
Abstract
Background and aims
To form nitrogen-fixing nodules on pea roots, Rhizobium leguminosarum biovar viciae must be competitive in the rhizosphere. Our aim was to identify genes important for rhizosphere fitness.
Methods
Signature-tagged mutants were screened using microarrays to identify mutants reduced for growth in pea rhizospheres. Candidate mutants were assessed relative to controls for growth in minimal medium, growth in pea rhizospheres and for infection of peas in mixed inoculants. Mutated genes were identified by DNA sequencing and confirmed by transduction.
Results
Of 5508 signature-tagged mutants, microarrays implicated 50 as having decreased rhizosphere fitness. Growth tests identified six mutants with rhizosphere-specific phenotypes. The mutation in one of the genes (araE) was in an arabinose catabolism operon and blocked growth on arabinose. The mutation in another gene (pcaM), encoding a predicted solute binding protein for protocatechuate and hydroxybenzoate uptake, decreased growth on protocatechuate. Both mutants were decreased for nodule infection competitiveness with mixed inoculants, but nodulated peas normally when inoculated alone. Other mutants with similar phenotypes had mutations predicted to affect secondary metabolism.
Conclusions
Catabolism of arabinose and protocatechuate in the pea rhizosphere is important for competitiveness of R.l. viciae. Other genes predicted to be involved in secondary metabolism are also important.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rhizobia are soil bacteria studied primarily because of their ability to infect the roots of leguminous plants, producing nitrogen-fixing nodules. Prior to infecting legume roots, rhizobia grow in the rhizosphere (the region of soil in close proximity to roots) using nutrients secreted from the plant roots. Rhizobia in the rhizosphere are motile and can attach to roots and root hairs where they can grow to form a biofilm. These attached rhizobia are well positioned to detect flavonoids, isoflavonoids and related compounds that induce the bacterial genes required for legume nodulation (Downie 2010).
Each nodule produced on legume roots is usually the result of a clonal infection event and typically a legume such as pea grown in the field produces around 150 nodules (Bourion et al. 2007). This corresponds to about 150 successful infections, but since there can be around 104–106 pea-nodulating bacteria (Rhizobium leguminosarum biovar viciae) per g of soil (and at least a Kg of soil will be occupied by a mature pea root system), it is clear that the vast majority of R.l. viciae bacteria in the soil will not infect peas in any given growing season. Therefore rhizobia must be able to survive and grow well in the rhizosphere, without necessarily infecting legumes.
Although a great deal is known about rhizobial genes required for nodule infection and nitrogen fixation (Downie 2010), less is known about genes required for growth and survival in the rhizosphere, because their identification is relatively difficult. Different approaches have been taken to identify genes required for successful growth in the rhizosphere. For example, promoter trapping approaches (referred to as in vivo expression technology, IVET) have been used to identify rhizosphere-expressed promoters in Pseudomonas fluorescens (Varivarn et al. 2013) and in R.l. viciae (Barr et al. 2008) and a recombination-based variation of IVET has been used to analyse gene expression in S. meliloti during rhizosphere and symbiotic growth of S. meliloti (Gao and Teplitski 2008). Microarray analyses of bacterial RNA has given insights into genes expressed following growth of Pseudomonas aeruginosa in a root exudate (Mark et al. 2005) and of R.l. viciae grown in root exudate and in different rhizospheres (Ramachandran et al. 2011). These studies led to the identification of genes induced in these environments and were followed up by the construction of targeted mutations, several of which decreased rhizosphere fitness. An alternative reverse genetics approach has been to use comparative genomics, in an attempt to identify genes prevalent in bacteria that grow in the rhizosphere (Redondo-Nieto et al. 2013; Silby et al. 2009). These reverse genetics approaches can identify many genes likely to be involved in rhizosphere growth or survival, but they require targeted mutagenesis to determine whether the genes are required for the bacteria to be competitive in the rhizosphere.
Direct approaches to identification of mutations that decrease the growth of bacteria in specific environments include signature-tagged mutagenesis (Pobigaylo et al. 2008) and direct sequencing of transposon insertions in large populations of transposon-mutagenized bacteria (Barquist et al. 2013). In both these approaches poorly growing representatives can be identified based on the depletion of bacteria containing specific transposon insertions within a population of bacteria in the tested environment. The depleted bacteria can be detected based on decreased levels of specific transposon insertions identified from parallel DNA sequencing of populations or by using microarrays to identify depletion of tagged transposon insertions in defined groups of mutants. To complement our previous work on IVET selection of rhizosphere-expressed genes (Barr et al. 2008) and microarray analysis and mutagenesis of genes induced in the rhizosphere of peas (Ramachandran et al. 2011), here we have used signature tagged mutagenesis to identify genes which, when mutated, decrease the fitness of R.l. viciae in the pea rhizosphere.
Materials and methods
Bacterial strains and growth conditions
R.l. viciae and Escherichia coli strains used in this work are listed in Table 1 . E. coli strains were grown at 37 °C in L medium (Sambrook et al. 1989) and R.l. viciae strains were grown at 28 °C either in TY medium (Beringer 1974) or in AMS minimal medium containing 10 mM NH4Cl and carbon sources as indicated (Poole et al. 1994). Antibiotics were used at the following concentrations (μg ml−1): spectinomycin (Spc: 100); streptomycin (Str: 500) and neomycin (Nm: 160) unless specified otherwise. Growth was monitored at 28 °C using an InfiniteF200 microtitre plate shaker/reader (Tecan, Reading, RG7 5AH, UK), measuring absorbance at 600 nm every 40 min. Transductions were done using phage RL38 as described previously (Buchanan-Wollaston 1979). All plant tests were done with Pisum sativum cv. Avola peas in a growth chamber at 22 °C with a 16 h-light 8 h-dark light cycle.
