
Abstract
Background and aims
Jacobaea vulgaris plants grow better in sterilized than in live soil. Foliar application of SA mitigates this negative effect of live soil on plant growth. To examine what causes the positive effect of SA application on plant growth in live soils, we analyzed the effects of SA application on the composition of active rhizosphere bacteria in the soil.
Methods
We studied the composition of the microbial community over four consecutive plant cycles (generations), using mRNA sequencing of the microbial communities in the rhizosphere of J. vulgaris. We initiated the experiment with an inoculum of live soil collected from the field, and at the start of each subsequent plant cycle, we inoculated a small part of the soil from the previous plant cycle into sterile bulk soil.
Results
Application of SA did not significantly increase or decrease the Shannon diversity at genus level within each generation, but several specific genera were enriched or depleted after foliar SA application. The composition of bacterial communities in the rhizosphere significantly differed between plant cycles (generations), but application of SA did not alter this pattern. In the first generation no genera were significantly affected by the SA treatment, but in the second, third and fourth generations, specific genera were significantly affected. 89 species out of the total 270 (32.4%) were present as the “core” microbiome in all treatments over four plant cycles.
Conclusions
Overall, our study shows that the composition of bacterial genera in the rhizosphere significantly differed between plant cycles, but that it was not strongly affected by foliar application of SA on J. vulgaris leaves. Further studies should examine how activation of the SA signaling pathway in the plant changes the functional genes of the rhizosphere bacterial community.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plants encounter a myriad of threats from the surrounding environment, including both abiotic and biotic stresses (Suzuki et al. 2014). Biotic stresses are mostly due to herbivory and pathogen infestation both below- and above-ground (Adair and Douglas 2017; Pieterse and Dicke 2007). Microbes in the soil can have a beneficial, pathogenic or neutral effect on the host plant. For example, soil bacteria such as Rhizoctonia species, often strongly negatively affect plant growth and survival (Issac et al. 1971). On the other hand, Plant growth-promoting rhizobacteria (PGPR), such as Pseudomonas and Burkholderia species are beneficial for the plant, e.g., via suppressing the growth of soil-borne pathogens or increasing nutrient availability (Bhattacharyya and Jha 2012). However, the overall net effect of soil microbial communities on plant growth is often negative (Nijjer et al. 2007). Most plant species grow less well in soils that contain a natural microbial community than in sterilized soils. This might be due to e.g., competition between plants and microbes for available nutrients or due to soil-borne plant pathogens (Berendsen et al. 2012; Callaway et al. 2004; Cesarano et al. 2017; Mazzoleni et al. 2015).
To counteract the effects of microbial pathogens, plants have evolved a broad range of defensive mechanisms, which are partly regulated via hormonal signaling pathways (Fujita et al. 2006). So far, studies have shown that foliar application of salicylic acid to plant tissues can activate Systemic acquired resistance (SAR) and boost the innate immune system of a plant (Peng et al. 2021). SAR is one of the most common defensive strategies of plants against biotrophic pathogenic microbes. Infection of plant tissues with biotrophic pathogens leads to activation of SA-responsive genes. Once SAR is induced by application of SA to plant leaves, Pathogenesis-related proteins (PRPs) including proteins like β-1, 3-glucanase and chitinases will be instantly expressed (Van Loon and Van Strien 1999). Commonly, both chitinases and glucanases show antimicrobial activities. Once PRPs are induced by SA, they act in several ways to assist plants against soil pathogens. Antifungal and antibacterial properties of PRPs can directly impact soil pathogens. PRPs can also indirectly boost host plant defenses through hydrolytic released compounds, such as ochitin and glucan fragments, and these oligosaccharides can further stimulate a series of defensive responses in the host plant (Lawrence et al. 2000; Edreva, 2005; Rashid, 2016).
Cultivars with a higher sensitivity to SA are often better defended against pathogens. For example, in tomato, exogenous application of SA can be effective against the pathogens Oidium neolycopersici and Botrytis cinerea, which cause powdery mildew and grey mould disease (Achuo et al. 2004; Seskar et al. 1998). In agriculture, application of SA is now used to suppress pathogenic microbial effects in e.g. tomato, pepper and pea crops (Barilli et al. 2010; Choi and Hwang 2011; Esmailzadeh et al. 2008). How SA application to the plant affects the microbial community in the soil is not fully uncovered.
Because plants alter the composition of the microbial community in the soil in which they grow, and SAR protects plants against pathogens, an important question is how activation of SAR alters the plant’s influence on the soil microbial community. Several studies have demonstrated that the activation of SAR indeed altered the composition of the soil microbial community and that SA can play a key role in shaping root bacterial communities (Hein et al. 2008; Kniskern et al. 2007; Lebeis et al. 2015). Furthermore, as plant species differ greatly in the way and magnitude in which they influence the soil bacterial community (Hannula et al. 2019; Pineda et al. 2020; Wubs and Bezemer. 2018), we may expect that the effects of SA application on the soil microbial community also differ among plant species.
Several studies have shown that the composition of the soil bacterial community varies greatly over time (e.g., Hannula et al. 2019). In a study on temporal variation in soils exposed to three different types of land use, the number of taxa present in the soil showed strong temporal variability, and these changes over time were considerably larger than the variation associated with land-use types (Lauder et al. 2013). Shade et al. (2012) demonstrated that soil microbial communities have clear successional trajectories. If generally true this would imply that application of SA to plants could also cause directed changes in the soil microbial community over time (Li et al. 2021). An important question is therefore how activation of SAR will alter the soil microbial community over time.
