampling time point (V2-V4), had greater levels of dentification genes during the final (R1-R3) sample point, and finally, had lower numbers of denitrification genes compared to inbred and hybrid maize by the end of season (LS means, p < 0.05, Tables S5-6). Full analysis of plant classification effects and additional genotype models on denitrification genes is presented in Supplemental Tables S5-6. Conversely, we found variation in the potential (N2O) DEA and (N2O + N2) DEA rates (log (ng N g d.w soil −1 h −1)) of root zone soils to be consistently influenced by genotype and plant classification, but not growth stage (Fig. 3b, S3b, potential (N2O) DEA: p < 0.001, potential (N2O + N2) DEA: p < 0.001, Table S8.1–4). Complete DEA (N2) did not significantly differ across genotypes in this study (p = 0.8, Table S8.7), but did across plant classification (p < 0.001, Table S8.8). For potential (N2O) DEA (log (ng N g d.w soil −1 h −1)), teosinte genotypes had lower activity by 75% and inbred genotypes on average stimulated potential (N2O) DEA denitrification by 32% (mean difference 102%, Fig. 3e, Table S9.3). On average, teosinte genotypes had lower potential (N2O + N2) DEA by 59% compared to inbred maize, which stimulated it by 4% (mean difference 63%, Fig. 3f, Table S9.2).
Static N2O flux chambers
To estimate whether our potential denitrification and nitrification rates were reflected in ecosystem fluxes, we placed static flux chambers in blocks with three of our genotypes inbred, hybrid, and teosinte. From these static chambers, we found that over the growing season, that teosinte genotype plots produced significantly less cumulative N2O production (mg L−1; linear interpolation of N2O over the season) (t = 2.01, df = 29, p = 0.05, Fig. 4a), and had lower N2O flux rate (mg m−2 min−1) (t = 2.09, df = 33, p = 0.04) compared to the inbred genotype. Hybrid plots were not significantly different in N2O production from inbred and teosinte (B73: t = 1.26, df = 29, p = 0.22; Teosinte: t = -0.76, df = 30, p = 0.25) or in N2O flux (B73: t = 1.74, df = 30, p = 0.09; Teosinte: t = -0.07 df = 30, p = 0.95). In addition to this, we observed a dynamic pattern in N2O flux (mg m−2 min−1) across the season – where early season fluxes were similar but diverged by the end of the growing season (Fig. 4b).

Cumulative N2O static flux chamber results over the growing season. A. LS means + 95% CI of cumulative N2O produced (mg L−1) averaged over the season comparing teosinte and B73 maize. B. LS means + 95% CI of N2O production flux (mg m−2 min−1) comparing teosinte and B73 maize displaying all timepoints. C. Carbon substrate usage (AWD) Z transformed compared among inbred B73, hybrid check1, and teosinte PI566677 (4 replicates per genotype) soil microbiomes measured using a BIOLOG-Eco microarray plates
Soil nutrient analysis
We found no significant difference in organic matter, estimated nitrogen release as estimated by SOM, cation exchange capacity, pH, sodium, iron, and boron between maize, hybrid, and teosinte plots. Teosinte plots had significantly higher levels of nitrate, phosphorus, potassium, sulfur, and manganese compared to inbred and hybrid maize plots (Wald’s test: p < 0.05; Table S11). Increased nitrate in teosinte plots may be indicative of the suppressed denitrification. Inbred maize plots had higher levels of buffer pH, copper, and calcium compared to teosinte and hybrid plots (Wald’s test: p < 0.05; Table S11).
Carbon substrate utilization
We found that plant classification was not statistically significant in explaining variation in microbial carbon substrate utilization (PerMANOVA: DF = 2, R2 = 0.25, p = 0.10). Between inbred B73, hybrid check1, and teosinte PI566677 genotypes, we found that teosinte root zone microbiomes had greater overall levels of substrate utilization compared to inbred and hybrid microbial communities (t-test; p < 0.05, Fig. 4c). We found no significant differences in utilization of individual substrates between inbred maize and teosinte microbial communities, significant differences in utilization of 3 substrates between inbred and hybrid maize, and significant differences in utilization of 2 substrates between hybrid and teosinte microbiomes (ANOSIM, p < 0.05). Inbred-hybrid differences include glycogen, D-cellobiose, and L-serine. Teosinte-hybrid differences include L-phenylalanine and tween-80.
Root zone microbiome
In this field experiment, we identified 37,596 different 16S rRNA operational taxonomic units (OTUs, 97% similarity, (rarefied to 100,000 reads per sample), and 2236 fungal OTUs (rarefied to 10,000) were identified from the ITS2 region (Table S2).
Within the prokaryotic community (based on 16S rRNA gene sequences), we found that plant genotype, genotype × time interactions, the location of the block, and sampling time explained 74% of the variation within the root zone microbiome; 26% of the variation within the microbial community was unexplained ( DF = 26, p < 0.001; Fig. 1, S7; Table S10). In total, 34% of the variation in the prokaryotic microbiome was explained by plant genetics; 18% of this 34% variation was independent of temporal effects while 16% was highly linked to the time of sampling (genetics × sampling time). Interestingly, 20% of the variation in the soil microbial community was explained by block location alone. This would mean that across time, 20% of the microbiome was unchanged across the season. Sampling time (independent of genotype) explained 12% of the variation within the microbiome. Roughly, these results suggest that plant genetics explained about a third of the variation in the root zone microbiome. Respectively, spatial and temporal effects seem to explain a third of the variation within the microbiome. Finally, an additional third of variation within the soil microbiome was unexplained.
Across plant classification (inbred, hybrid, teosinte), the most divergent genotype points showed the greatest differences in microbiome recruitment (Fig. S7-8; Table S10.1–3). Specifically, teosinte and hybrid maize treatments have the strongest effect on the composition of the soil microbiome agroecosystem. Teosinte root zone soils contained greater relative abundance of Actinobacteria and Proteobacteria (specifically, Actinomycetales, Burkholderiales) and less Acidobacteria (iii1-15, Solibacteres) compared to modern maize (Fig. S9-10). Additional analysis was carried out within plant category (i.e., within inbred, within hybrid, within teosinte), and inbred maize was the only category where genotype did not significantly contribute to differential microbiome recruitment.