Bacterial growth in pea rhizospheres was done with pea seedlings grown in 50 ml screw-capped Falcon centrifuge tubes (Fisher Scientific UK Ltd Loughborough LE11 5RG). Washed vermiculite was added to the 30 ml mark on each tube, 10 ml of nitrogen-free FP medium (Fahraeus 1957) was added and the tubes were capped and autoclaved. A single sterile germinating pea seed was added to each tube and after 1 week in a growth chamber, the tubes were inoculated with about 104 bacteria. One week later, bacteria were recovered from the rhizosphere by cutting off the plant shoots, adding 18 ml of phosphate buffered saline, vigorously mixing the tubes for 20 min and then making serial dilutions to count the bacteria. In the absence of pea seedlings there was very little bacterial growth and so the increase in the rhizosphere population (from about 104 to about 108 bacteria) was due to the pea roots.
Molecular biology techniques
DNA cloning, ligations, transformation, restriction enzyme digestions and DNA hybridizations were done using standard methods (Sambrook et al. 1989). Plasmid DNA was isolated using the Promega (Southampton, SO16 7NS UK) Wizard Plus SV Miniprep DNA Purification System following the recommended protocol. Genomic DNA for DNA hybridization was isolated as described previously (Chen and Kuo 1993). DNA for cloning, PCR, or from the rhizosphere bacteria was isolated using the QIAGEN (UK, Manchester M15 6SH) DNeasy Tissue kit.
Transposon insertion sites were identified either by inverse PCR or arbitrary primed PCR. For inverse PCR, DNA was digested with EcoRI and ligated and inverse PCR reactions were carried out using 1 μl ligated genomic DNA, 10 pmol of each of the primers GCGATCCAGACTGAATGCCC and TCGACCTGCATCTAGCCCGC, 7 μl H2O, 10 μl Sigma ReadyMix (redtaq PCR mix) Sigma-Aldrich Dorset SP8 4XT, UK) using a PCR protocol as follows; 94 °C for 3 min; 94 °C for 45 s, 58 °C for 45 s, 72 °C for 5 min repeated for 30 cycles. Arbitrary primed PCR was done as described (Das et al. 2005) using the primers GGCCACGCGTCGACTAGTCA-NNNNNNNNNN-CGATC and GGCCACGCGTCGACTAGTCA. Sequencing was done at The Genome Analysis Centre (Norwich UK). Database searches were done at NCBI using the BLAST program (Altschul et al. 1990) and compared to the published sequence of R.l. viciae 3841 (Young et al. 2006).
Construction of signature tagged mutant libraries
A library of pG18-STM plasmids (Pobigaylo et al. 2006) was obtained from Anke Becker from the University of Bielefeld, Germany. These plasmids contain a modified form of mTn5-GNm from pCRS487 (Reeve et al. 2006), with additional linker cassettes containing two signature tags and primer binding sites for amplification of the tags. For mutagenesis, each E. coli S17-1 λpir carrying a pG18-STM plasmid was cultured at 37 °C in LB Km Gm to OD600 0.4–0.5, mixed (300 μl of E. coli and 700 μl of 3841) with freshly TY-grown 3841, the bacteria were pelleted by centrifugation, resuspended in 50 μl TY and spread onto a sterile nitrocellulose filter on a TY plate. Plates were incubated at 30 °C for 40 h, the bacteria on the filters were resuspended in 1 ml AMS and 0.9 ml was inoculated into 10 ml TY Nm, Str and incubated at 26 °C, shaking for 8 h. These bacteria were pelleted by centrifugation and resuspended in 600 μl AMS, 400 μl 50 % glycerol. Prior to storage at −70 °C, 100 μl was plated onto AMS plates containing 10 mM succinate, 20 mM pyruvate, 500 μg/ml Nm, 500 μg/ml Str. After 4 days at 28 °C colonies were picked and stored in 20 % glycerol. Pools of different mutants carrying different tags were arranged as described by Pobigaylo et al. (2006).
Microarray screening of signature tagged mutants
For each pool of mutants, six pea seedlings were inoculated with a pool of 102 signature-tagged mutant bacteria each with a different tag. Cultures were grown overnight at 26 °C in 10 ml TY Nm Str and 1 ml at OD600 0.1 (approximately 108 CFU) was pelleted, resuspended in 1 ml AMS and diluted (10 μl in 10 ml AMS, to about 105 CFU ml−1) and 1 ml of this was inoculated onto to each of six 7-day-old seedlings. Genomic DNA was extracted from the washed bacterial suspension to represent the inoculant pool. A week after inoculation, mutant pools of bacteria from the six rhizospheres were combined into two samples, each derived from the rhizosphere of three plants and DNA was isolated from both samples.
Microarrays were printed onto Corning UltraGAPS slides (Fisher Scientific UK Ltd Loughborough LE11 5RG) by Antony Jones at the Functional Genomics & Proteomics Unit at The University of Birmingham. The signature tags used were 24-mer oligonucleotides. Arrays were printed using a BioRobotics TAS microarraying robot, with eight blocks of 24 × 24 spots. This allows for a minimum of 20 replicas of each experimental tag, and 12 replicas of each “landing light” control tag.