Previous studies showed that inoculation of a sterilized soil with natural, live soil reduced plant growth in comparison with that in sterilized soil for the plant species Jacobaea vulgaris (Joosten et al. 2009; Kos et al. 2015). Interestingly, applying SA to the leaves mitigated these negative effects (Jing et al. 2021). This implies that activation of SA-induced resistance may potentially suppress microbial pathogens present in live soil. If this is the case, an important question is whether the repeated foliar application of SA during consecutive generations of plant growth will increase this effect and hence, whether there is a selection for a more beneficial bacterial community. Conceptually, the temporal dynamics of foliar application of SA can follow different trajectories (Fig. 1). First, it is possible that both foliar application of SA and the effect of different generations do not alter the soil bacterial composition (Fig. 1i). Second, foliar application of SA may lead to different bacterial communities independent of time (Fig. 1ii). Third, bacterial communities may differ among generations but are not influenced by the SA application (Fig. 1iii). Fourth, foliar application of SA may influence bacterial communities, but these effects may differ among generations (Fig. 1iv).

Conceptual figure showing the potential effects of SA application on J. vulgaris over four generations. (i) No effect of SA and time. The bacterial community does not differ between SA-treated and control plants, and does not differ over time. (ii) SA effect only. The bacterial community is affected by SA application but the effect does not differ over time. (iii) Time effect only. The bacterial community changes over time, but is not affected by the SA treatment. (iv) SA × time effect. The bacterial community is affected by SA-application but these effects differ among generations
In this study, we sequenced the mRNA from rhizosphere soil samples of both SA-treated and control plants during four consecutive generations of growth of J. vulgaris. In each consecutive generation soil from the previous plant growth period was used. Using mRNA instead of DNA or rRNA enabled us to focus on the active soil microbial community (Gilbert et al. 2008). In this study, we focus on the bacterial community. Twenty-four rhizosphere soils were sequenced with an Illumina sequencing platform. The goal of this study is to answer the following questions: (1) How does the foliar application of SA in J. vulgaris affect the bacterial composition in the rhizosphere and is there a time effect or an interactive time × SA effect on the bacterial community? (2) What is the “core” bacterial community in the soils of plants exposed to the SA treatment and of control plants? (3) How does the application of SA influence the bacterial community in each generation? Are the SA effects consistent over time?
Materials and methods
Multi-generational plant growth experiment
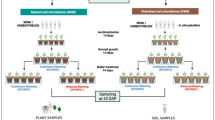
The current study focused on the effect of foliar SA application on the composition of the bacterial community in the rhizosphere in the inoculated soil. The effects of foliar application of SA on plant growth in the four generations were described elsewhere (Jing et al. 2021). A summary of the effects of foliar SA application on the growth of J. vulgaris are shown in Fig. S1. Details of the experiment are described below.
Jacobaea vulgaris (common ragwort) seeds were collected at the dunes of Meijendel (a calcareous sandy area from a coastal dune area north of The Hague, The Netherlands, 52°11′N, 4°31′E). Prior to germination, all seeds were surface sterilized (shaken for 2 min in 70% ethanol, then rinsed with sterilized water, put for 12 min in 2% bleach, and then rinsed again four times with sterilized water to minimize influences of seed-borne microbes (Bakker et al. 2015). The soil was also collected at Meijendel. The topsoil was collected to a depth of 15 cm after removing the grassland vegetation and the organic layer of the surface. The soil was sieved using a 5 mm sized mesh, homogenized with a concrete mixer, and then stored into 20-L plastic bags (Nasco Whirl–Pak Sample Bag). Bags were either sterilized by 35-K Gray gamma-irradiation (Synergy Health Company, Ede, The Netherlands) or kept at 4 °C for inoculation.
Surface sterilized seeds were germinated in sterile Petri dishes on moisturized filter paper. After one week, seedlings were randomly transferred individually to 500 mL pots consisting of a mixture of 90% sterilized soil and 10% live soil. Prior to potting but after mixing, the soil was kept in bags and left in the climate room for 14 days so that the mixed soil could settle and microbial communities could colonize the sterilized soil. After potting the seedlings, pots were randomly distributed over a climate room (relative humidity 70%, light 16 h at 20 °C, dark 8 h at 20 °C). Plants were watered regularly with Milli-Q and 5 mL Steiner nutrient solution (Steiner 1979) was added per plant on day seven after planting. 10 mL Steiner nutrient solution was added on day 13, and 20 mL Steiner nutrient solution was added on days 19, 28, 37, 42. The Steiner nutrient solution was prepared from seven different stock solutions (106.2 g Ca(NO3)2·4H2O, 29.3 g KNO3, 13.6 g KH2PO4, 49.2 g MgSO4·7H2O, 25.2 g K2SO4 and 2.24 g KOH, 3.29 g Fe-EDTA added to 1 L demineralized water), and a stock solution with micro elements (a mixed solution of 0.181 g MnCl2·4H2O, 0.286 g H3BO3, 0.022 g ZnSO4·7H2O, 0.0078 g CuSO4·5H2O and 0.0126 g NaMoO4·2H2O added to 1 L demineralized water). Ten mL of each stock solution was diluted in 1 L of demineralized water before use.
Plants were allocated to either a hormonal treatment (SA) or served as control (only solvent). Both treatments were replicated ten times. During plant growth, the phytohormone SA was applied to leaves three times a week for four consecutive weeks. The first application was given when plants were 14 days old. About 0.75 mL of 100 μM SA was sprayed on the leaves while carefully avoiding spillover to the soil. One week later, the treatment was repeated with 1.50 mL of SA. In the next week, the treatment was repeated with 2.25 mL of SA. SA solvent (purchased from Sigma-Aldrich, ≥ 99.0%) was made by dissolving 6.91 mg in 69.10 μl of ethanol (Wei et al. 2021). To avoid that SA application to the leaves would spoil to the soil, the soil was covered with sterilized aluminum foil during the application. Milli-Q water was then added until a final volume of 500 mL. Control plants were sprayed with sterile water with the same solvent (ethanol in Milli-Q water).