Fungal communities showed similar results as the prokaryotic communities, except for notably weaker effects of space. This may indicate that fungi are less dispersal-limited than bacterial communities (Table S10).
Relationship between the microbial community and N-cycling function
To further understand the differential contribution of the root zone microbiome to the potential function of a soil sample, we used Weighted Gene Correlation Network Analysis (WGCNA) (Langfelder and Horvath 2008) to identify four unique co-correlated clusters of OTUs (modules) with a significant response to potential function (3 modules that were correlated to potential nitrification, and one module that was correlated to potential (N2O) DEA (Fig. 5). WGCNA taxa Module 2 was positively correlated to potential nitrification (r = 0.20, p < 0.001), while two modules were negatively correlated to potential nitrification (Module 4: r = -0.25, p < 0.001; Module 7: r = -0.23, p < 0.001). Module 2 contained 129 OTUs and was dominated by the presence of Acidobacteria. Interestingly, the second most dominant phylum in this module – Cholorflexi – was recently shown to have the ability to carry out potential nitrification (Spieck et al. 2020). Module 4 contained 290 OTUs and Module 7 contained 38 OTUs, both modules were dominated by Actinobacteria.

Weighted Gene Correlation Network Analysis (WGCNA) results between potential denitrification and potential nitrification and microbial community composition. WGCNA starts by clustering microbial OTUs into modules of highly correlated taxa (based on abundance). These modules are then regressed against our explanatory factor (here that is denitrification and nitrification. A. Strength and significance of correlation between microbial modules and nitrogen cycling function. B. Correlations among microbial modules generated in the clustering process. C. Phylum-level composition of modules that were significantly correlated to changes in potential nitrification and denitrification
Discussion
Microbial N-cycling processes are modulated by crop genetics
Across a variety of environmental conditions and plant species, it has been shown that genetic variation within a plant population can consistently shape the rhizosphere microbiome (Peiffer et al. 2013a, b; Xu et al. 2018; Deng et al. 2021). Additionally, previous studies have also shown that changes in rhizosphere microbiome functional groups resulting from host plant genetics are important to ecosystem processes (i.e., carbon and nitrogen cycling) (Bulgarelli et al. 2015; Mwafulirwa et al. 2021a, b). Furthermore, for decades, researchers have known that plant carbon contributions to soils play a significant role in modulating microbial nitrogen transformations (Haller and Stolp 1985; Qian et al. 1997). However, there is a lack of data showing how host genotype-specific changes in microbiome functional groups influence nitrogen cycling ecosystem processes. This field study demonstrates that the effect of plant genotype extends to modulating functions of the root zone microbiome. Specifically, we observed that the plant genotype influenced the recruitment of functional groups related to nitrification and denitrification along with the potential rates of those ecosystem processes (Figs. 1, 2 and 3). These findings highlight the feasibility of breeding crops for microbiome-associated phenotypes (MAPs) to influence N-cycling microbes and their functions (Oyserman et al. 2018). Ultimately, our results suggest that we can select genetic haplotypes linked to MAPs within populations of agricultural cultivars to promote sustainable ecosystem processes within agroecosystems.
Our study demonstrates that genetic variation within Zea mays plays a significant role in both the assembly of the microbiome and the nitrogen cycling capability of the community, even in a stochastic field setting. Our most genetically heterogeneous treatments (wild and domesticated hybrid) had the strongest effects on the composition and function of the microbiome (Smýkal et al. 2018; Favela et al. 2021, 2022; Ren et al. 2022). Our hybrids showed patterns of microbial community recruitment distinct from their inbred parents, suggesting that heterosis plays a role in recruitment and function of the root zone microbiome. Interestingly, others have found that the expression of heterosis for root biomass and germination can be modulated by the presence of a soil microbiome (Wagner et al. 2020, 2021). Understanding the genetic basis of heterosis for MAPs is critical for designing efficient breeding programs for optimizing soil microbiome functions. Furthermore, we found that teosinte genotypes had the strongest influence on the activity of soil microbial taxa. We hypothesize that these differences in the teosinte microbiome were largely driven by a diverse set of belowground phenotypes (Gaudin et al. 2011), which facilitate changes that shape biotic interactions (via, exudate production, composition of metabolites, and root morphology) and abiotic characteristics (pH, moisture, NO3−, NH4+) which we know to shape nitrogen cycling taxa (Haller and Stolp 1985; Qian et al. 1997). These results suggest that selective reincorporation of traits important to N-cycling would be key MAPs for “rewilding” modern breeding programs for sustainability (Perino et al. 2019; Razzaq et al. 2021). Importantly, this field study confirms that N-cycling functions, as well as composition of the rhizosphere microbiome, are responsive to genetic variation within the plant host.
Potential mechanisms underlying maize genotypes nitrification and denitrification differences
We observed that teosinte and hybrid maize root zones had communities with a lower potential to carry out nitrification compared to inbred maize and shifts in the residing amoA genes (Fig. 2a, 3d, 3c-d). While it is clear the function of these nitrifiers is modulated by variation in genotype, it is unclear whether this is a direct or indirect process. It is possible that this alteration to function is caused by an indirect change to the abiotic environment (i.e., mineralization, ammonium, soil moisture, carbon) (Mwafulirwa et al. 2021b). Alternatively, these changes in function could be caused by direct phytochemically mediated biological suppression nitrification. Biological nitrification inhibition (BNI) has been seen across a variety of different grass species (particularly in wild varieties) (Coskun et al. 2017a, b) with recent work showing that modern maize does have the phytochemical capacity for BNI (Otaka et al. 2022). It has already been shown that teosinte’s metabolome and nutrient up take differs from modern maize, perhaps some of these physiological changes are related to changes in nitrification capacity (Wang et al. 2018; Xu et al. 2019). As nitrification can result in major losses of N fertilizer, investigating whether teosinte characteristics that confer lowered nitrification can be reincorporated into modern maize maybe of interest to future breeders (Nair et al. 2020; Subbarao et al. 2021). Evaluating the vast diversity of maize cultivars for the capacity to alter the nitrogen cycle either directly or indirectly using genetic information is a valuable first step toward incorporating and improving these novel MAPs in agriculturally viable germplasm.
