
Abstract
The maize Activator/Dissociation (Ac/Ds) transposable element system was introduced into sugar beet. The autonomous Ac and non-autonomous Ds element excise from the T-DNA vector and integrate at novel positions in the sugar beet genome. Ac and Ds excisions generate footprints in the donor T-DNA that support the hairpin model for transposon excision. Two complete integration events into genomic sugar beet DNA were obtained by IPCR. Integration of Ac leads to an eight bp duplication, while integration of Ds in a homologue of a sugar beet flowering locus gene did not induce a duplication. The molecular structure of the target site indicates Ds integration into a double strand break. Analyses of transposase transcription using RT–PCR revealed low amounts of alternatively spliced mRNAs. The fourth intron of the transposase was found to be partially misspliced. Four different splice products were identified. In addition, the second and third exon were found to harbour two and three novel introns, respectively. These utilize each the same splice donor but several alternative splice acceptor sites. Using the SplicePredictor online tool, one of the two introns within exon two is predicted to be efficiently spliced in maize. Most interestingly, splicing of this intron together with the four major introns of Ac would generate a transposase that lacks the DNA binding domain and two of its three nuclear localization signals, but still harbours the dimerization domain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Members of the Ac/Ds transposable element system of maize are class II elements that transpose via a cut and paste mechanism (Kunze and Weil 2002). Ac/Ds belong to the family of hAT transposable elements named after their founding members hobo from Drosophila melanogaster, Ac from Zea mays, and Tam3 from Antirrhinum majus (Calvi et al. 1991; Hehl et al. 1991). The Ac/Ds transposable element system has been stably introduced into numerous heterologous host plants such as tobacco, Arabidopsis thaliana, carrot, tomato, potato, rice, and barley (Baker et al. 1986; Knapp et al. 1988; Murai et al. 1991; Scholz et al. 2001; Van Sluys et al. 1987; Yoder et al. 1988). Before the elements can be used as molecular tools in novel host plants, it is important to learn if they are transpositionally active and approach transposition frequencies useful for their applications. For example, in tobacco the elements transpose over at least five generations, transactivate Ds transposition, show a positive effect of Ac copy numbers on Ds transactivation frequencies, and induce an eight bp duplication upon integration (Hehl and Baker 1989, 1990). The high transposition activity of Ac in tobacco may have for instance facilitated the isolation of the TMV resistance gene N by transposon tagging (Dinesh-Kumar et al. 1995; Whitham et al. 1994). Transposon tagging with the Ac/Ds system has since become applicable in a large number of transformable heterologous host plants either with the wild type elements or with modified elements necessary to boost transposition frequencies (Hehl 1994; Osborne and Baker 1995).
Examples for host plants with low transposition frequencies of the wild type Ac/Ds system are Lactuca sativa, Brassica napus, and, initially, A. thaliana (Babwah and Waddell 2002; Schmidt and Willmitzer 1989; Yang et al. 1993). Transposition frequencies in A. thaliana were improved by employing different transposase promoters and by shortening the untranslated leader of the transposase gene (Honma et al. 1993; Lawson et al. 1994; Osborne et al. 1995; Scofield et al. 1992; Swinburne et al. 1992).
Despite the large number of heterologous host plants, there are still crop plants in which the introduction of Ac and Ds may be desirable. One of these host plants is sugar beet (Beta vulgaris). Sugar beet is an important crop plant for sugar production (Elliott and Weston 1993). Currently, breeding programs aim to reduce impurities such as potassium salts, amino acids, and betain that negatively affect sugar extraction (Bosemark 1993). Recent biotechnological improvements using transformation technologies for sugar beet include salt tolerance and the delay of bolting time (Liu et al. 2008; Mutasa-Gottgens et al. 2009). Furthermore, tissue specific and storage induced genes and their promoters were identified (Kloos et al. 2002; Oltmanns et al. 2006; Rotthues et al. 2008; Stahl et al. 2004). These may be applicable for the spatial and temporal expression of genes to achieve desired biotechnological improvements.