After six weeks, plants were gently removed from the pots. The rhizosphere soil for each treatment was harvested for each pot individually by gently shaking three times to remove the loosely adhering soil, after which rhizosphere soil was collected onto a sterile filter paper by removing the remnant soil with a fine sterilized brush. Rhizosphere soil was put in a 2 mL Eppendorf tube and stored at − 80 °C for further RNA extraction. The remaining rhizosphere soil and adhering soil of the ten pots were mixed and used as inoculum (live soil) for the next generational of plant growth. The inoculum soil (10%) was mixed with 90% sterilized soil.
The set-up was repeated for another three generations under the same conditions as described above so that there were four generations of plant growth. For the second, third, and fourth generation, the soil inoculum was derived from the previous generation from the same treatment and was a mixture of rhizosphere soil and the loosen adhering soil surrounding the roots. Again, after mixing, the soil was kept in bags and left in the climate room for 14 days. Hereafter, pots were filled with soil and a J. vulgaris seedling was planted into each pot. All replicate soils from the SA or control treatment were mixed before the inoculation. The SA treatment was carried out as described above in each generation. Fifty-four days after planting, all plants were harvested for each set-up per generation.
RNA extraction and meta-transcriptomic sequencing
For each treatment, the three successively labeled samples (Nos. 1, 2, 3, Nos. 4, 5, 6 and Nos. 7, 8, 9) were mixed and used as one composed replicate, Hence, three composed replicates were generated and used for RNA extraction for each treatment in each generation and a total of 24 soil samples were used for RNA extraction (3 replicates × 2 treatments × 4 generations). Total RNA was extracted with the RNeasy PowerSoil Total RNA kit (Qiagen). RNA concentration and quality were assessed by running 1 µl of the extracted raw RNA on the 4200 TapeStation (Agilent). Subsequently, unwanted DNA, salts and buffers were removed with the RNeasy minElute Cleanup Kit (Qiagen). The Ribo-Zero Magnetic kit for bacteria (Illumina) was used for mRNA enrichment and an RNA Clean and Concentrator kit (Zymoresearch) was used to clean additional buffers and proteins of the rRNA-depleted RNA. All the steps in extracting and cleaning RNA were according to the supplier’s instructions. Double-stranded cDNA was generated from the cleaned RNA obtained in the final step. Library preparation (Illumina Nextera XT DNA library), processing and sequencing were performed by FG Technologies (Leiden, The Netherlands) with paired-end (PE) 150 bp templates. Raw sequencing reads were deposited in NCBI database (Accession number: SUB8738755). Twenty-four metatranscriptomic libraries were generated, the size of each library is indicated in Table S1 and Fig. S2.
Data processing
Trimmomatic 0.39 was used for the removal of adapters of paired-end raw reads (Bolger et al. 2014). FastQC was applied to check the qualities, the bases with a threshold lower than 30 were cut off with Trimmomatic (Andrews 2010). Ribosomal RNAs of all 24 metatranscriptomic libraries were filtered with SortMeRNA (Sorting ribosomal RNA) (Kopylova et al. 2012). Eight rRNA representative databases (silva-bac-16 s-id90, silva-arc-16 s-id95, silva-euk-18 s-id95, silva-bac-23 s-id98, silva-arc-23 s-id98, silva-euk-28 s-id98 rfam-5 s-id98, rfam-5.8 s-id98) were derived from the SILVA SSU and LSU databases (release 119) and the RFAM databases with HMMER 3.1b1 and SumaClust v1.0.00 were used for fast filtering of rRNA from eukaryote, prokaryote and archaea. Then, all reads of the 24 metatranscriptomic libraries were combined into one set, which was the input of a de novo assembly. Trinity with default parameters was used for the metatranscriptomic assembly (Haas et al. 2013). Later, the quality of assembled contigs was assessed with Trinity scripts. The CD-HIT-EST algorithm was used to remove the duplicates of each transcript and reads with shorter than 300 bps were removed with a script modified from Li and Godzik (2006), after which reads of each library were mapped back to transcriptome with Bowtie2 (Langmead and Salzberg 2012). The isoform IDs per sample were extracted with Seqkit (Shen et al. 2016). Contigs of each sample were generated and then aligned against the NCBI NR (non-redundant) database by DIAMOND with a cut off e-value at 1e-5 (Buchfink et al. 2015). The closest match with an identity higher than 80% was kept for mapping. The output file of Blastx was further analyzed with the Lowest common ancestor (LCA) algorithm in MEGAN (version 6.0) with all default parameters (Camon et al. 2005; Huson et al. 2016). MEGAN was used to compute the data at different taxonomic levels and in this process NCBI taxonomy was employed to summarize results. The detailed workflow is described in Huson et al. (2007). A count table of microbial species was obtained with read counts assigned directly to taxon for the 24 samples. The number of assigned reads per taxa was extracted at species, genus, family and phylum level respectively. The number of identified phyla, families, genera and species were counted, and the composition and the percentage of reads used for each classification level were calculated. Trinity script was used to estimate the SA application and time effects on bacterial gene expression and a principal component analysis (PCA) was employed using the normalized number of genes to examine the composition of rhizosphere soil samples of SA-treated and control plants for the four generations. The analyses and results of gene expression are not part of this manuscript and are presented in the Appendix Gene expression.
Statistical analysis
Differences in the number of total reads and the number of non-rRNA reads over four generations were presented as mean ± SD. A Heinrich’s triangle figure was generated to visualize microbial composition at different phylogenetic levels of all the identified microbes from the 24 rhizosphere soil samples. Log10-transformed hit numbers of each genus were plotted as a function of the ranked genus abundance numbers, including all species, and a cut-off was performed with an abundance larger than 0.01% of the total reads. A Shapiro–Wilk test was used to test for differences between the distribution of abundance between the SA and control treatment.