Signature tag oligonucleotides were amplified from genomic DNA isolated from inoculant and rhizosphere bacteria by PCR, using primers fluorescently labelled with 5′ Cy3 or Cy5 marker dyes respectively. PCR reactions for amplifying the signature tags contained 2 μl isolated genomic DNA template, 25 pmol of each primer, 50 μl 2x Promega GoTaq Green Master Mix and made up to 100 μl with H2O. The primers STM-P1 (AAAGGACGTGGTTTACGGGGC), STM-P2 (TATATGAATGCCGCCACCCCC), STM-P3 (ATTTTAACTCCCCTCCGCCGC), and STM-P4 TAGTCCTGGTGCATTGAGCCC were as described (Pobigaylo et al. 2006) and the PCR cycle was as described above. The reactions were passed through Macherey-Nagel NucleoSpin Extract II PCR Cleanup kit, and four replica samples were eluted in 74 μl. Samples were eluted into amber Eppendorf 1.5 ml microcentrifuge tubes, to limit the exposure of fluorescently-labelled PCR products to light. The amounts of DNA, and Cy3- or Cy5- incorporation were quantified at 260, 550 and 650 nm respectively.
Aliquots of 5 pmol of each of the Cy3- or Cy5-labelled products were mixed, dried under vacuum, and resuspended in 20 μl H2O. Control tags (amplified tags represented on the array but not in the mutant population) were added at a concentration of 500 fmol per control signature tag. Arrays were hybridized using an Advalytix Array Booster DNA Microarray Incubator (Olympus America Inc., Concord, MA, USA).
Microarray slides were scanned using an Axon GenePix 4200A scanner, with GenePixPro 6 software (Molecular Devices Wokingham, RG41 5TS). Slide images were analysed using BlueFuse software, using channel 1 as the control (532, Cy3, inoculant pool) channel and channel 2 as the rhizosphere extract (635, Cy5) channel. BlueFuse output files were imported into the GeneSpring (Agilent Technologies UK, Shropshire SY7 8NR) software package, where replica data for each spot were collated and normalized using Lowess normalizations to show mean values for all oligonucleotide spots. Arrays were compared for Cy3 and Cy5 balance both as a whole and when separated into tag pair groups. Significance was tested using Bonferroni corrected P values as described in the Genespring software.
Tests of rhizosphere fitness and competitive nodule infection
Fifty mutants showing a statistically significant decrease of at least 2-fold in one tag and at least 1.5 fold decrease in the other tag from the microarrays (Table 2) were mixed with the control strain RU3940 at a ratio (mutant:control) of 10:1 (or 100:1 as specified); strain RU3940 is equally competitive with WT for rhizosphere growth (Tett et al. 2014). To set this up, each strain was grown overnight at 26 °C in a shaking incubator, centrifuged and resuspended in 1 ml of AMS at OD600 0.1. Each mutant (90 or 990 μl) was mixed with the control strain (10 μl) in 10 ml AMS and serially diluted to about 104 CFU per ml and plated in triplicate on selective (Spc or Nm) TY medium to count the bacteria in inoculants. Then 1 ml was inoculated onto each of 9 pea seedlings grown and inoculated as for the mutant screen. After 7 days, rhizosphere bacteria were recovered and the bacteria from three rhizospheres were combined, thereby generating three independent pools each from three pea seedlings. Bacteria in each rhizosphere sample were counted in triplicate using selective (Sp or Nm) TY plates. Only data for those mutants showing significantly poorer growth than the control strain are presented. For those primary mutants that were outcompeted with WT, the mutations were transduced into WT R.l. viciae strain 300 and the transductants were retested as described above except that instead of using RU3940 as the competitor strain, 3841 was used. In these experiments the control and mutant bacteria were counted using TY Str or TY Nm respectively; it would have been inappropriate to use RU3940 in the nodule competitiveness tests described below, because it is defective for nitrogen fixation.
Competitive nodule infection experiments were done with inoculants grown as described above except that the control strain was 3841 and mixed with each mutant at a ratio of 1:1. The bacteria (about 104 in 10 ml H2O) were inoculated onto germinated pea seedlings in 250 ml flasks containing sand and vermiculite as described previously (Williams et al. 2008). Nodules were collected 25 days after inoculation, surface sterilized and then exudate from crushed nodules was plated on TY Str and TY Nm plates to identify nodule occupancy.
Results
Identification of signature-tagged mutants of R.l. viciae potentially attenuated for rhizosphere growth
A signature-tagged transposon mutant library was constructed using the pG18-STM plasmids described previously for mini-Tn5 mutagenesis of S. meliloti (Pobigaylo et al. 2008). Each mini-Tn5 contains two different DNA tags and all of the tags used can be distinguished on the basis of DNA hybridisation. We used a subset of the plasmids to generate pools of mutants and each pool contained 102 mutants all with different mini-Tn5 insertions. In this approach each mutant is present at a ratio of about 1:100 relative to the population. We used 54 pools of mutants corresponding to 5508 individual mutants and inoculated each pool separately onto the roots of six individual pea seedlings that had been grown for 1 week in sand/vermiculite. A portion of the inoculum was frozen and stored.
Rhizobia were recovered from the rhizosphere a week after inoculation and the signature-tagged oligonucleotides on the mini-Tn5 insertions were amplified from genomic DNA isolated from both the stored inoculants and from bacteria recovered from the rhizospheres. The DNA was fluorescently labelled using primers labelled with 5′ Cy3 or Cy5 dyes respectively. After clean-up and quantification of DNA and determination of the level of Cy5 or Cy5 incorporation, the labelled DNA was used to hybridise (at high stringency) microarrays containing multiple replicates of the signature tags. Analysis of the data identified 50 mutants that showed a decrease of at least 2-fold in one tag and at least a 1.5-fold decrease in the other tag (Table 2).