Interactions of plant genotypes with denitrifying microorganisms followed a similar pattern to nitrifiers, except with considerably more variation. Over the growing season, genotype and cultivar classification played a significant role in shaping potential denitrification activity and denitrification gene composition (Figs. 2b-c, 3e-f, 3e-f). These results were surprising, as maize is typically grown in aerobic soils and denitrification is an anaerobic process. These results suggest that the genotype can interact with the highly variable hot spots and hot moments typical of denitrification (Krichels and Yang 2019). Interestingly, teosinte appears to support lower potential (N2O) DEA and (N2O + N2) DEA rates leading to the hypothesis that teosinte contains some indirect (e.g., soil moisture, nitrate loads) or direct mechanism to shape denitrification not previously reported. One possible direct mechanism could be, biological denitrification inhibition (BDI), which is hypothesized to have evolved in plants as a mechanism to compete with denitrifying microbes for soil nitrates. BDI work is still in early stages with only a single class of metabolites, procyanidins, being shown to mediate BDI (Bardon et al. 2014, 2016; Galland et al. 2019). Interestingly, outside the context of BDI, a considerable amount of work has focused on a maize depolarized procyanidin (cyanidin) and anthocyanin (a glucoside cyanidin), showing that maize genotypes have considerable variation in cyanidin and anthocyanin production (Sharma et al. 2011; Paulsmeyer et al. 2017). While not quantified in this study, the BDI differences observed here among maize genotypes could potentially be related to differences in cyanidin and anthocyanin exudation in the rhizosphere. It is possible that these rhizosphere cyanidins and anthocyanin (Tselas et al. 1979; Hawes et al. 1998) are acting as anti-reductants, which is known to occur at low pH (Becker 2016), and are competitive inhibitors of denitrification or allosteric inhibitors like procyanidins. Further research needs to be done to determine the abiotic and biotic drivers of these alterations in population level denitrification differences.
Outcomes of plant modulation of N-cycling processes
From a sustainability perspective, this study highlights a potential avenue to reduce agricultural N losses and GHG emissions generated by soil microorganisms. N2O static chamber results (Fig. 4a-b) provide support that the potential nitrification and denitrification assays are representative of more variable ecosystem processes. It should be noted that these potential assays and N2O chambers, have their limitations (Nannipieri et al. 2018; Grace et al. 2020). Potential assays indicate that the maximum function of these N-cycling communities has changed in response to plant host in the root zone. While the N2O chambers shows that changes in microbiome functional potential may be related to differences in N fluxes at the ecosystem scale. The capability for crop genotypes to reduce N2O losses is an exciting finding, as agriculture is a major producer of N2O emissions (Vitousek et al. 1997; Reay et al. 2012), and these results suggest an additional tool to potentially curb production of this potent GHG. While these N2O results presented are interesting, a major limitation of this study is that we only examined these cultivars in a single field experiment and static flux chambers are known to be extremely variable (Waldo et al. 2019). Further support for our potential nitrification and denitrification assays translating to actual differences in field processes can be seen in our end of season soil nutrient analysis. From this analysis for a single end of season timepoint we observed that teosinte plots had higher levels of nitrates (Table S11) compared to hybrid and inbred plots. These higher nitrate levels could be driven by the plant, but this claim is difficult to support with the current dataset, since we only collected a single physiochemical timepoint. Teosinte’s enhanced ability to mine nutrients may be facilitated by changes in root zone pH driven by carbon exudation, and root density. This conclusion is supported by our finding showing that teosinte plots have a lower buffer pH (residual or reactive pH) compared to hybrid and inbred maize (p < 0.05, Table S11). Alteration of soil buffer pH is likely underpinning many of the functional changes we observed in this study. In addition, we observed modest differences in microbial community carbon metabolism between plant classifications (Fig. 4c). Teosinte-derived microbial communities were better at degrading phytochemical precursors (e.g., phenylalanine, oleic acid) in the flavonoid and linoleic acid synthesis pathways (Kaur-Bhambra et al. 2022), while hybrid-derived microbial communities were well-equipped to consume carbohydrates. This result is interesting, as microbes differ in their ability to metabolize different photosynthates exuded into the rhizosphere (Fan et al. 2022), lending support to the hypothesis that differences we are observing across microbiomes may be caused by altered amount and composition of plant exudates. This hypothesis is further supported by work in barley showing that genotypes can vary in rhizodeposition-derived carbon and this variation shapes soil microbial mineralization (Mwafulirwa et al. 2016). The next steps toward incorporating potential nitrogen conservation MAPs into agricultural practices would be research to explore whether suppression of nitrogen transformations is consistent across a wide range of biogeographic environments.