The Shannon-diversity index was calculated for each of the 24 samples and differences between the Shannon-diversity of soils of SA treated plants and soils of control plants were tested with a student t-test. Subsequently, abundance at genus level was used to construct NMDS (nonmetric multidimensional scaling), PCA (Principal component analysis), OPLS-DA (orthogonal projection to latent structures discriminant analysis), and Venn diagrams, and Pearson distance and the Ward clustering algorithm statistical analysis was calculated since most of the reads were identified at the genus level.
Two-factor Venn diagrams were constructed to illustrate the numbers of unique and common genera in soil samples within each generation for the SA and control treatments, and a four-factor Venn diagram including all generations was performed for the SA and the control treatment separately (Heberle et al. 2015).
PCA and OPLS-DA were performed with SIMACA 15.0 using relative abundance at genus level. The relative abundance was calculated using the absolute abundance number of one genus divided by the total abundance of all bacterial genera in the sample. Before performing OPLS-DA analysis, we checked that our data fitted the model with a cross‐validated residual (CV)-ANOVA significance testing (n = 270, P < 0.02).
To visualize the compositional changes among different treatment and time categories, a NMDS using the Bray–Curtis index as a measure of dissimilarity was generated using relative abundances. To verify changes in composition due to the SA treatment and time effect, a PERMANOVA test was performed using the Adonis function (number of permutations = 999) in R (version 3.4.0) within the “vegan” package.
Local “immigration” and “extinction” in the rhizosphere soil of SA-treated or control plants over generations at genus level were calculated and the numbers were presented in Venn diagrams. A Student’s t-test was used to identify bacterial genera that were significantly enriched in soil samples of SA-treated or control plants. P values were adjusted for False discovery rates (FDR).
A Spearman’s rank correlation was performed to identify the genera that were significantly positively or negatively correlated with generation within the SA or control treatment. Genera with P values smaller than 0.05 (without a correction for multiple comparisons) were selected to create a heatmap for all 24 samples. Hierarchical clustering analysis was done for the 24 samples together, based on the relative abundance to show the similarity. The row-centered relative abundance of each genus was used to construct the color key (Chong et al. 2018). Heatmaps for only SA and only control treatments were also generated.
Results
Metatranscriptomic sequence data
A total of 898.4 million raw sequence reads were obtained from the 24 metatranscriptomic libraries, the smallest and largest library contained 25.0 and 52.0 million raw sequence reads, respectively (supplementary data Table S1). 846.9 million reads were kept after removing adapters and quality filtering control with FastQC. In total, 775.3 million reads were removed with the SortMeRNA program as ribosomal RNA (rRNA) reads when aligning them against eight rRNA representative databases (silva-bac-16 s-id90, silva-arc-16 s-id95, silva-euk-18 s-id95, silva-bac-23 s-id98, silva-arc-23 s-id98, silva-euk-28 s-id98 rfam-5 s-id98, rfam-5.8 s-id98), and 71.6 million reads were used as non-rRNA reads for further de novo assembly with Trinity (Fig. S2). For this set, the smallest library contained 1.5 million reads and the largest one 5.9 million reads. Reads for de novo assembly were normalized with the Trinity in silico normalization algorithm. The average Guanine-cytosine (GC) content for the 24 libraries was 60.10%. After assembly, 0.99 million contigs were removed because their length was shorter than 300 bps. A total of 1.3 million unique contigs were identified after removing duplicates with CD-HIT-EST. In total, 392.4 million bases were assembled. After we checked the quality of the contigs in all samples by realigning all contigs back to the assemblies using Bowtie2, the average mapping rate for proper pairs was 45.41%.
Overview of the assigned reads at differential microbial classification levels
When we aligned the 1.3 million unique contigs against the NR (non-redundant) database with DIAMOND and MEGAN 6.0, 0.39 million contigs were taxonomically classified, while the others did not provide a match with the available taxonomic information. Based on the analysis in MEGAN, the identified contigs were assigned at different classification levels. Twenty-two different bacterial phyla were identified, 283 families and 382 bacterial genera and 1081 bacterial species (Fig. S3). At the phylum, family, genus and species level 23.4%, 23.4%, 20.4% and 14.9% of the total number of contigs were assigned, respectively. Bacteria were the most prevalent in the microbial community taking up 98.3% of the total number of reads (Fig. S4a). Eukaryotes, with algae taking the largest proportion, were the second dominant, but Eukaryotes only covered 1.5% of the total number of reads (Fig. S4b).
SA application and time effects on bacterial community diversity and composition
From the total of 408 bacterial genera, 270 genera were included in the analysis (contigs with a relative abundance of more than 0.01%, Fig. S5). The genera in both soils showed significantly different abundance curves (Shapiro–Wilk test, df = 407, P < 0.0001; Fig. S5), the abundance curve in the SA soil was lower than that in the control soil. Application of SA did not significantly increase or decrease the Shannon diversity at genus level within each generation (t-test for the 1st generation: t = − 0.63, df = 5, P = 0.27; 2nd generation: t = 0.07, df = 5, P = 0.47; 3rd generation: t = 0.67, df = 5, P = 0.26; 4th generation: t = 0.50, df = 5, P = 0.31).
The NMDS plot showed that the bacterial communities of the same generation clustered together (Fig. 2a), PERMANOVA R2 = 0.30, P = 0.001). The SA and control separated in the NMDS plot (Fig. S7) but the separation was not significant (PERMANOVA R2 = 0.05, P = 0.18). Similar patterns were observed in a principal component analysis (PCA; Fig. S8). The OPLS-DA analysis showed clusters for replicates within each generation, and clear separation for the SA effect but only in the 2nd, 3rd and 4th generation (Fig. 2b). However, the generation effect was more evident than the SA effect.