For the improvement of traits and for the study of crop plant biology, transposable elements are useful tools. Such approaches comprise, for example, transposon tagging, activation tagging, or the generation of marker gene free transgenic plants (Aarts et al. 1993; Wilson et al. 1996; Yoder and Goldsborough 1994). In recent years several sugar beet inherent transposable elements such as Vulmar1, En/Spm-like elements, MITEs, and retrotransposon-like elements have been identified (Jacobs et al. 2004; Kuykendall et al. 2008; Menzel et al. 2006). Transposon tagging with transposable elements inherent to the host species often poses a problem because of their high copy numbers and the associated difficulty to identify the element at the gene of interest. Therefore it may be useful to have a well defined and low copy number element system in sugar beet.
To make sugar beet accessible for biotechnological improvements involving transposable elements, the maize Ac/Ds system was introduced into transgenic hairy roots and fertile transgenic plants. Transgenic tissue and plants were subjected to a variety of molecular investigations to gain insight into transposition and transposase transcription in this novel heterologous host.
Materials and methods
Plasmid constructs
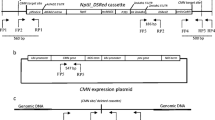
The plasmid pVKHbar::DspOp is based on the binary T-DNA vector pVKH-35S-pA (Reintanz 1997). The Ds element has been described earlier (Wirtz et al. 1997). Ds consists of 254 bp and 320 bp of the Ac 5′ and 3′ termini and exists as a SalI fragment in plasmid pUC19. From this plasmid Ds was recovered by SalI digest, the ends were filled in, and the fragment was integrated into a HindIII digested and filled in plasmid pUC19. This results in the abolishment of the SalI sites but the HindIII sites on either side of Ds were retained. Ds harbours a unique MscI site in which a filled in SacI/HindIII lac operator element harbouring two modified lac operator elements and a 35S minimal promoter (pOp) was inserted (Moore et al. 1998). Both orientations of the operator were obtained. Both DspOp fragments were then recovered as HindIII fragments, filled in and ligated into the BamHI digested and filled in plasmid pCBN. pCBN harbours a cauliflower mosaic virus 35S promoter, the coding region of the phosphinothricin or bar resistance gene and the 3′ polyA addition signal from the nopaline synthase gene. BamHI cuts between the 35S promoter and the coding region of the bar gene and HindIII can be used to release the complete selectable reporter gene. The resulting plasmids were designated pbar::DspOp1 and pbar::DspOp2. Both plasmids harbour Ds in the same orientation but are different with respect to the orientation of the pOp element. Both bar::DspOp constructs were then cloned into the binary T-DNA vector pVKH-35S-pA (Reintanz 1997). To remove the 35S promoter, pVKH-35S-pA was digested with SstI/BamHI, the ends were filled in and the vector was religated. The resulting plasmid was digested with HindIII and ligated to the HindIII fragments harbouring bar::DspOp1 and bar::DspOp2, respectively. The resulting plasmids are pVKHbar::DspOp1 and pVKHbar::DspOp2. Figure 1 shows a schematic drawing of the bar::DspOp insert in pVKHbar::DspOp.

Schematic representations of Ac and Ds constructs introduced on T-DNA vectors into sugar beet. The positions of all primers used are indicated. Primer numbers correspond to primers in Table 1
For transactivation of Ds, a T-DNA construct expressing the Ac transposase gene under the control of a double 35S promoter was employed (Osborne et al. 1995). The double 35S promoter controls the Ac gene on the Ac-18 element that is incapable to transpose because of a 3′ terminal deletion (Hehl and Baker 1989). Figure 1 shows a schematic drawing of this stable Ac construct (Ac st)
pOCA28 bar::Ac9 is a binary T-DNA vector that harbours a wild type Ac element in the selectable bar marker gene. Ac9 was cloned from the wx-m9 locus and has been modified to contain adjacent SalI restriction sites for further cloning of the element (Fedoroff et al. 1983; Laufs et al. 1990; Pohlman et al. 1984). As a T-DNA vector pOCA28 was used (Honma et al. 1993; Olszewski et al. 1988). First, the unique BamHI site of pOCA28 was removed by digesting the vector with BamHI, a fill in reaction and subsequent ligation. This plasmid was then digested with HindIII and the HindIII insert of pCBN (see above) was inserted. The resulting vector is designated pOCA28bar. The Ac9 element on a SalI fragment was inserted into the BamHI site of pOCA28bar after a partial fill in reaction with C and T (Ac9, SalI) or A and G (pOCA28bar, BamHI), respectively. Figure 1 shows a schematic drawing of the bar::Ac9 insert in pOCA28bar::Ac9. For all recombinant DNA techniques standard protocols were employed (Sambrook et al. 1989).