Confirmation of impairment of rhizosphere growth and identification of mutations
Each of the 50 mutants identified as being lower in rhizosphere abundance based on the microarray analysis was cultured and mixed in a ratio (10:1 or 100:1) with a Spc-resistant strain (RU3940), which was previously shown to be unaffected for rhizosphere growth in competitive assays (Tett et al. 2014). The ratios of control and tagged mutant bacteria in the inoculants were checked by colony counts selecting with Nm (mutant) or Spc (control) and each mixture of bacteria was inoculated onto multiple pea seedlings grown in sand/vermiculite. After 1 week, the bacteria were recovered from the rhizospheres and dilutions were plated onto TY plates containing Nm or Spc to count the relative numbers of mutant and control bacteria in the rhizospheres. In this assay, the expectation is that if the mini-Tn5-tagged mutant is defective for rhizosphere colonisation, then following a week of growth in the rhizosphere, the proportion of the control strain will significantly increase from the level at which it was inoculated. Based on this assay (Fig. 1) ten of the mutants (Table 2) showed a significant increase in the relative numbers of the control bacteria relative to mutants. This confirms the microarray data suggesting that these mutants are less fit for growth in the rhizosphere than the control strain.

Tests of increased growth of a control strain relative to primary mutants decreased for rhizosphere competitiveness. Mutants identified from microarray experiments as being likely to be defective for rhizoshere competitiveness were mixed with strain RU3940 (which is equally competitive with WT) in a ratio of about 10:1 (except for X365 which was mixed at a ratio of about 100:1) and inoculated into the rhizospheres of one-week-old pea seedlings. After 1 week of growth bacteria were isolated from the rhizospheres. Also shown (control) is a strain which carries mini Tn5 but showed no defect for rhizosphere growth. The black and open bars respectively show the % of strain RU3940 in the initial inoculum and in the rhizosphere as determined by bacterial counts. The data are averages (± SE) and all comparisons (except the control) were significantly different (p = <0.05) based on Tukey’s HSD Test
The sites of the mini-Tn5 insertions in the ten mutants were determined by DNA sequencing (Table 3). Nine of the mutations were confirmed to have been caused by insertion of mini-Tn5, but one of the mutations (in X385) appeared to have been caused by integration of the whole plasmid carrying mini-Tn5. Five of the mutations are in genes associated with intermediate metabolism with the following predicted functions: RL0031, an S-adenosyl homocysteine hydrolase involved in methionine biosynthesis; RL0855, a hydrolase acting on an unknown substrate; RL0634, probably a sugar alcohol dehydrogenase; RL3613, an α-ketoglutarate semialdehyde dehydrogenase; RL4123 a component of a sarcosine oxidase. Two of the mutations are in genes encoding predicted components of transporters, one (pRL90234) an inner membrane permease component and another (RL3906) in a periplasmic solute binding protein, which falls into the family of hydrophobic amino acid uptake transporters (HAAT) and is homologous to S. meliloti PcaM involved in protocatechuate transport (MacLean et al. 2008). Two of the mutations appear to be associated with the synthesis of polysaccharides; RL0811A, a predicted lipopolysaccharide glycosyl transferase and RL0079 related to inner-membrane polysaccharide synthases. The mutation in RL1109 is in a gene of unknown function encoding a conserved hypothetical protein.
Tests of growth rates of mutants
The selection of the tagged mutants on minimal medium should have excluded auxotrophs, but the mutation in X365 is in RL0031, a gene predicted to be involved in methionine biosynthesis. Therefore, we checked the growth rates of all the mutants in liquid minimal medium (AMS containing 10 mM mannitol). This revealed that X365 (mutation in RL0031) grows more slowly than WT (Table 3); this longer doubling time of 8.3 h was reduced to 6.0 (±0.1) h by the addition of 10 μM methionine. The observation that there was growth in the absence of methionine indicates that the mutation affects, but does not block methionine biosynthesis. The mutation in X385 within a predicted polysaccharide synthase (RL0079) also significantly reduced growth (Table 3). It seems very likely that the reason that these two strains are not competitive in the rhizosphere is due to their poor growth in minimal medium and in the rhizosphere. Most of the other strains grew with growth rates that were not significantly different from WT (Table 3) although X379 carrying a mutation in a predicted LPS glycosyl transferase (RL0811A) also grew slightly slower than WT (Table 3).
Tests of rhizosphere competitiveness of transductants
The identification of a plasmid integration into RL0079 alerted us to the possibility that although we had identified genes mutated by mini-Tn5 insertions, we could not exclude the possibility of secondary mutations causing the decreased rhizosphere competitiveness. Therefore we transduced the mutations back into the WT (strain 300) and all the transductants were confirmed by PCR to have the mutations in the expected genes. Despite repeated attempts, we were unable to transduce the mutations in the genes RL1109 (strain X378) or RL0079 (strain X385) into WT.
The eight transductants were tested for growth in pea rhizospheres essentially as described for the original mutants, except that the control strain (present at about 10 % in the inoculum) was 3841. With the exception of A1399, and A1402, the transductants showed a statistically significant decrease in rhizosphere competitiveness based on increased levels of recovery of the control strain (Fig. 2). This demonstrates that mutations in these genes decrease rhizosphere competitiveness. However, there was not significantly enhanced recovery of the control strain relative to the transductants A1399 (carrying the mutation in pRL90234), and A1402 (with the mTn5 inserted in RL4123). Since we confirmed by DNA sequencing that in A1399 and A1402 the mini Tn5 insertions were in the predicted genes, these mutations appear not to reduce competitiveness in the rhizosphere. This indicates that the rhizosphere defects in the original mutants were probably caused by mutations in other genes.