The effects of seasonal phenology and Zea genotype × sampling time interaction over the growing season played a major role in microbiome recruitment and function. Maize has different nutrient requirements across the growing season, and these nutrients are extracted from the soil environment (Bender et al. 2013), so, along with genotype-specific biochemistry, plant growth and development likely influence interactions with the soil microbial community. In addition, previous studies have shown that the complexity of the microbiome is built through time (Shi et al. 2016; Emmett et al. 2020; Ajilogba et al. 2022). These temporal effects are important to consider, as they can dramatically influence the conclusions drawn about the interaction between plants and their microbiome. We observed that potential nitrification and potential denitrification were dependent on plant growth stage (Fig. 3). Potential nitrification and denitrification, for example, seemed to peak in the middle of the season, coinciding with plant primary productivity (Fig. 3). Perhaps, during this high productivity phase Zea is releasing greater quantities of fresh exudate resulting in the priming of soil organic matter by microorganisms. Furthermore, rhizosphere C exudation has been shown to enhance the release of N (Phillips et al. 2011; Dijkstra et al. 2013; Emmett et al. 2020), perhaps explaining the overall increase of nitrification at this timepoint. Furthermore, these temporal impacts highlight a limitation in this study, whereby we may be overestimating our genotype differences because of differences in growth phenology among the classes of plants. In addition, temporal growth patterns are likely interacting with the measurability of effects in the root zone as root density of the plant is anticipated to change through plant development (Chaparro et al. 2013; Vetterlein et al. 2020; Tkacz and Poole 2021).
Relationship between microbial diversity and N-cycling
We found that different microbial taxa were correlated with functional changes in the microbiome. WGCNA identified four modules of microbial taxa that were significantly associated with both changes of potential nitrification and denitrification (Fig. 5). These results suggest that specific taxa and their interactions play a role in driving the function of the microbiome and that, to some degree, plants can influence the activities of specific microbial groups. Interestingly, we observed that modules positively correlated with higher potential nitrification rates were dominated by gram-negative bacteria within the phylum Acidobacteria. In contrast, those that were negatively correlated with nitrification were dominated by gram-positive bacteria within the Actinobacteria phylum. Surprisingly, these modules of correlated OTUs were not dominated by known nitrifying taxa, suggesting that nitrification processes may be, in part, dependent on the metabolism of other microbial community members, that nitrification is controlled by the level of transcriptional regulation rather than nitrifier population size, nitrifiers or nitrifier ideal conditions are facilitating habitat alteration that is influencing microbiome structure (e.g. altering pH to enrich for Acidobacteria), or that some other microbial interaction (i.e. predation, competition) is controlling nitrification (Baskaran et al. 2020; Spieck et al. 2020; Burian et al. 2021). Determining how ecological interactions within the microbiome influence microbial functions is critical for understanding and predicting microbially-mediated ecosystem functions (Kuypers et al. 2018). In addition, we carried out analysis to determine how functional gene diversity, abundance, and composition related to potential functions. Among all functional gene comparisons between bacterial amoA, and archaeal amoA. We only found bacterial amoA diversity to be correlated to potential function, while abundance and composition was not (Fig. 3a-b). These results suggest that at least for potential nitrification the regulation of diversity is important to function. Overall, it is clear that understanding the relationship between microbial characteristics and functions is complex and will require further research to determine the rules of these relationships.
Conclusion
The ability of plants to influence microbial functions in the rhizosphere likely evolved as a mechanism for nutrient retention, enhancing plant competition for available nutrients from the soil matrix (Philippot et al. 2013b; Delaux and Schornack 2021; Lata et al. 2022). It is becoming increasingly clear that plant genetic variation (within and among species) modulates the activities of the soil-associated microbiome and that these alterations can impact soil biogeochemical functions (Falkowski et al. 2008; Morris et al. 2020). Identification of the plant genomic regions that direct recruitment of N-cycling microorganisms or modulation of their activities will enable progress on re-engineering the agroecosystem to reduce its contributions to N pollution (Johnson 2006; Subbarao and Searchinger 2021). That is not to say we suggest growing teosinte in the field, but that these wild cultivars are reservoirs of critical genetic variation that are potentially important to enhancing sustainability. This study thus contributes an important advancement by showing that maize, an agronomically important crop, has genetic variation that contributes to alterations in the microbiomes and N-cycling function – potentially enough variation to breed and incorporate these extended phenotype ecosystem traits into modern hybrid cultivars. Furthermore, we have demonstrated that a small subset of hybrids does appear to have regulation of N-cycling microbiome in a way similar to teosinte. In addition, it should be noted that a limitation of the work presented is that we did not characterize the mechanisms by which we are observing these effects. A great deal of mechanistic work is needed in this area of research. Furthermore, it should be noted that while plant genotypic control of function plays an active role in the growing season other historic edaphic and abiotic effects will play a major role in managing and predicting the agroecosystem N cycle. Integrating sustainability-related microbiome associated phenotypes into our agricultural systems is a way forward to address many agronomic challenges facing society (York et al. 2022; Favela et al. 2023).
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. Sequences and code are available via Github and SRA database. Please references methods for details.