Plant transformation
For hairy root transformation, two Agrobacterium tumefaciens C58C1 strains (Deblaere et al. 1985) harbouring a binary T-DNA vector with a stabilized Ac transposase construct (Osborne et al. 1995), and the construct pVKHbar::DspOp were used together with Agrobacterium rhizogenes strain Ri15834 (Lippincott and Lippincott 1969). Sugar beet seedlings of the genotype 2B0035 (PLANTA GmbH) were grown under sterile conditions on MS medium (4.9 g/l Murashige Skoog from Duchefa Biochemie B. V., Haarlem, The Netherlands) for 3–4 weeks until they have reached a height of 5 cm. From a 5 ml over night culture of the three Agrobacterium lines under selective conditions, 50 μl were transferred each to 50 ml LB-medium without antibiotics and incubated for further 21 h. The cultures were diluted with LB-medium to yield an optical density of OD600nm = 0.4–0.6. 4 ml from each culture were mixed in a petri dish. Petioles (0.5–1 cm) of sugar beet seedlings were dipped for several seconds in this bacterial solution, dried on filter paper and transferred to MS-BAP plates (4.9 g/l Murashige Skoog, complemented with 10 g/l sucrose, 0.25 mg/l N6-Benzyladenine, pH 5.9–6.0). The plates were incubated at 24°C for 2 days (16 h light, 8 h dark). After this, the petioles were transferred to MS-BAP/C/T/H plates (MS-BAP complemented with 350 mg/l carbenicillin, 100 mg/l timentin, 20 mg/l hygromycin, pH 5.9–6.0). Hygromycin selects for T-DNA transfer of the Ac and Ds containing constructs while carbenicillin and timentin select against Agrobacterium. The plates were incubated at 24°C and petioles were transferred every 2–3 days onto fresh MS-BAP/C/T/H plates. After about 2 weeks hairy roots were visible at the sites where the petioles have been cut. In a subsequent second selection phase the hairy roots were cut from the petioles and transferred to plates containing ½ B5/C/T/H medium (half strength Gamborg B5 medium including vitamins from Duchefa, complemented with 350 mg/l carbenicillin, 100 mg/l timentin, 50 mg/l hygromycin). After about 4 weeks the roots were cut again and transferred to fresh plates with the same medium. After this time the selection for Ds excision was initiated by cutting the roots again and transferring them to fresh ½ B5/C/T/H plates containing 3 mg/l phosphinothricin. After 4 weeks the roots were transferred again to fresh ½ B5/C/T/H plates containing 6 mg/l phosphinothricin. All selections and root regenerations were done at 24°C in the dark. Phosphinothricin resistant roots from independent transformations were subjected to molecular analysis using PCR and IPCR (see below).
Transgenic sugar beet harbouring the wild type Ac9 element were generated at the PLANTA GmbH using pOCA28bar::Ac9. Seed of two transgenic lines (001R, 002R) that were backcrossed with non-transgenic sugar beet were kindly provided by the PLANTA GmbH for analysis. Seed of these backcrosses were germinated in soil and kept in a greenhouse with a 12 h day and 12 h night period. These plants were subjected to PCR, IPCR, and RT–PCR analysis.
Transgenic tobacco harbouring a stabilized Ac transposase construct (Osborne et al. 1995) were generated according to standard protocols (Hehl and Baker 1989). One transgenic line, designated A3 was subjected to RT–PCR analysis of Ac transcription and splicing.