Multivariate analysis of the bacterial community in soil samples from SA-treated and control plants grown in four generations. Shown are sample scores from a Nonmetric Multi-Dimensional Scaling (NMDS) plot (a), and an Orthogonal Projections to Latent Structures Discriminant analysis (OPLS-DA) plot (b) from the 24 rhizosphere soil samples
Core bacterial community
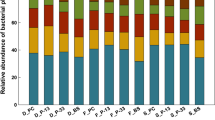
Eighty-nine species out of the total of 270 (32.4%) were present in all generations in at least two out of the three replicates of the soils of SA-treated and control plants suggesting that these make up the “core” microbiome (Fig. 3a). On average in each generation, 72.9% of all the genera were present in both soils (Fig. 3b). In the first generation, both soils shared about 74.2% of the genera while 7.7% only occurred in the SA-treatment and 18.0% only in the control (Fig. 3b-1). The percentage of shared genera by SA-soil and control soil in the 2nd, 3rd and 4th generation was 67.6%, 72.9% and 76.8% (Figs. 3, 4). For soils of the control treatment, 49.5% of the genera were shared over all four generations; while 45.1% of genera were shared in soils of the SA treated plants over four generations (Fig. 4c; Table S2). Immigration (i.e., genera not present in the soil in the previous generation) was somewhat higher in the SA treatment (on average 42 new genera) than in the control (on average 34 new genera) while the opposite was true for extinction rates (genera present in the previous but not in the current generation). On average, 31 genera went extinct in the SA treatment and 33 in the control treatment (Fig. 4). The information on Archaea, virus and eukaryote is listed in the supplementary information (Fig. S6).

Venn diagrams showing the number of unique and shared bacterial genera in the rhizosphere soil samples of SA-treated and control plants. The diagram in a is based on an analysis of genera that occur in all generations of the SA treatment, in b each generation is analyzed separately, c shows the diagram for all generations combined for the SA and control treatment

Local “immigration” and “extinction” of bacterial genera in the rhizosphere soil of SA-treated and control plants over time. For each of two consecutive generations, shown are the number of genera present only in the first of those generations (i.e., representing genera that go extinct), present in both generations, and present only in the second of those generations (i.e., representing generate that immigrate). Genera were considered present in a treatment when present in at least two of the tree replicates. G1, G2, G3 and G4 represent the 1st, 2nd, 3rd and 4th generation
SA selection of rhizosphere bacteria
When analyzed per generation, in total eight genera differed among the SA treatment and control (Fig. 5). No genus was significantly affected in more than one generation and no genera were significantly affected in the first generation. Most of the significant genera were only present in either the control or SA treatment. A Spearman’s rank correlation showed that 41 (out of 240) genera in the rhizosphere soil of SA-treated plants were significantly increasing and 31 genera were decreasing over generations. For the control soils these numbers were 47 and 27, respectively out of a total of 239 genera (Table S3). The heatmap including all 24 samples showed a clear generation effect, but no clear SA effect (Fig. 6). A heatmap representing the patterns of all identified genera in the 12 rhizosphere soils of SA-treated plants showed that replicates within a generation clustered and that the 2nd, 3rd and 4th generation showed a higher similarity than the 1st generation (Fig S8a). For the control plants, the samples from the first generation differed from the three other generations (Fig S8b). The variation in gene expression of bacteria followed the same pattern as that observed for composition (see Appendix Gene expression).

Bar chart showing the relative abundance expressed as % (mean ± SE) in control (left) or SA treated (right) plants of the genera that were significantly up or down regulated in at least one generation. The relative abundance is shown for all four generations. The significance is based on a student t-test with P values (< 0.05) adjusted based on False discovery rates (FDR). Genus names and generation code are listed on the left. RA represents relative abundance of read counts

Heatmap with a hierarchical clustering analysis of all genera of rhizosphere soil of SA-treated plants and control plants in the 24 samples. The hierarchical clustering was calculated with Pearson distance and the Ward clustering algorithm based on the relative abundance of each genus. The color code represents the row-centered relative abundance. SA1, SA2, SA3 and SA4 represent SA treatments from the 1st generation, 2nd generation, 3rd generation and 4th generation. Control1, control2, control3 and control4 represent control treatments from the 1st generation, 2nd generation, 3rd generation and 4th generation
Discussion
In this study, we examined how the activation of SA-induced resistance in the plant impacts the microbial composition in the rhizosphere, and how this effect changes over generations of plant growth. Our study shows that the composition of the active rhizosphere bacteria communities of the plant J. vulgaris changed significantly over generations, but that neither the effects of activation of SA-associated plant defense pathways nor the interaction between generation number and SA on the bacterial composition had a significant impact on the composition. Within generations the application of SA selected for different bacterial genera in the rhizosphere soil, but these selected genera differed from generation to generation. There were no SA-mediated changes in active bacterial genera in the first generation, suggesting that there are no immediate effects of activation of the SA defense pathway on the soil microbial composition.
Our study showed that aboveground activation of SA-associated plant defense pathways influenced different bacterial genera in the second, third and fourth generations. Effects of SA-induced resistance on the soil microbial community were reported in several other studies. For example, Hein et al. (2008) compared the effect of SA application on the composition of rhizosphere bacterial communities in several Arabidopsis mutants with Terminal restriction fragment length polymorphism (T-RFLP) analysis. They found that SA-induced resistance changed the structure of bacterial communities in the rhizosphere. In addition, Lebeis et al. (2015) demonstrated that SA application modulates colonization of the root microbiome by specific bacterial taxa. SA in plants is associated with the expression of Pathogenesis-related proteins (PRPs). We hypothesize that PRPs, consisting of proteins like β-1, 3-glucanase and chitinases are expressed at a higher level after the application of SA to plant leaves. Consequently, soil bacterial pathogens will be affected by the antibacterial activities of the released PRPs. It has been shown in several studies that these PRPs possess antimicrobial activities resulting in suppression of microbial pathogens, consequently changing the microbial composition (Qu et al. 2020; Dos Santos and Olivares 2021; Rashid 2016). In our research we did not find a significant expression of pathogenesis-related genes in the soil after foliar application of SA on plant leaves.