References
Ajilogba CF, Olanrewaju OS, Babalola OO (2022) Plant Growth Stage Drives the Temporal and Spatial Dynamics of the Bacterial Microbiome in the Rhizosphere of Vigna subterranea. Front Microbiol 13:1–10. https://doi.org/10.3389/fmicb.2022.825377
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Baggs EM, Cairns JE, Mhlanga B, Petroli CD, Chamberlin J, Karwat H, Gowda MS (2023) Exploiting crop genotype-specific root-soil interactions to enhance agronomic efficiency. Front Soil Sci 3:1125604. https://doi.org/10.3389/fsoil.2023.1125604
Bardon C, Piola F, Bellvert F et al (2014) Evidence for biological denitrification inhibition (BDI) by plant secondary metabolites. New Phytol 204:620–630. https://doi.org/10.1111/nph.12944
Bardon C, Poly F, Piola F et al (2016) Mechanism of biological denitrification inhibition: Procyanidins induce an allosteric transition of the membrane-bound nitrate reductase through membrane alteration. FEMS Microbiol Ecol 92:1–11. https://doi.org/10.1093/femsec/fiw034
Baskaran V, Patil PK, Antony ML et al (2020) Microbial community profiling of ammonia and nitrite oxidizing bacterial enrichments from brackishwater ecosystems for mitigating nitrogen species. Sci Rep 10:1–11. https://doi.org/10.1038/s41598-020-62183-9
Becker PM (2016) Antireduction: An ancient strategy fit for future. Biosci Rep 36:.https://doi.org/10.1042/BSR20160085
Bender RR, Haegele JW, Ruffo ML, Below FE (2013) Nutrient uptake, partitioning, and remobilization in modern, transgenic insect-protected maize hybrids. Agron J 105:161–170. https://doi.org/10.2134/agronj2012.0352
Bouffaud ML, Renoud S, Moenne-Loccoz Y, Muller D (2016) Is plant evolutionary history impacting recruitment of diazotrophs and nifH expression in the rhizosphere? Sci Rep 6:. https://doi.org/10.1038/srep21690
Bowles TM, Atallah SS, Campbell EE et al (2018) Addressing agricultural nitrogen losses in a changing climate. Nat Sustain 1:399–408. https://doi.org/10.1038/s41893-018-0106-0
Brisson VL, Schmidt JE, Northen TR, et al (2019) Impacts of Maize Domestication and Breeding on Rhizosphere Microbial Community Recruitment from a Nutrient Depleted Agricultural Soil. Sci Rep 9:. https://doi.org/10.1038/s41598-019-52148-y
Bulgarelli D, Garrido-Oter R, Münch PC et al (2015) Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17:392–403. https://doi.org/10.1016/j.chom.2015.01.011
Burian A, Pinn D, Peralta-Maraver I, et al (2021) Predation increases multiple components of microbial diversity in activated sludge communities. ISME J 1–9. https://doi.org/10.1038/s41396-021-01145-z
Canarini A, Kaiser C, Merchant A, et al (2019) Root exudation of primary metabolites: Mechanisms and their roles in plant responses to environmental stimuli. Front Plant Sci 10 https://doi.org/10.3389/fpls.2019.00157
Caporaso JG, Kuczynski J, Stombaugh J et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Chaparro JM, Badri DV, Vivanco JM (2013) Rhizosphere Microbiome Assemblage is Affected by Plant Development 8:790–803. https://doi.org/10.1038/ismej.2013.196
Classen AT, Boyle SI, Haskins KE et al (2003) Community-level physiological profiles of bacteria and fungi: Plate type and incubation temperature influences on contrasting soils. FEMS Microbiol Ecol 44:319–328. https://doi.org/10.1016/S0168-6496(03)00068-0
Compant S, Samad A, Faist H, Sessitsch A (2019) A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J Adv Res 19:29–37. https://doi.org/10.1016/j.jare.2019.03.004
Coskun D, Britto DT, Shi W, Kronzucker HJ (2017a) Nitrogen transformations in modern agriculture and the role of biological nitrification inhibition. Nat Plants 3:17074
Coskun D, Britto DT, Shi W, Kronzucker HJ (2017b) How plant root exudates shape the nitrogen cycle. Trends Plant Sci 22:661–673. https://doi.org/10.1016/j.tplants.2017.05.004
Davidson EA, David MB, Galloway JN et al (2012) Excess nitrogen in the U.S. Environment: Trends, Risks, and Solutions. Issues in Ecology 15:1–16
Delaux P-M, Schornack S (2021) Plant evolution driven by interactions with symbiotic and pathogenic microbes. Science (1979) 371:eaba6605-undefined. https://doi.org/10.1126/science.aba6605
Deng S, Caddell DF, Xu G et al (2021) Genome wide association study reveals plant loci controlling heritability of the rhizosphere microbiome. ISME J 15:3181–3194. https://doi.org/10.1038/s41396-021-00993-z
DeSantis TZ, Hugenholtz P, Larsen N et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. https://doi.org/10.1128/AEM.03006-05
Dijkstra FA, Carrillo Y, Pendall E, Morgan JA (2013) Rhizosphere priming: A nutrient perspective. Front Microbiol 4:1–8. https://doi.org/10.3389/fmicb.2013.00216
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Edwards JD, Pittelkow CM, Kent AD, Yang WH (2018) Dynamic biochar effects on soil nitrous oxide emissions and underlying microbial processes during the maize growing season. Soil Biol Biochem 122:81–90. https://doi.org/10.1016/j.soilbio.2018.04.008
Emmett BD, Buckley DH, Drinkwater LE (2020) Plant growth rate and nitrogen uptake shape rhizosphere bacterial community composition and activity in an agricultural field. New Phytol 225:960–973. https://doi.org/10.1111/nph.16171
Falkowski PG, Fenchel T, Delong EF (2008) The microbial engines that drive Earth's biogeochemical cycles. Science 320(5879):1034–1039. https://www.science.org/doi/10.1126/science.1153213
Fan K, Holland-Moritz H, Walsh C et al (2022) Identification of the rhizosphere microbes that actively consume plant-derived carbon. Soil Biol Biochem 166:108577. https://doi.org/10.1016/j.soilbio.2022.108577