PCR and IPCR analyses of Ds transposition in transgenic hairy roots
Excision of Ds in phosphinothricin resistant transgenic hairy roots was analysed by PCR. Genomic DNA was prepared according to the following protocol. A 4–5 cm long root piece was used for genomic DNA extraction with the ‘REDEXtract-N-AmpTM Plant PCR Kit’ from Sigma–Aldrich GmbH (Taufkirchen, Germany) according to the protocol from the manufacturer. The sequences of all primers used in the PCR and IPCR analyses are shown in Table 1. The positions of the primers are shown schematically in Figs. 1 and 2. For the amplification of the empty donor site of Ds, the primers number 1 and 2 were employed. PCR amplifications were performed in a final volume of 20 μl harbouring 2 μl template DNA (1–100 ng), 11.8 μl dH2O, 2 μl 10xExtension-buffer (ExTaqTMBuffer, Appligene Oncor, Illkirch, France), 2 μl dNTP-mix (2.5 mM each), 1 μl of each primer (20 μM), and 0.2 μl Taq polymerase (5 u/μl, Qbiogene—MP Biomedicals, Illkirch, France). The PCR program used was: 3 min 94°C, 35× (1 min 94°C, 1 min 55°C, 2 min 72°C), 10 min 72°C, subsequently 4°C. Results of PCR analyses were always analysed gel electrophoretically.

Schematic representaion of Ac and Ds integration events in sugar beet. Three IPCR products, 35–43(Ds), 36–43(Ds), and 001R-20(Ac) were obtained with primer pairs number 5/6 and 11/12 (arrows). Genomic integration sites (WT) were amplified with primer pairs number 7/8, 13/14, and 15/16. The hatched boxes represent Ac or Ds terminal sequences and the white boxes genomic sugar beet DNA. Primer numbers correspond to primers in Table 1
For IPCR analysis genomic DNA was isolated with the ‘DNeasy Plant Mini Kit’ (Qiagen, Hilden, Germany). Approximately 100 mg of plant material was employed for extraction. Approximately 400–800 ng genomic DNA was digested with HincII. After purification of the genomic DNA fragments using the ‘QIAquick Nucleotide Removal Kit’ (Qiagen) fragments were ligated under conditions favouring circularization of the molecules. After inactivation of the T4 DNA ligase two PCR reactions were performed. For the first amplification primers number 3 and 4 were employed (Table 1; Fig. 1). PCR amplifications were performed in a final volume of 25 μl harbouring 5 μl template DNA (1–100 ng), 15.3 μl dH2O, 2.5 μl 10xPCR-buffer (100 mM Tris–HCl pH 8.3, 500 mM KCl, 2 mM dNTP, 15 mM MgCl2), 1 μl of each primer (20 μM), and 0.2 μl Taq polymerase (5 u/μl, Qbiogene). The PCR program used was: 4 min 95°C, 30× (30 s 95°C, 30 s 60°C, 2 min 72°C), 10 min 72°C, subsequently 4°C. A second PCR amplification followed using 1 μl of the first amplification, and nested primers number 5 and 6 (Table 1; Figs. 1, 2). The PCR conditions were the same as those for the first amplification.
The fragments amplified in the PCR and IPCR analyses were cloned into pCR®2.1 using the ‘TA Cloning®-Kit’ (Invitrogen GmbH, Karlsruhe, Germany). Sequence analysis of the inserts was performed with M13 universal and reverse primers using the ‘ALFexpressTMAutoreadTMSequencing Kit’ and an A.L.F-Sequencer (Amersham Biosciences, Freiburg, Germany).
For amplification of a genomic insertion site of Ds, primers number 7 and 8 were used (Table 1; Fig. 2). Sugar beet DNA from a non-transgenic plant was employed for amplification. PCR amplifications were performed in a final volume of 25 μl harbouring 1 μl template DNA (1–100 ng), 17.35 μl dH2O, 2.5 μl 10xPCR-buffer (Peqlab, Erlangen, Germany), 0.5 μl dNTP-mix (10 mM), 1.5 μl MgSO4 (25 mM), 1 μl of each primer (20 μM), and 0.15 μl Pwo DNA polymerase (5 u/μl, Peqlab). The PCR program used was: 4 min 94°C, 35× (30 s 94°C, 30 s 55°C, 90 s 70°C), 5 min 70°C, subsequently 4°C. The amplified fragment was cloned into pCR®2.1 using the ‘TA Cloning®-Kit’ (Invitrogen). Sequence analysis of the insert was performed with M13 reverse primer using the ‘BigDye®Terminator v1.1 Cycle Sequencing Kit’ and an ‘ABI Prism Genetic Analyzer’ (Applied Biosystems, Weiterstadt, Germany).