Tests of increased rhizosphere growth of a wild-type strain relative to transductants. Mutations potentially decreasing rhizosphere competitiveness were transduced into R.l. viciae strain 300 and the transductants were mixed with the wild-type strain (3841) in a ratio of 10:1 and inoculated into the rhizspheres of one-week-old pea seedlings. After 1 week bacteria were isolated from the rhizospheres. The black and open bars respectively show the % of the wild-type in the initial inoculum and in the rhizospheres. The data are averages (± SE) and those tests showing significant differences (p = <0.05 based on Tukey’s HSD Test) between numbers of the wild-type recovered from the inoculants and rhizospheres are marked with asterisks
Tests of competitiveness for nodule infection
Decreased growth of mutants in the rhizosphere should give rise to decreased competitiveness during nodule infection on the basis that decreased rhizosphere growth should result in reduced numbers associated with the roots and therefore decreased competitiveness for infection. Firstly we tested nodule induction on peas by all the transductants shown in Table 2. None of the mutants tested showed any significant difference in nodulation during the 21 days after inoculation; the plants produced an average of around 70 pink nodules. These observations demonstrate that none of the mutations causing decreased rhizosphere competitiveness blocked nodulation. The presence of pink nodules indicates that the mutants can also reduce nitrogen in nodules and this was confirmed with acetylene reduction assays (data not shown). Since the mutation that we failed to transduce from X385 is in a predicted polysaccharide synthase, and some such mutations can block nodule infection, we also tested this strain for nodulation. X385 formed an average of 60 nodules per plant (not significantly different from WT); again the nodules were pink and of a normal size indicating that the putative polysaccharide made by RL3906 is not required for the symbiosis with pea.
We then tested those transductants (Table 2) that showed a relative decrease in rhizosphere growth for competitiveness in nodule infection tests. Each mutant was inoculated at a ratio of 1:1 together with the control strain 3841 onto peas and then bacteria were extracted from nodules to determine the proportions of nodules infected by mutant or WT. Strains RU4372, A1398, A1401, A1404, A1405, A1406 and X385 were significantly reduced for frequency of nodule infection compared with WT (Fig. 3). As a control we also analysed competitive nodule infection with A1402 and as would be predicted from the lack of a difference in rhizosphere competition, it was also unaffected for competitive nodule infection (Fig. 3).

Tests of nodulation competitiveness of mutants with decreased rhizosphere growth. Mutants and the wild-type strain (3841) were mixed 1:1 and inoculated onto pea seedlings grown on sand/vermiculite. Three weeks after inoculation, bacteria were isolated from nodules and the different strains were identified on the basis of their different antibiotic resistances. Mutants showing nodule occupancies significantly lower than expected (chi squared test) are marked with asterisks. Data were obtained using at least three different plants and at least 50 nodules per plant. The black bars show the % of nodules occupied by 3841 (StrR), the white bars show the % of the nodules occupied by each mutant (KanR) and the hatched bars show the % of nodules from which there was growth on both antibiotics
Analysis of the gene expression patterns using data published previously (Ramachandran et al. 2011) revealed that gene RL0031 (mutated in X365 and RU4372) was not specifically induced in the rhizosphere and so in line with our earlier conclusion, the decreased infection competitiveness of RU4372 is most probably due to the reduced growth caused by partial methionine auxotrophy. Similarly, the putative LPS biosynthesis gene RL0811A (mutated in X39 and A1401) appeared not to be induced in the rhizosphere, and so the decreased rhizosphere and nodule infective fitness of this strain may be caused by the reduced growth rate (Table 3). In contrast, RL0634 (X377 and A1399) was induced in both pea and sugarbeet rhizospheres, RL1109 (X378), RL0079 (X385) and RL4123 (X380 and A1402) were all induced in pea rhizospheres, while RL0085 (X383 and A1404) was induced by pea root exudate (Ramachandran et al. 2011). These patterns of gene expression are in concordance with potential rhizosphere fitness reductions caused by these mutations in genes of undefined functions. The analysis of the genes of defined functions is described below.
Characterisation of arabinose and protocatechuate utilisation mutants
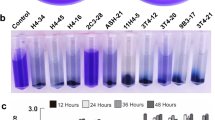
The product of RL3613 (mutated in strain X382 and its transductant A1404) is homologous (84 % identity) to AraE (accession CAC49585) within the araABCDEF operon required for arabinose catabolism and uptake in S. meliloti 1021 (Poysti et al. 2007). A homologous operon is present in R.l. viciae equivalent to RL3617-RL3612 and this operon is induced by arabinose (Ramachandran et al. 2011). The araE mutant A1404 (carrying mini-Tn5 in RL3613) was unable to grow on arabinose (Fig. 4). Arabinose catabolism can occur via different pathways in bacteria: e.g., in E. coli it is catabolised via sugar phosphorylation by being isomerised to ribulose, which is phosphorylated to ribulose−5-phosphate, which is then epimerised into xylulose−5-phosphate. However an alternative pathway occurs in Azospirilum brasilense in which arabinose is catabolised sequentially via D-arabinose-1,4 lactone, D arabinoate and 2-dehydro-3-deoxy-D arabinoate to α-ketoglutaric semialdehyde, the last of these steps being catalysed by an enzyme referred to as ketoglutarate semialdehyde dehydrogenase (KGSADH) (Watanabe et al. 2006). There are three types of KGSADHs: types I, II and III are involved in the catabolism of arabinose, glucarate/galactarate and hydroxyproline respectively (Watanabe et al. 2007). RL3613 (araE) is similar (33 % identity over the full protein) to the A. brasilense Type-I KGDASH (Accession BAE94276) that has been shown (Watanabe et al. 2007) to convert α-ketoglutaric semialdehyde to alpha-ketoglutarate with the concomitant reduction of NADP+. The resulting alpha-ketoglutarate from this reaction then enters the TCA cycle. The lack of growth of the araE mutants X382 and A1404 is presumably due to the loss of this gene product. However, downstream of RL3613 is RL3612 which shows 78 % identity to arabinonate dehydratase (AraC) from A. brasilense, and so it is possible that the mutation could be polar on this gene. The upstream genes RL3617-RL3615; araABD) are predicted to encode components of an arabinose transporter as demonstrated previously in S. meliloti (Poysti et al. 2007).