Even though the experimental conditions and plant genotypes remained the same throughout the experiment, the effects of SA application on the bacterial community differed among generations. In this context, it is important to note that for each generation we used an inoculum, which means that we placed a subset of the microbial community in a sterile background. This may explain why there was so much variation temporally, as in each generation a different subset of the microbial community may have been activated. It is also possible that the composition of the bacterial community is variable over time within each generation and as a consequence also among generations (Hannula et al. 2019; Hickey et al. 2013; Lauber et al. 2013). Moreover, in this study we only focused on the microbial community from the rhizosphere samples of J. vulgaris, and we did not examine the endophytic microbial community which can be strongly affected by application of SA to plant leaves (Noman et al. 2021). Future studies should examine the effects of belowground plant defense responses of J. vulgaris on endophytic bacteria in the roots.
Of the four potential models, our data matched with the third one (Fig. 1iii), showing that the bacterial communities did differ among generations but were not strongly influenced by SA application. This is line with studies showing that the composition of the soil bacterial community fluctuates greatly during plant growth (Hannula et al. 2019; Lauder et al. 2013). Moreover, the application of SA selects for different bacterial genera in the rhizosphere soil from generation to generation. This suggests that the effects of SA application to plants on the soil microbial community are not consistent over time. If generally true it will be difficult to predict the effects of activation of plant defenses on soil microbes, and ultimately how this will influence the interactions between plants and microbes in the rhizosphere (Li et al. 2021).
Interestingly, in soils of SA-treated plants, we found an increase of Caballeronia, unclassified Cytophagaceae, Crinalium and Candidatus Thermofonsia Clade 2. The Caballeronia genus is often reported to play an important role in fixing nitrogen and promoting plant growth. Species in this genus are predominantly endophytic diazotrophic bacteria and N-fixing bacteria (Padda et al. 2018; Puri et al. 2018, 2020). This suggests that activation of SA signaling pathways in J. vulgaris plants may benefit bacteria that are beneficial to plant growth, but further studies are needed to confirm this. The functions of the other species of which the abundance differentially increased are poorly understood. It is noteworthy, though, that Crinalium is a genus that is often isolated from sandy dune soils, so it is not surprising that we detected this genus as we used dune soils in our experiment (Mikhailyuk et al. 2019). Interestingly, the gene expression pattern followed the same pattern as the compositional changes of soil bacteria communities. Therefore, we suggest that further studies should extract the information of these detected genera at both the species level and gene functional level.
In conclusion, we provide evidence that the composition of bacterial communities in the rhizosphere significantly differed between plant cycles (generations), but we found no evidence that application of SA altered this pattern. However, application of SA influenced different bacterial genera in the rhizosphere, but the responsive genera varied between generations. No bacterial genera were detected that responded to SA application in the first generation suggesting that there are no immediate responses of bacteria in the rhizosphere to SA application to plants. However, further studies are required before we can make firm conclusions about this. Our results provide a new perspective on the effects of plant hormones on temporal changes in the soil microbial community.
References
Achuo EA, Audenaert K, Meziane H, Höfte M (2004) The salicylic acid-dependent defence pathway is effective against different pathogens in tomato and tobacco. Plant Pathol 53:65–72
Adair KL, Douglas AE (2017) Making a microbiome: the many determinants of host-associated microbial community composition. Curr Opin Microbiol 35:23–29
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Fast QC, 532:1. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Bakker MG, Chaparro JM, Manter DK, Vivanco JM (2015) Impacts of bulk soil microbial community structure on rhizosphere microbiomes of Zea mays. Plant Soil 392:115–126
Barilli E, Sillero JC, Rubiales D (2010) Induction of systemic acquired resistance in pea against rust (Uromyces pisi) by exogenous application of biotic and abiotic inducers. J Phytopathol 158:30–34
Berendsen RL, Pieterse CM, Bakker PA (2012) The rhizosphere microbiome and plant health. Trends Plant Sci 17:478–486
Bhattacharyya PN, Jha DK (2012) Plant growth-promoting rhizobacteria (PGPR): emergence in agriculture. World J Microb Biot 28:1327–1350
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59
Callaway RM, Thelen GC, Rodriguez A, Holben WE (2004) Soil biota and exotic plant invasion. Nature 427:731–733
Camon EB, Barrell DG, Dimmer EC, Lee V, Magrane M, Maslen J, Binns D, Apweiler R (2005) An evaluation of GO annotation retrieval for BioCreAtIvE and GOA. BMC Bioinformatics 6:S17
Cesarano G, Zotti M, Antignani V, Marra R, Scala F, Bonanomi G (2017) Soil sickness and negative plant-soil feedback: a reappraisal of hypotheses. J Plant Pathol 99:545–570
Choi HW, Hwang BK (2011) Systemic acquired resistance of pepper to microbial pathogens. J Plant Pathol 159:393–400
Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46:W486–W494
Dos Santos LF, Olivares FL (2021) Plant microbiome structure and benefits for sustainable agriculture. Curr Plant Biol 26:100198