The sequences were further analysed using software programs Chromas 2.31 (Technelysium Pty Ltd., Helensvale, Australia), DNA-StriderTM (Douglas 1995), BioEdit Sequence Alignment Editor, Version 5.0.9 (Hall 1999), and BLAST (Altschul et al. 1997).
PCR and IPCR analyses of Ac transposition in transgenic sugar beet plants
Transposition of Ac9 in sugar beet was analysed by PCR and IPCR. Genomic DNA from transgenic sugar beet plants (kindly provided by the PLANTA GmbH) transformed with pOCA28bar::Ac9 was prepared according to a previously published method (Hehl and Baker 1990). For some analyses genomic DNA was also isolated according to the following protocol. Approximately 100 mg of leaf material was homogenized in liquid nitrogen in a micro test tube. Subsequently the homogenate was vigorously resuspended in 500 μl CTAB DNA extraction buffer (0.1 M Tris–HCl, 20 mM EDTA, 2% w/v CTAB, 1.4 mM NaCl, 1% w/v PVP-10, pH 8.0) supplemented with 5 μl 2-mercaptoethanol and 4 μl RNase (5 mg/ml). The suspension was incubated for 30 min at 65°C and precipitated by centrifugation (12,000g). The supernatant was collected and extracted with an equal volume chloroform/isoamyl alcohol (24:1 v/v). After centrifugation (11,000g) the aqueous phase was collected and supplemented with 1/10 volume 3 M sodium acetate (pH 5.2). Genomic DNA was precipitated by adding an equal volume cold isopropanol. After incubation for 60 min at 4°C the DNA was sedimented by centrifugation (15,000g) for 15 min. The pellet was washed with 70% ethanol and after removal of the ethanol and air drying of the pellet, the DNA was dissolved in 50 μl LTE (10 mM Tris–HCl, 0.1 mM EDTA, pH 7.5).
For the amplification of the empty donor site of Ac, the primers number 1 and 2 were employed (Table 1; Fig. 1). PCR amplifications were performed in a final volume of 25 μl harbouring 2 μl template DNA (100 ng), 17.1 μl dH2O, 2.5 μl 10xPCR-buffer, 0.75 μl MgCl2 (25 mM), 0.5 μl dNTP-mix (10 mM), 1 μl of each primer (20 μM), and 0.15 μl Taq polymerase (5 u/μl ‚omni-taq-OLS®’, OMNI Life Science, Eidelstedt, Germany). The PCR program for amplification was: 4 min 94°C, 35× (30 s 94°C, 30 s 55°C, 90 s 70°C), 5 min 70°C, subsequently 4°C.
Inverse PCR (IPCR) was used for the isolation of an integrated Ac element. Approximately 200 ng genomic DNA was digested with NcoI. After heat inactivation of the enzyme, NcoI fragments were ligated under conditions favouring circularization of the molecules. After inactivation of the T4 DNA ligase two PCR reactions were performed. For the first amplification primers number 9 and 10 were employed (Table 1; Fig. 1). PCR amplifications were performed in a final volume of 25 μl harbouring 5 μl template DNA (1–100 ng), 13.55 μl dH2O, 2.5 μl 10xPCR-buffer, 0.75 μl MgCl2 (50 mM), 1 μl dNTP-mix (10 mM), 1 μl of each primer (20 μM), and 0.2 μl Taq polymerase (5 u/μl ‘omni-taq-OLS®’, OMNI Life Science). The PCR program used was: 3 min 95°C, 33x (30 s 95°C, 30 s 56°C, 3 min 70°C), 5 min 70°C, subsequently 4°C. A second PCR amplification followed using 2.5 μl of a 1:100 dilution of the first amplification, and nested primers number 11 and 12 (Table 1; Figs. 1, 2). The PCR conditions were the same as those for the first amplification.
For amplification of a genomic insertion site of Ac, primer pairs 13/14, and 15/16 (nested primers for reamplification) were used (Table 1; Fig. 2). PCR amplifications were performed as described for the IPCR above.