Favela A, Bohn MO, Kent AD (2021) Maize germplasm chronosequence shows crop breeding history impacts recruitment of the rhizosphere microbiome. ISME J 15:2454–2464. https://doi.org/10.1038/s41396-021-00923-z
Favela A, Bohn M, Kent A (2022) N-Cycling Microbiome Recruitment Differences Between Modern and Wild Zea mays . Phytobiomes J 1–10. https://doi.org/10.1094/pbiomes-08-21-0049-r
Favela A, Bohn MO, Kent AD (2023) Application of plant extended phenotypes to manage the agricultural microbiome belowground. Frontiers in Microbiomes 2:1–13. https://doi.org/10.3389/frmbi.2023.1157681
Fish JA, Chai B, Wang Q et al (2013) FunGene: The functional gene pipeline and repository. Front Microbiol 4:1–14. https://doi.org/10.3389/fmicb.2013.00291
Galland W, Piola F, Burlet A, et al (2019) Biological denitrification inhibition (BDI) in the field: A strategy to improve plant nutrition and growth. Soil Biol Biochem 136:. https://doi.org/10.1016/j.soilbio.2019.06.009
Gaudin ACM, McClymont SA, Raizada MN (2011) The nitrogen adaptation strategy of the wild teosinte ancestor of modern maize. Zea Mays Subsp Parviglumis Crop Sci 51:2780–2795. https://doi.org/10.2135/cropsci2010.12.0686
Gentry LE, Below FE (1993) Maize Productivity as Influenced by Form and Availability of Nitrogen. Crop Sci 33:491–497. https://doi.org/10.2135/cropsci1993.0011183x003300030015x
Gilmour AR, Gogel BJ, Cullis BR, Welham S, Thompson R (2015) ASReml user guide release 4.1 structural specification. VSN international Ltd, Hemel hempstead
Grace PR, van der Weerden TJ, Rowlings DW et al (2020) Global Research Alliance N2O chamber methodology guidelines: Considerations for automated flux measurement. J Environ Qual 49:1126–1140. https://doi.org/10.1002/jeq2.20124
Haller T, Stolp H (1985) Quantitative estimation of root exudation of maize plants. Plant and Soil 86:207–216. https://doi.org/10.1007/BF02182895
Hannon GJ (2014) FASTX-Toolkit. [Online] https://hannonlab.cshl.edu/fastx_toolkit. Accessed 10/01/2017
Hawes MC, Brigham LA, Wen F et al (1998) Function of root border cells in plant health: Pioneers in the rhizosphere. Annu Rev Phytopathol 36:311–327. https://doi.org/10.1146/annurev.phyto.36.1.311
Hayatsu M, Katsuyama C, Tago K (2021) Overview of recent researches on nitrifying microorganisms in soil. Soil Sci Plant Nutr 67(6):619–632. https://doi.org/10.1080/00380768.2021.1981119
Huang AC, Jiang T, Liu YX, et al (2019) A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science (1979) 364:. https://doi.org/10.1126/science.aau6389
Ishii S, Kitamura G, Segawa T et al (2014) Microfluidic quantitative PCR for simultaneous quantification of multiple viruses in environmental water samples. Appl Environ Microbiol 80:7505–7511. https://doi.org/10.1128/AEM.02578-14
Johnson DW (2006) Progressive N Limitation In Forests: Review And Implications For Long-Term Responses To Elevated Co 2. Ecology 87:64–75. https://doi.org/10.1890/04-1781
Kaur-Bhambra J, Wardak DLR, Prosser JI, Gubry-Rangin C (2022) Revisiting plant biological nitrification inhibition efficiency using multiple archaeal and bacterial ammonia-oxidising cultures. Biol Fertil Soils 58:241–249. https://doi.org/10.1007/s00374-020-01533-1
Krichels AH, Yang WH (2019) Dynamic Controls on Field-Scale Soil Nitrous Oxide Hot Spots and Hot Moments Across a Microtopographic Gradient. J Geophys Res Biogeosci 124:3618–3634. https://doi.org/10.1029/2019JG005224
Kuypers MMM, Marchant HK, Kartal B (2018) The microbial nitrogen-cycling network. Nat Rev Microbiol 16:263–276. https://doi.org/10.1038/nrmicro.2018.9
Langfelder P, Horvath S (2008) WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9:1–13. https://doi.org/10.1186/1471-2105-9-559
Lata JC, Le Roux X, Koffi KF et al (2022) The causes of the selection of biological nitrification inhibition (BNI) in relation to ecosystem functioning and a research agenda to explore them. Biol Fertil Soils 58:207–224. https://doi.org/10.1007/s00374-022-01630-3
Lucas M, Gil J, Robertson GP et al (2023) Changes in soil pore structure generated by the root systems of maize, sorghum and switchgrass affect in situ N2O emissions and bacterial denitrification. Biol Fertil Soils. https://doi.org/10.1007/s00374-023-01761-1
Mag T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. 27:2957–2963. https://doi.org/10.1093/bioinformatics/btr507
Moreau D, Pivato B, Bru D et al (2015) Plant traits related to nitrogen uptake influence plant- microbe competition. Ecology 96:2300–2310. https://doi.org/10.1890/14-1761.1
Moreau D, Bardgett RD, Finlay RD et al (2019) A plant perspective on nitrogen cycling in the rhizosphere. Funct Ecol 33:540–552. https://doi.org/10.1111/1365-2435.13303
Morris A, Meyer K, Bohannan B (2020). Opinion Piece Linking Microbial Communities to Ecosystem Functions: What We Can Learn from Genotype-Phenotype Mapping in Organisms. https://doi.org/10.1098/rstb.2019.0244
Mwafulirwa L, Baggs EM, Russell J et al (2016) Barley genotype influences stabilization of rhizodeposition-derived C and soil organic matter mineralization. Soil Biol Biochem 95:60–69. https://doi.org/10.1016/j.soilbio.2015.12.011
Mwafulirwa L, Baggs EM, Russell J et al (2021a) Identification of barley genetic regions influencing plant–microbe interactions and carbon cycling in soil. Plant Soil. https://doi.org/10.1007/s11104-021-05113-6
Mwafulirwa L, Paterson E, Cairns JE et al (2021b) Genotypic variation in maize (Zea mays) influences rates of soil organic matter mineralization and gross nitrification. New Phytol 231:2015–2028. https://doi.org/10.1111/nph.17537