Mutants A1404 or A1406 are defective for growth on arabinose or protocatechuate respectively. a: Strains 3841 (WT) and A1404 (araE) were inoculated into AMS medium containing 10 mM arabinose. b: Strains 3841(WT) and A1406 (pcaM) were inoculated into AMS medium containing 5 mM p-hydroxybenzoate c: Strains 3841(WT) and A1406 (pcaM) were inoculated into AMS medium containing 3 mM protocatechuate. Strains were precultured to stationary phase on AMS mannitol medium, washed and inoculated to an OD600 of about 0.1 into the growth medium containing defined sole sources of carbon. Growth was measured at 28 °C in microtitre plates was monitored at 600 nm every 40 min. The data shown are logarithmic plots of average OD600 values (± SE) of three cultures derived from different primary inoculants
The product of gene RL3906 (mutated in X384 and its transductant A1406) is 88 % identical to the periplasmic solute binding protein Smb20568 previously identified in S. meliloti strain 1021 (MacLean et al. 2011). The corresponding gene is pcaM within the pcaMNVWX operon involved in the transport of protocatechuate. This operon is regulated by the LysR-type regulator PcaQ whose DNA-binding site is known (MacLean et al. 2008). A PcaQ-like binding site was predicted upstream of RL3906 (MacLean et al. 2011) and the products of the genes downstream of RL3906 (RL3907-RL3910) are all homologous (>75 % identity) to PcaN, PcaV, PcaW and PcaX respectively. Upstream of (and in the same orientation as) RL3906 is RL3905 which is predicted to encode parahydroxybenzoate hydroxylase, that catabolises 4-hydroxybenzoate to protocatechuate. By analogy with S. meliloti, all of these genes are probably regulated by the LysR-type transcriptional regulator PcaQ, which is homologous to RL3904. Protocatechuate induced the expression of RL3906 and the genes RL3095-RL3910 were all induced by 4-hydroxybenzoate (Ramachandran et al. 2011). Furthermore, the gene most strongly induced in WT R.l. viciae (strain 3841) in the pea rhizosphere is RL3016 encoding a predicted protocatechuate oxidoreductase (Ramachandran et al. 2011), indicating that this pathway is important for rhizosphere fitness. Growth of R.l. viciae WT on protocatechuate as sole C source was poor in WT and was significantly reduced in the mutant (A1406) carrying the mutation in pcaM (RL3906) (Fig. 4). We tested growth on p-hydroxybenzoic acid as sole carbon source; unlike S. meliloti strain 1021 we observed growth on this substrate, but we saw no difference between the pcaM (RL3906) mutant A1406 and WT.
Discussion
There are some important issues that came to light in this study aimed at identifying rhizobial genes important for rhizosphere growth. One is that it may be important to determine that the identified mutation caused the observed phenotype; one of the cleanest phenotypes was seen with the primary mutants X377 and X380, but the phenotype was not seen with the transductants A1399 and A1402, indicating that the identified mutations did not cause the rhizosphere phenotype. Using phage-mediated transduction of the mutations into the wild-type has two advantages: (a) it demonstatrates that the transposon caused the phenotype of decreased rhizosphere competitiveness and (b) it reconfirms the phenotype in completely independent tests. Relatively few of the previous signature-tagged-mutagenesis screens have confirmed that the genes identified were caused by the transposon insertions that were characterised. This issue of secondary mutations potentially leading to false conclusions is not a concern in the saturation mutagenic approaches used to analyse mutants decreased for growth in specific environments.
Another issue was that there can be significant variability in the bacterial counts and sometimes these may obscure real differences caused by the mutations. In this regard, we believe that measurements of the nodule infection gave good corroboration, because decreased growth in the rhizosphere would be predicted to reduce the rhizosphere population of mutants, thereby decreasing their probability of infecting nodules. Clearly assays of competitive nodule infection reflect more than rhizosphere colonisation, but it would be surprising if a mutant decreased for rhizosphere growth nodulated at equivalent levels to WT. Therefore, although initially there appeared to be some decrease of rhizosphere colonisation caused by mutation of RL4123 (a predicted sarcosine oxidase), the observation that A1402 carrying the mutation was unaffected for competitive nodule infection suggests that this gene is not critical for rhizosphere growth. This contrasts with e.g., strains A1398 and A1406 where the rhizosphere growth decrease is relatively small, but their competitive nodule infection is clearly compromised. The mutations affecting arabinose and protocatechuate utilisation also caused decreased competitive nodule infection.
Arabinose is a likely substrate that could be used by rhizobia in the rhizosphere; it is a component of the plant cell wall and is present in arabinogalactan proteins secreted by plant root cells. Utilising such arabinose would require additional enzymes that would release it from the polysaccharides in which it is found. When a reporter-GFP construct induced by arabinose, was used to determine if arabinose was present on barley roots, induction of the reporter occurred mainly at the root-seed boundary and primary roots, but not at root tips (Casavant et al. 2002). It seems likely that arabinose released directly or indirectly from pea roots can enable R.l. viciae 3841 to be more competitive in the rhizosphere, leading to a modest increase in nodule competitiveness. In S. meliloti, arabinose transport and catabolism has been analysed and arabinose catabolism mutants nodulated normally on alfalfa; however no significant effect was observed on competitiveness for infection of alfalfa nodules (Poysti et al. 2007).