Edreva A (2005) Pathogenesis-related proteins: research progress in the last 15 years. Gen Appl Plant Physiol 31:105–124
Esmailzadeh M, Soleimani MJ (2008) Exogenous applications of salicylic acid for inducing systemic acquired resistance against tomato stem canker disease. J Biol Sci 8:1039–1044
Fujita M, Fujita Y, Noutoshi Y, Takahashi F, Narusaka Y, Yamaguchi-Shinozaki K, Shinozaki K (2006) Crosstalk between abiotic and biotic stress responses: a current view from the points of convergence in the stress signaling networks. Curr Opin Plant Biol 9:436–442
Gilbert JA, Field D, Huang Y, Edwards R, Li W, Gilna P, Joint I (2008) Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE 3:e3042
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494
Hannula SE, Kielak AM, Steinauer K, Huberty M, Jongen R, Jonathan R, Heinen R, Bezemer TM (2019) Time after time: temporal variation in the effects of grass and forb species on soil bacterial and fungal communities. Mbio 10:e02635-e2719
Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R (2015) InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16:169
Hein JW, Wolfe GV, Blee KA (2008) Comparison of rhizosphere bacterial communities in Arabidopsis thaliana mutants for systemic acquired resistance. Microb Ecol 55:333–343
Hickey RJ, Abdo Z, Zhou X, Nemeth K, Hansmann M, Osborn TW III, Wang F, Forney LJ (2013) Effects of tampons and menses on the composition and diversity of vaginal microbial communities over time. BJOG 120:695–706
Huson DH, Auch AF, Qi J, Schuster SC (2007) MEGAN analysis of metagenomic data. Genome Res 17:377–386
Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, Ruscheweyh HJ, Tappu R (2016) MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol 12:e1004957
Isaac I, Fletcher P, Harrison JAC (1971) Quantitative isolation of Verticillium spp. from soil and moribund potato haulm. Ann Appl Biol 67:177–183
Jing Z, Klinkhamer PGL, Vrieling K, Bezemer TM (2021) Foliar application of SA alters the effects of live soils on plant performance BAAE https://doi.org/10.21203/rs.3.rs-159617/v1
Joosten L, Mulder PP, Klinkhamer PG, van Veen JA (2009) Soil-borne microorganisms and soil-type affect pyrrolizidine alkaloids in Jacobaea vulgaris. Plant Soil 325:133
Kniskern JM, Traw MB, Bergelson J (2007) Salicylic acid and jasmonic acid signaling defense pathways reduce natural bacterial diversity on Arabidopsis thaliana. Mol Plant-Microbe Interact 20:1512–1522
Kopylova E, Noé L, Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28:3211–3217
Kos M, Tuijl MA, de Roo J, Mulder PP, Bezemer TM (2015) Plant-soil feedback effects on plant quality and performance of an aboveground herbivore interact with fertilisation. Oikos 124:658–667
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357
Lauber CL, Ramirez KS, Aanderud Z, Lennon J, Fierer N (2013) Temporal variability in soil microbial communities across land-use types. ISME J 7:1641–1650
Lawrence CB, Singh NP, Qiu J, Gardner RG, Tuzun S (2000) Constitutive hydrolytic enzymes are associated with polygenic resistance of tomato to Alternaria solani and may function as an elicitor release mechanism. Physiol Mol Plant Pathol 57:211–220
Lebeis SL, Paredes SH, Lundberg DS, Breakfield N, Gehring J, McDonald M, Malfatti S, Del Rio TG, Jones CD, Tringe SG, Dangl JL (2015) Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Sci 349:860–864
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Li E, de Jonge R, Liu C, Jiang H, Friman VP, Pieterse CM, Bakker PA, Jousset A (2021) Rapid evolution of bacterial mutualism in the plant rhizosphere. Nat Commun 12:1–13
Mazzoleni S, Bonanomi G, Incerti G, Chiusano ML, Termolino P, Mingo A, Senatore M, Giannino F, Cartenì F, Rietkerk M, Lanzotti V (2015) Inhibitory and toxic effects of extracellular self-DNA in litter: a mechanism for negative plant-soil feedbacks? New Phytol 205:1195–1210
Mikhailyuk T, Vinogradova O, Holzinger A, Glaser K, Samolov E, Karsten U (2019) New record of the rare genus Crinalium Crow (Oscillatoriales, Cyanobacteria) from sand dunes of the Baltic Sea, Germany: epitypification and emendation of Crinalium magnum Fritsch et John based on an integrative approach. Phytotaxa 400:165–179
Nijjer S, Rogers WE, Siemann E (2007) Negative plant-soil feedbacks may limit persistence of an invasive tree due to rapid accumulation of soil pathogens. Proc Royal Soc 274:2621–2627
Noman M, Ahmed T, Ijaz U, Shahid M, Li D, Manzoor I, Song F (2021) Plant-microbiome crosstalk: dawning from composition and assembly of microbial community to improvement of disease resilience in plants. Inter J Mol Sci 22:6852
Padda KP, Puri A, Chanway CP (2018) Isolation and identification of endophytic diazotrophs from lodgepole pine trees growing at unreclaimed gravel mining pits in central interior British Columbia, Canada. Can J for Res 48:1601–1606
Peng Y, Yang J, Li X, Zhang Y (2021) Salicylic acid: biosynthesis and signaling. Annu Rev Plant Biol 23:72
Pieterse CM, Dicke M (2007) Plant interactions with microbes and insects: from molecular mechanisms to ecology. Trends Plant Sci 12:564–569
Pineda A, Kaplan I, Hannula SE, Ghanem W, Bezemer TM (2020) Conditioning the soil microbiome through plant-soil feedbacks suppresses an aboveground insect pest. New Phytol 226:595–608
Puri A, Padda KP, Chanway CP (2018) Evidence of endophytic diazotrophic bacteria in lodgepole pine and hybrid white spruce trees growing in soils with different nutrient statuses in the West Chilcotin region of British Columbia, Canada. For Ecol Manage 430:558–565
Puri A, Padda KP, Chanway CP (2020) Can naturally-occurring endophytic nitrogen-fixing bacteria of hybrid white spruce sustain boreal forest tree growth on extremely nutrient-poor soils? Soil Biol Biochem 140:107642