Nair D, Baral KR, Abalos D, et al (2020) Nitrate leaching and nitrous oxide emissions from maize after grass-clover on a coarse sandy soil: Mitigation potentials of 3,4-dimethylpyrazole phosphate (DMPP). J Environ Manage 260:. https://doi.org/10.1016/j.jenvman.2020.110165
Nannipieri P, Trasar-Cepeda C, Dick RP (2018) Soil enzyme activity: a brief history and biochemistry as a basis for appropriate interpretations and meta-analysis. Biol Fertil Soils 54:11–19. https://doi.org/10.1007/s00374-017-1245-6
Nielsen RL (2002) Corn growth and development: What goes on from planting to harvest. Purdue University, University Extension, West Lafayette, IN
Oksanen J, Kindt R, Legendre P, et al (2007) The vegan Package Title Community Ecology Package. In: http://cran.r-project.org/, http://r-forge.r-project.org/projects/vegan/. http://ftp.uni-bayreuth.de/math/statlib/R/CRAN/doc/packages/vegan.pdf. Accessed 19 Feb 2018
Oksanen FJ et al (2017) Vegan: community ecology package. R package Version 2.4-3. https://CRAN.Rproject.org/package=vegan
Otaka J, Subbarao GV, Ono H, Yoshihashi T (2022) Biological nitrification inhibition in maize—isolation and identification of hydrophobic inhibitors from root exudates. Biol Fertil Soils 58:251–264. https://doi.org/10.1007/s00374-021-01577-x
Otaka J, Subbarao GV, MingLi J et al (2023) Isolation and characterization of the hydrophilic BNI compound, 6-methoxy-2(3H)-benzoxazolone (MBOA), from maize roots. Plant Soil 489:341–359. https://doi.org/10.1007/s11104-023-06021-7
Oyserman BO, Medema MH, Raaijmakers JM (2018) Road MAPs to engineer host microbiomes. Curr Opin Microbiol 43:46–54. https://doi.org/10.1016/j.mib.2017.11.023
Parkin TB (2008) Effect of Sampling Frequency on Estimates of Cumulative Nitrous Oxide Emissions. J Environ Qual 37:1390–1395. https://doi.org/10.2134/jeq2007.0333
Parkin TB, Venterea RT (2010) USDA-ARS GRACEnet project protocols, chapter 3. Chamber-based trace gas flux measurements. Sampling protocols. Beltsville, MD, pp 1–39
Paulsmeyer M, Chatham L, Becker T et al (2017) Survey of Anthocyanin Composition and Concentration in Diverse Maize Germplasms. J Agric Food Chem 65:4341–4350. https://doi.org/10.1021/acs.jafc.7b00771
Peiffer JA, Spor A, Koren O et al (2013a) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci 110:6548–6553. https://doi.org/10.1073/pnas.1302837110
Peiffer JA, Spor A, Koren O et al (2013b) Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci U S A 110:6548–6553. https://doi.org/10.1073/pnas.1302837110
Peralta AL, Johnston ER, Matthews JW, Kent AD (2016) Abiotic correlates of microbial community structure and nitrogen cycling functions vary within wetlands. Freshwater Science 35:573–588. https://doi.org/10.1086/685688
Pérez-Izquierdo L, Zabal-Aguirre M, González-Martínez SC et al (2019) Plant intraspecific variation modulates nutrient cycling through its below ground rhizospheric microbiome. J Ecol 107:1594–1605. https://doi.org/10.1111/1365-2745.13202
Perino A, Pereira HM, Navarro LM, et al (2019) Rewilding complex ecosystems. Science (1979) 364:. https://doi.org/10.1126/science.aav5570
Petroli CD, Subbarao G V, Burgueño JA, et al (2023) Genetic variation among elite inbred lines suggests potential to breed for BNI-capacity in maize. Sci Rep 13:. https://doi.org/10.1038/s41598-023-39720-3
Philippot L, Hallin S, Schloter M (2007) Ecology of Denitrifying Prokaryotes in Agricultural Soil. Adv Agron 96:249–305
Philippot L, Raaijmakers JM, Lemanceau P, Van Der Putten WH (2013a) Going back to the roots: The microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789–799. https://doi.org/10.1038/nrmicro3109
Philippot L, Raaijmakers JM, Lemanceau P, Van Der Putten WH (2013b) Going back to the roots: The microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789–799
Phillips RP, Finzi AC, Bernhardt ES (2011) Enhanced root exudation induces microbial feedbacks to N cycling in a pine forest under long-term CO2fumigation. Ecol Lett 14:187–194. https://doi.org/10.1111/j.1461-0248.2010.01570.x
Qian JH, Doran JW, Walters DT (1997) Maize plant contributions to root zone available carbon and microbial transformations of nitrogen. Soil Biol Biochem 29:1451–1462. https://doi.org/10.1016/S0038-0717(97)00043-6
Raglin SS, Soman C, Ma Y, Kent AD (2022) Long term influence of fertility and rotation on soil nitrification potential and nitrifier communities. Front Soil Sci 2:838497. https://doi.org/10.3389/fsoil.2022.838497
Razzaq A, Wani SH, Saleem F et al (2021) Rewilding crops for climate resilience: Economic analysis and de novo domestication strategies. J Exp Bot 72:6123–6139. https://doi.org/10.1093/jxb/erab276
Reay DS, Davidson EA, Smith KA, et al (2012) Global agriculture and nitrous oxide emissions. Nat Clim Chang 2:. https://doi.org/10.1038/NCLIMATE1458
Ren W, Zhao L, Liang J et al (2022) Genome-wide dissection of changes in maize root system architecture during modern breeding. Nat Plants 8:1408–1422. https://doi.org/10.1038/s41477-022-01274-z
Schinner F, Ohlinger R, Kandeler E, Margesin R (1996) Methods in Soil Biology. Springer
Schmidt JE, Mazza Rodrigues JL, Brisson VL et al (2020) Impacts of directed evolution and soil management legacy on the maize rhizobiome. Soil Biol Biochem 145:107794. https://doi.org/10.1016/j.soilbio.2020.107794
Sharma M, Cortes-Cruz M, Ahern KR et al (2011) Identification of the Pr1 Gene Product Completes the Anthocyanin Biosynthesis Pathway of Maize. Genetics 188:69–79. https://doi.org/10.1534/genetics.110.126136
Shi S, Nuccio EE, He Z, et al (2016) The interconnected rhizosphere: High network connectivity dominates rhizosphere assemblages. 926–936. https://doi.org/10.1111/ele.12630