Whereas arabinose is a good carbon source for rhizobial growth, protocatechuate and 4-hyroxybenzoate are poorer substrates. Protocatechuate is catabolised via the β-ketoadipate pathway in S. meliloti (MacLean et al. 2006) and similar genes are present in two clusters in R.l. viciae 3841: pRL110086 (pcaG); pRL110087 (pcaH1) pRL110088 (pcaC), pRL110089 (pcaD), pRL110090 (pcaQ); and pRL110286 (pcaR), pRL110287 (pcaI), pRL110288 (pcaJ) and pRL110289 (pcaF). Although in S. meliloti the protocatechuate transporter genes pcaMNVWX (Smb20568–Smb20784) are linked to the degradation genes (MacLean et al. 2011), these genes appear not to be linked in R.l. viciae (Mauchline et al. 2006). The mutation in RL3609 affects a predicted protocatechuate solute binding protein gene on the chromosome whereas the catabolism genes are on plasmid pRL11. It has been suggested, based on inhibitor studies that there is one transporter that is specific for dihydroxy-benzoic acids (like protocatechuate) and one for mono-hydroxy benzoic acids, like 4-OH benzoate (Wong et al. 1991) and our observation that the mutation in RL3609 affects growth on protocatechuate but not 4-OH-benzoate is consistent with that conclusion. Protocatechuate at concentrations above 5 mM can inhibit growth and so two roles for protocatechuate degradation can be envisaged; one for catabolism of low amounts enabling growth of R.l. viciae on protocatechuate and another for degradation of toxic metabolites such as proptocatechuate (or related compounds). Our data indicate that protocatechuate or related metabolites may be important for rhizosphere fitness. One possible source of this substrate is from lignin degradation which can produce metabolites like vanillate and syringate which are further catabolized through the protocatechuate catabolism pathway (Kamimura et al. 2010).
Conclusions
Screening for mutants of R.l. viciae with reduced growth or survival in the pea rhizosphere showed that the ability to catabolise arabinose and protocatechuate is important for rhizosphere fitness. The reduced rhizosphere fitness of these mutants was correlated with decreased nodule infection competitiveness. Other genes of undefined function were also identified as reducing rhizosphere fitness and again this was correlated with decreased competitiveness for nodule infection. .
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Barquist L, Boinett CJ, Cain AK (2013) Approaches to querying bacterial genomes with transposon-insertion sequencing. RNA Biol 10:1161–1169
Barr M, East AK, Leonard M, Mauchline TH, Poole PS (2008) In vivo expression technology (IVET) selection of genes of Rhizobium leguminosarum biovar viciae A34 expressed in the rhizosphere. FEMS Microbiol Lett 282:219–227
Beringer JE (1974) R factor transfer in Rhizobium leguminosarum. J Gen Microbiol 84:188–198
Bourion V, Laguerre G, Depret G, Voisin AS, Salon C, Duc G (2007) Genetic variability in nodulation and root growth affects nitrogen fixation and accumulation in pea. Ann Bot 100:589–598
BuchananWollaston V (1979) Generalized transduction in Rhizobium leguminosarum. J Gen Microbiol 112:135–142
Casavant NC, Beattie GA, Phillips GJ, Halverson LJ (2002) Site-specific recombination-based genetic system for reporting transient or low-level gene expression. Environ Microbiol 68:3588–3596
Chen WP, Kuo TT (1993) A simple and rapid method for the preparation of Gram-negative bacterial genomic DNA. Nucleic Acids Res 21:2260
Das S, Noe JC, Paik S, Kitten T (2005) An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J Microbiol Methods 63:89–94
DeLorenzo V, Fernandez S, Herrero M, Jakubzik U, Timmis KN (1993) Engineering of alkyl-responsive and haloaromatic-responsive gene-expression with mini-transposons containing regulated promoters of biodegradative pathways of Pseudomonas. Gene 130:41–46
Downie JA (2010) The roles of extracellular proteins, polysaccharides and signals in the interactions of rhizobia with legume roots. FEMS Microbiol Rev 34:150–170
Fahraeus G (1957) The infection of clover root hairs by nodule bacteria studied by a simple glass technique. J Gen Microbiol 16:374–381
Gao MS, Teplitski M (2008) RIVET - A tool for in vivo analysis of symbiotically relevant gene expression in Sinorhizobium meliloti. Mol Plant Microbe Interact 21:162–170
Johnston AWB, Beringer JE (1975) Identification of the Rhizobium strains in pea root nodules using genetic markers. J Gen Microbiol 87:343–350
Kamimura N, Takamura K, Hara H, Kasai D, Natsume R, Senda T, Katayama Y, Fukuda M, Masai E (2010) Regulatory system of the protocatechuate 4,5-cleavage pathway genes essential for lignin downstream catabolism. J Bacteriol 192:3394–3405
MacLean AA, MacPherson G, Aneja P, Finan TM (2006) Characterization of the beta-ketoadipate pathway in Sinorhizobium meliloti. Appl Environ Microbiol 72:5403–5413
MacLean AM, Anstey MI, Finan TM (2008) Binding site determinants for the LysR-type transcriptional regulator PcaQ in the legume endosymbiont Sinorhizobium meliloti. J Bacteriol 190:1237–1246
MacLean AM, Haerty W, Golding GB, Finan TM (2011) The LysR-type PcaQ protein regulates expression of a protocatechuate-inducible ABC-type transport system in Sinorhizobium meliloti. Microbiology 157:2522–2533