Cloning, sequencing, and analysis of PCR fragments were carried out as described in the previous chapter.
RT–PCR analysis of Ac transcripts
For RNA extraction up to 100 mg of leaf material was homogenized in liquid nitrogen in a micro test tube. All further steps were performed according to the manual of the ‘NucleoSpin® RNA Plant’ kit (Macherey & Nagel GmbH & Co KG, Düren, Germany). The final volume of the RNA solution after preparation was 60 μl. A test gel electrophoresis confirmed the integrity of the RNA. For reverse transcription 11 μl of the RNA solution was used. First strand cDNA was synthesized with the ‘RevertAid H Minus First Strand cDNA Synthesis Kit’ from Fermentas (St. Leon-Rot, Germany) according to the manufacturers protocol. For PCR amplification primers 17 through 26 were employed (Table 1). L and R designate corresponding primer pairs for each amplification (Table 2). PCR amplifications were performed in a final volume of 25 μl harbouring 2 μl template cDNA, 16.75 μl dH2O, 2.5 μl 10xPCR-buffer, 1.5 μl MgCl2 (25 mM), 0.5 μl dNTP-mix (10 mM), 0.8 μl of each primer (20 μM), and 0.15 μl Taq polymerase (5 u/μl, New England BioLabs, Frankfurt a. M., Germany). The PCR program for amplification was: 3 min 94°C, 31× (30 s 94°C, 30 s 57°C, 90 s 72°C), 5 min 72°C, subsequently 4°C. PCR fragments were analysed gel electrophoretically. Cloning, sequencing, and analysis of PCR fragments were carried out as described above.
Bioinformatic analysis of splice sites
To investigate the efficiency with which the identified splice donor and splice acceptor sites would be spliced in a dicot (Arabidopsis thaliana) and in a monocot plant (Zea mays) SplicePredictor at http://deepc2.psi.iastate.edu/cgi-bin/sp.cgi was employed (Brendel and Kleffe 1998). To investigate splice sites, the sequence of the Ac element (Acc. X05424) was submitted online (SplicePredictor Version of July 4, 2007). Among the parameters selected, Arabidopsis and maize were chosen. Furthermore, the fields “*-value” and “Display highest scoring sites only” were changed to “all sites”. To receive values for all sites “Cutoff” was set to −99.9. The results are summarized in Table 3 for all splice sites identified experimentally in sugar beet.
Results
Ac trans-activation of Ds in transgenic sugar beet hairy root lines
Excision of Ds from the T-DNA vector in the presence of a stabilized Ac transposase was investigated in transgenic hairy roots. Transgenic hairy root lines were enriched for Ds excision by phosphinothricin selection. A total of ten independently generated hairy root lines were analysed molecularly. PCR analysis of Ds donor sites revealed a fragment of around 240 bp that is expected when Ds is excised from the T-DNA in seven of these lines (data not shown). Fragments from four lines were cloned and sequenced. Figure 3a shows the sequence at the Ds donor site obtained from each of these four fragments. For comparison, the sequence directly adjacent to the Ds element in the T-DNA vector is also shown (WTDs). The sequences reveal footprints that either show a one bp deletion (36–48, 35–43, 35–66), or a one bp deletion together with a one bp exchange (35–19).