Smýkal P, Nelson MN, Berger JD, Von Wettberg EJB (2018) The impact of genetic changes during crop domestication. Agronomy 8:1–22. https://doi.org/10.3390/agronomy8070119
Spieck E, Spohn M, Wendt K et al (2020) Extremophilic nitrite-oxidizing Chloroflexi from Yellowstone hot springs. ISME J 14:364–379. https://doi.org/10.1038/s41396-019-0530-9
Subbarao GV, Searchinger TD (2021) A “more ammonium solution” to mitigate nitrogen pollution and boost crop yields. Proc Natl Acad Sci U S A 118:1–5. https://doi.org/10.1073/pnas.2107576118
Subbarao GV, Sahrawat KL, Nakahara K et al (2013) A paradigm shift towards low-nitrifying production systems: The role of biological nitrification inhibition (BNI). Ann Bot 112:297–316
Subbarao GV, Kishii M, Bozal-Leorri A et al (2021) Enlisting wild grass genes to combat nitrification in wheat farming: A nature-based solution. Proc Natl Acad Sci 118:e2106595118. https://doi.org/10.1073/pnas.2106595118
Tkacz A, Poole P (2021) The plant microbiome: The dark and dirty secrets of plant growth. Plants People Planet 3:124–129. https://doi.org/10.1002/ppp3.10167
Trivedi P, Leach JE, Tringe SG, et al (2020) Plant–microbiome interactions: from community assembly to plant health. Nat Rev Microbiol 18:. https://doi.org/10.1038/s41579-020-0412-1
Tselas SK, Georghiou KC, Thanos CA (1979) Anthocyanin formation in maize roots. Plant Sci Lett 16:81–86. https://doi.org/10.1016/0304-4211(79)90011-7
Vetterlein D, Carminati A, Kögel-Knabner I et al (2020) Rhizosphere Spatiotemporal Organization–A Key to Rhizosphere Functions. Frontiers in Agronomy 2:1–22. https://doi.org/10.3389/fagro.2020.00008
Vitousek PM, Aber JD, Howarth RW, et al (1997) Human Alteration of the Global Nitrogen Cycle: Sources and human alteration of the global nitrogen cycle: sources and consequences. Source: Ecological Applications Ecological Applications Ecological Applications 7:737–750. https://doi.org/10.1890/1051-0761(1997)007[0737:HAOTGN]2.0.CO;2
Vitousek PM, Menge DNL, Reed SC, Cleveland CC (2013) Biological nitrogen fixation: Rates, patterns and ecological controls in terrestrial ecosystems. Philosophical Trans Royal Soc b: Biol Sci 368:20130119. https://doi.org/10.1098/rstb.2013.0119
Wagner MR, Roberts JH, Balint-Kurti P, Holland JB (2020) Heterosis of leaf and rhizosphere microbiomes in field-grown maize. New Phytol 228:1055–1069. https://doi.org/10.1111/nph.16730
Wagner MR, Tang C, Salvato F et al (2021) Microbe-dependent heterosis in maize. Proc Natl Acad Sci U S A 118:1–8. https://doi.org/10.1073/pnas.2021965118
Waldo S, Russell ES, Kostyanovsky K et al (2019) N2O Emissions From Two Agroecosystems: High Spatial Variability and Long Pulses Observed Using Static Chambers and the Flux-Gradient Technique. J Geophys Res Biogeosci 124:1887–1904. https://doi.org/10.1029/2019JG005032
Walters WA, Jin Z, Youngblut N et al (2018) Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc Natl Acad Sci U S A 115:7368–7373. https://doi.org/10.1073/pnas.1800918115
Wang X, Chen Q, Wu Y et al (2018) Genome-wide Analysis of Transcriptional Variability in a Large Maize-Teosinte Population. Mol Plant 11:443–459. https://doi.org/10.1016/j.molp.2017.12.011
Weier KL, Macrae IC, Myers RJK (1993) Denitrification in a clay soil under pasture and annual crop: estimation of potential losses using intact soil cores. Soil Biol Biochem 25(8):991–997. https://doi.org/10.1016/0038-0717(93)90145-2
Xu J, Zhang Y, Zhang P et al (2018) The structure and function of the global citrus rhizosphere microbiome. Nat Commun 9:4894. https://doi.org/10.1038/s41467-018-07343-2
Xu G, Cao J, Wang X et al (2019) Evolutionary metabolomics identifies substantial metabolic divergence between maize and its wild ancestor, teosinte. Plant Cell 31:1990–2009. https://doi.org/10.1105/TPC.19.00111
York LM, Cumming JR, Trusiak A et al (2022) Bioenergy Underground: Challenges and opportunities for phenotyping roots and the microbiome for sustainable bioenergy crop production. The Plant Phenome Journal 5:1–25. https://doi.org/10.1002/ppj2.20028
Zhang X, Davidson EA, Mauzerall DL et al (2015). Managing Nitrogen for Sustain Dev. https://doi.org/10.1038/nature15743
Acknowledgements
We thank Savannah Henderson and Sierra Raglin for assistance in the field and lab; Christopher Mujjabi and the Crop Sciences Research and Education Center for assistance with field planting and maintenance; Mark Band at the University of Illinois Functional Genomics Center for assistance with the development of multiplex PCR using the Fluidigm system; and Chris Wright and the staff of the DNA Services Lab at the Roy J. Carver Biotechnology Center for assistance with DNA sequencing.
Funding
Support was provided by the National Science Foundation Integrative Graduate Education and Research Traineeship (NSF IGERT) grant 1069157 and the NSF Graduate Research Fellowship Program. This work was funded by the DOE Center for Advanced Bioenergy and Bioproducts Innovation (U.S. Department of Energy, Office of Science, Biological and Environmental Research Program under Award Number DE-SC0018420). Any opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the U.S. Department of Energy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Responsible Editor: Eric Paterson.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Favela, A., Bohn, M.O. & Kent, A.D. Genetic variation in Zea mays influences microbial nitrification and denitrification in conventional agroecosystems. Plant Soil (2024). https://doi.org/10.1007/s11104-024-06720-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11104-024-06720-9