Mark GL, Dow JM, Kiely PD, Higgins H, Haynes J, Baysse C, Abbas A, Foley T, Franks A, Morrissey J, O’Gara F (2005) Transcriptome profiling of bacterial responses to root exudates identifies genes involved in microbe-plant interactions. Proc Natl Acad Sci U S A 102:17454–17459
Mauchline TH, Fowler JE, East AK, Sartor AL, Zaheer R, Hosie AHF, Poole PS, Finan TM (2006) Mapping the Sinorhizobium meliloti 1021 solute-binding protein-dependent transportome. Proc Natl Acad Sci U S A 103:17933–17938
Pobigaylo N, Wetter D, Szymczak S, Schiller U, Kurtz S, Meyer F, Nattkemper TW, Becker A (2006) Construction of a large signature-tagged mini-Tn5 transposon library and its application to mutagenesis of Sinorhizobium meliloti. Appl Environ Microbiol 72:4329–4337
Pobigaylo N, Szymczak S, Nattkemper TW, Becker A (2008) Identification of genes relevant to symbiosis and competitiveness in Sinorhizobium meliloti using signature-tagged mutants. Mol Plant Microbe Interact 21:219–231
Poole PS, Schofield NA, Reid CJ, Drew EM, Walshaw DL (1994) Identification of chromosomal genes located downstream of dctD that affect the requirement for calcium and the lipopolysaccharide layer of Rhizobium leguminosarum. Microbiology 140:2797–2809
Poysti NJ, Loewen ED, Wang Z, Oresnik IJ (2007) Sinorhizobium meliloti pSymB carries genes necessary for arabinose transport and catabolism. Microbiology 153:727–736
Ramachandran VK, East AK, Karunakaran R, Downie JA, Poole PS (2011) Adaptation of Rhizobium leguminosarum to pea, alfalfa and sugar beet rhizospheres investigated by comparative transcriptomics. Genome Biol 12:R106. doi:10.1186/gb-2011-12-10-r106
Redondo-Nieto M, Barret M, Morrissey J, Germaine K, Martinez-Granero F, Barahona E, Navazo A, Sanchez-Contreras M, Moynihan JA, Muriel C, Dowling D, O’Gara F, Martin M, Rivilla R (2013) Genome sequence reveals that Pseudomonas fluorescens F113 possesses a large and diverse array of systems for rhizosphere function and host interaction. BMC Genomics 14:54. doi:10.1186/1471-2164-14-54
Reeve WG, Brau L, Castelli J, Garau G, Sohlenkamp C, Geiger O, Dilworth MJ, Glenn AR, Howieson JG, Tiwaril RP (2006) The Sinorhizobium medicae WSM419 lpiA gene is transcriptionally activated by FsrR and required to enhance survival in lethal acid conditions. Microbiology 152:3049–3059
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor, New York
Silby MW, Cerdeno-Tarraga AM, Vernikos GS et al (2009) Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol 10:R51. doi:10.1186/gb-2009-10-5-r51
Tett AJ, Karunakaran R, Poole PS (2014) Characterisation of SalRAB a salicylic-acid-inducible positively-regulated efflux System of Rhizobium leguminosarum bv. viciae 3841. PLoS One 9:e103647. doi:10.1371/journal.pone.0103647
Varivarn K, Champa LA, Silby MW, Robleto EA (2013) Colonization strategies of Pseudomonas fluorescens Pf0-1: activation of soil-specific genes important for diverse and specific environments. BMC Microbiol 13:92. doi:10.1186/1471-2180-13-92
Watanabe S, Kodaki T, Makino K (2006) Cloning, expression, and characterization of bacterial L-arabinose 1-dehydrogenase involved in an alternative pathway of L-arabinose metabolism. J Biol Chem 281:2612–2623
Watanabe S, Yamada M, Ohtsu I, Makino K (2007) alpha-ketoglutaric semialdehyde dehydrogenase isozymes involved in metabolic pathways of D-glucarate, D-galactarate, and hydroxy-L-proline. Molecular and metabolic convergent evolution. J Biol Chem 282:6685–6695
Williams A, Wilkinson A, Krehenbrink M, Russo DM, Zorreguieta A, Downie JA (2008) Glucomannan-mediated attachment of Rhizobium leguminosarum to pea root hairs is required for competitive nodule infection. J Bacteriol 190:4706–4715
Wong CM, Dilworth MJ, Glenn AR (1991) Evidence for two uptake systems in Rhizobium leguminosarum for hydroxy-aromatic compounds metabolized by the 3-oxoadipate pathway. Arch Microbiol 156:385–391
Young JP, Crossman LC, Johnston AWB, Thomson NR, Ghazoui ZF, Hull KH, Wexler M, Curson ARJ, Todd JD, Poole PS, Mauchline TH, East AK, Quail MA, Churcher C, Arrowsmith C, Cherevach I, Chillingworth T, Clarke K, Cronin A, Davis P, Fraser A, Hance Z, Hauser H, Jagels K, Moule S, Mungall K, Norbertczak H, Rabbinowitsch E, Sanders M, Simmonds M, Whitehead S, Parkhill J (2006) The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol 7:R34. doi:10.1186/gb-2006-7-4-r34
Acknowledgments
We thank Anke Becker for providing the library of plasmids with signature-tagged transposons and Antony Jones for making the arrays. Marij Frederix, Alison East and Bouchra Mousine, gave advice, technical help and constructive discussion. The work was supported by a Postdoctoral Contract (to P G-F) financed by the Education Ministry of the Spanish Government, by a BBSRC studentship to JS and by the BBSRC with a grant to PSP (BB/C517025/2), a grant in aid (to PSP and JAD) and JAD was also supported in part by the John Innes Foundation.
Conflict of interest
The authors declare that there is not conflict of interest
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Katharina Pawlowski.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Garcia-Fraile, P., Seaman, J.C., Karunakaran, R. et al. Arabinose and protocatechuate catabolism genes are important for growth of Rhizobium leguminosarum biovar viciae in the pea rhizosphere. Plant Soil 390, 251–264 (2015). https://doi.org/10.1007/s11104-015-2389-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-015-2389-5