Qu Q, Zhang Z, Peijnenburg WJ, Liu W, Lu T, Hu B, Chen J, Chen J, Lin Z, Qian H (2020) Rhizosphere microbiome assembly and its impact on plant growth. J Agric Food Chem 7:5024–5038
Rashid A (2016) Defense responses of plant cell wall non-catalytic proteins against pathogens. Physiol Mol 94:38–46
Seskar M, Shulaev V, Raskin I (1998) Endogenous methyl salicylate in pathogen-inoculated tobacco plants. Plant Physiol 116:387–392
Shade A, Peter H, Allison SD, Baho D, Berga M, Bürgmann H, Huber DH, Langenheder S, Lennon JT, Martiny JB, Matulich KL (2012) Fundamentals of microbial community resistance and resilience. Front Microbiol 3:417
Shen W, Le S, Li Y, Hu F (2016) SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11:e0163962
Steiner AA (1979) The selective capacity of plants for ions and its importance for the composition and treatment of the nutrient solution. In Symposium on Research on Recirculating Water Culture 98:87–98
Suzuki N, Rivero RM, Shulaev V, Blumwald E, Mittler R (2014) Abiotic and biotic stress combinations. New Phytol 203:32–43
Van Loon LC, Van Strien EA (1999) The families of pathogenesis-related proteins, their activities, and comparative analysis of PR-1 type proteins. Physiol Mol Plant Pathol 55:85–97
Wei X, Vrieling K, Kim HK, Mulder PP, Klinkhamer PG (2021) Application of methyl jasmonate and salicylic acid lead to contrasting effects on the plant’s metabolome and herbivory. Plant Sci 303:110784
Wubs EJ, Bezemer TM (2018) Plant community evenness responds to spatial plant-soil feedback heterogeneity primarily through the diversity of soil conditioning. Funct Ecol 32:509–521
Acknowledgements
We would like to thank Connor Philippo for his practical assistance in extracting RNA and quality control of the raw reads, Bing Xie for his generous supporting in sharing his private server and help in troubleshooting, Fons Verbeek for sharing public resource of the server in the Leiden institute of advanced computer science (LIACS). We also thank Martine Huberty for discussing PCA statistical analysis. We thank Robin van Schendel for assisting in the Trinity assembly on Shark cluster, Meciael Liem and Alexander Kloosterman for troubleshooting coding errors in Linux, Niels van Tol for helping introducing people who can help us with performing Trinity assembly. Jing Zhang would also like to thank the China Scholarship Council for financial support.
Author information
Authors and Affiliations
Contributions
JZ, MB, KV and PK designed the experiment, JZ carried out the experiment, JZ, KV, MB and PK performed the data analysis. JZ, KV, MB and PK wrote the first draft of the manuscript, and all the authors contributed to revisions.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Responsible Editor: Xianyong Lin.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendices
Appendix Gene expression
Data processing
Trimmomatic 0.39 was used for the removal of adapters of paired-end raw reads (Bolger et al. 2014). FastQC was applied to check the qualities, the bases with a threshold lower than 30 were cut off with Trimmomatic (Andrews 2010). Ribosomal RNAs of all 24 metatranscriptomic libraries were filtered with SortMeRNA (Sorting ribosomal RNA) (Kopylova et al. 2012). Eight rRNA representative databases (silva-bac-16 s-id90, silva-arc-16 s-id95, silva-euk-18 s-id95, silva-bac-23 s-id98, silva-arc-23 s-id98, silva-euk-28 s-id98 rfam-5 s-id98, rfam-5.8 s-id98) were derived from the SILVA SSU and LSU databases (release 119) and the RFAM databases with HMMER 3.1b1 and SumaClust v1.0.00 were used for fast filtering of rRNA from eukaryote, prokaryote and archaea. Then, all reads of the 24 metatranscriptomic libraries were combined into one set, which was the input of a de novo assembly. Trinity with default parameters was used for the metatranscriptomic assembly (Haas et al. 2013). Later, A combined set of all quality filtered reads from all 24 rhizosphere libraries was combined into a single reference transcriptome assembly. Assembly and contig quality analysis was performed using Trinity scripts.
Statistical analysis
To determine the SA application and time effects on bacterial gene expression, a Principal component analysis (PCA) was employed using the normalized number of genes to examine the composition of rhizosphere soil samples of SA-treated and control plants for the four generations. A PERMANOVA test was performed using the adonis function (number of permutations = 999) in R within the “vegan” package to verify the effects of the SA treatment and time on the composition of all expressed genes.
Results
SA application and time effects on bacterial gene expression
A Principal component analysis (PCA) using log2 transformed normalized CPM showed that the read counts of contigs of the microbial community among generations were well separated (Fig. 7). This was in line with the permutation test (PERMANOVA R2 = 0.22, F = 19.6, df1 = 3, df2 > 999, p < 0.01). In addition, the effect of SA application was significant (PERMANOVA R2 = 0.07, F = 6.3, df1 = 1, df2 > 999, p < 0.05). Gene expression patterns of SA-treated J. vulgaris and control samples were better separated in the third and fourth generation than in the first and second generation (Appendix Fig. 7).

Scatter plot from a Principal component analysis (PCA) of TMM normalized Counts per million (CPM) representing the overall gene expression patterns of different rhizosphere soil samples of SA-treated and control J. vulgaris plants over generations. Shapes represent the treatments and colors represent generations
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Klinkhamer, P.G.L., Vrieling, K. et al. Belowground responses of bacterial communities to foliar SA application over four plant generations. Plant Soil 470, 65–79 (2022). https://doi.org/10.1007/s11104-021-05158-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-021-05158-7