Transposition of Ds in transgenic hairy roots of sugar beet. a Footprints after Ds excision. The sequence adjacent to the Ds element in the T-DNA vector (WTDs) is compared to four sequences obtained from four independently transformed hairy root lines (36–48, 35–19, 35–43, 35–66). Deleted nucleotides are indicated with an underlined space and the additional nucleotide shown as an exchange from A to T is indicated with a bold letter. The position of Ds in the WTDs transgenic line is indicated with a triangle. b Insertion of Ds causes a 2 bp deletion. The sequence adjacent to an integrated Ds element (triangle) from hairy root line 35–43 is compared with the same sequence amplified from untransformed sugar beet (WT). Two base pairs at the target site of Ds are deleted (underlined)
To isolate integration events of Ds, transgenic hairy root lines in which the excision of Ds has been confirmed by PCR were subjected to IPCR analysis. Two IPCR products with Ds adjacent DNA of 280 and 299 base pairs were obtained. In none of these PCR products an eight base pair duplication was found adjacent to Ds (data not shown). Upon BLAST sequence comparison with the GenBank database, only one of the Ds adjacent genomic DNAs showed homology with a sugar beet gene. 217 bp of the 299 bp adjacent to Ds cloned from hairy root line 35–43 show 86% sequence identity to the FLC-like1 gene of Beta vulgaris (Reeves et al. 2007). The Ds adjacent genomic sequences were further compared by a BLAST analysis with unpublished sugar beet genomic DNA sequences, confirming the integration of Ds into sugar beet genomic DNA (Juliane Dohm and Heinz Himmelbauer, personal communication). Interestingly, the 280 bp sequence is colinear with genomic DNA, indicating the identification of two adjacent Ds integration events. Figure 2 shows the amplified and closely adjacent Ds elements schematically.
None of the putative Ds integration events showed an eight bp target site duplication adjacent to Ds. In the case of the two adjacent Ds elements, this is explained by the lack of a complete integration event (Fig. 2, 36–43). To investigate the second case (Fig. 2, 35–43). the target site of the Ds element was amplified from non-transgenic sugar beet. A DNA fragment could be amplified, cloned, and sequenced (Fig. 2). Figure 3b shows a sequence comparison of both sugar beet alleles in the presence (35–43) and absence (WT) of Ds. This analysis confirmed the lack of an eight base pair duplication upon Ds integration and indicates that the nucleotides directly adjacent to Ds at the integration site were deleted.
Autonomous transposition of Ac in transgenic sugar beet plants
Transgenic sugar beet plants were generated by Agrobacterium tumefaciens T-DNA transformation using pOCA28 bar::Ac9. Primary transformants were backcrossed to non transgenic sugar beet. Seed of the offspring of two transgenic lines (001R and 002R) was germinated on soil under non-selective conditions and subjected to PCR analyses for the presence of Ac and the empty donor site. In two out of eleven (001R) and in seven out of twentyseven (002R) offspring plants the empty donor site were detected (data not shown). The PCR fragment for the empty donor site was cloned and sequenced from a total of six different plants from both backcrosses. Figure 4a shows the sequence at the empty donor sites compared to the sequence when Ac is still present in the T-DNA locus (WTAc). The sequences reveal footprints that show a one bp deletion together with a one bp exchange. Two different types of footprints were detected representing at least three independent molecular events. Footprints in offspring of line 001R and 002R have occured independently because they are from two different transgenic progenitor plants.

Transposition of Ac in transgenic sugar beet plants. a Footprints after Ac excision. The sequence adjacent to the Ac element in the T-DNA vector (WTAc) is compared to six sequences obtained from offspring of two Ac transgenic plants (001 and 002). Deleted nucleotides are indicated with an underlined space and nucleotide exchanges from T to A and G to C are indicated with a bold letter. The position of Ac in the WTAc transgenic line is indicated with a triangle. b Insertion of Ac causes an eight bp duplication. The sequence adjacent to an integrated Ac element (triangle) from sugar beet line 001R-20 is compared with the homologous sequence amplified from sugar beet (WT). The eight bp duplication adjacent to Ac is indicated in bold. This sequence is present only once in the sequence lacking Ac (bold and underlined)
To isolate transposed Ac elements, IPCR was employed. Several IPCR products were cloned and sequenced from plants in which the empty donor site and Ac were detected by PCR. Most of them harboured only a few nucleotides between the ends of Ac or were end to end joinings of the Ac termini. Only one IPCR product harboured Ac adjacent DNA that could be analysed further (Fig. 2, 001R-20). The IPCR product harboured 281 bp and 1169 bp on either side of the integration site. An eight bp sequence duplication was found directly adjacent to the integrated Ac element (Fig. 4b). Using the Ac adjacent genomic sequence in a BLAST analysis, a contiguous stretch of 296 bp within the 1169 bp long adjacent DNA shows 85% sequence identity to the second intron of the cwiwit gene from Beta vulgaris (Acc. AJ277458, data not shown).
To confirm that the eight bp duplication was generated upon integration, the integration site was amplified from the same transgenic plant from which the transposed Ac was isolated. Two different products were expected. Either, the Ac element has transposed further and left a footprint at the integration site, or the wild type sequence prior to Ac integration without the eight bp duplication. All eleven sequenced PCR fragments harboured the wild type sequence without the eight bp duplication. Figure 4b shows the sequence comparison at the integration site adjacent to Ac with the site void of Ac. The data indicates autonomous transposition of the maize Ac element in sugar beet.
Alternative splicing of the Ac transposase in sugar beet and tobacco
To gain more insight into expression and splicing of the Ac transcript, RT–PCR analyses were performed with RNA extracted from plants harbouring Ac. As control, RT–PCR analyses were performed for tobacco harbouring a stabilized Ac element (Acst). Figure 5a shows a schematic drawing of the Ac transposable element with the primary transcript and positions of introns. For RT–PCR analysis primer pairs were designed to comprise each intron individually (Tables 1, 2; Fig. 5a). These are represented by amplicons A through D. Amplicons E, F, and G were amplified with primer pairs from within intron four and outside of the intron. Amplicons H, I, and J cover two adjacent introns (Table 2; Fig. 5a). RT–PCR with primers for amplicons A, B, and C amplified expected fragments with a size of 493, 496, and 460 bp, respectively, in Ac containing sugar beet and tobacco (Fig. 5b). In contrast, amplicon D which comprises intron four shows, in addition to the expected 284 bp long fragment, at least three fragments of larger sizes in sugar beet but not in tobacco. To gain insight into the nature of the additional fragments, these were cloned and sequenced. As shown in Fig. 6, six different PCR products were obtained. The largest product of 671 bp harbours the complete intron four with splice donor and acceptor sites at positions 3616 and 4002 in the 4565 bp long Ac element (Acc. X05424). The 583 bp and 553 bp long products were generated by splicing of a 88 bp and a 118 bp intron, respectively, in which two alternative splice donor sites at positions 3915 and 3885 and the intron four splice acceptor site at position 4002 were used. The 551 bp long product was generated by splicing of a 120 bp intron in which the intron four splice donor site at position 3616 and a splice acceptor site at position 3735 were used. The fifth product of 433 bp was generated by simultaneously splicing of the 120 bp and the 118 bp introns mentioned above. In the smallest cloned PCR fragment of 284 bp the complete intron four is removed.

RT–PCR analyses of Ac transcripts. a Schematic representation of the 4565 bp Ac element with major transcription start site (TSS), polyadenylation site (PA), start and stop codon, and positions of all four introns. Below Ac a schematic representation of all PCR amplicons A through J amplified by RT–PCR is shown. PCR primer sequences and primer pairs for each amplicon are listed in Tables 1, 2. Positions of primers within Ac are reflected by the number in their designations. b Gel electrophoretic separation of RT–PCR products for Ac amplicons A through D from B. vulgaris and N. tabacum. Next to D a small range size standard with fragments of 100 bp size differences has been loaded on the gel. c Gel electrophoretic separation of RT–PCR products for Ac amplicons H through J from B. vulgaris and N. tabacum. Next to J a small range size standard has been loaded on the gel

Schematic representation of RT–PCR products cloned and sequenced from transgenic sugar beet. Sizes of all RT-PCR fragments obtained from amplicons D, H, and I are shown relative to their position in the Ac primary transcript below. Positions of splice donor sites (d) and splice acceptor sites (a) relative to the 4.565 bp Ac element are shown. Black boxes indicate introns and include their sizes in bp
All PCR products which were obtained from amplicon D are three base pairs shorter than one would expect from the sequence given by Kunze et al. (1987). This is due to a few sequence alterations in the 3′-UTR of the Ac element investigated here.
To confirm these novel alternative splice products and to potentially detect more alternatively spliced transcripts, RT–PCR experiments were performed to detect amplicons E, F, and G (Fig. 5a; Table 2). By using primers within intron four, this experiment was supposed to be more specific to detect unspliced and alternatively spliced products. After cloning and sequencing of PCR products the alternative splice sites described above were confirmed, but from these PCRs no additional splice products were detected in sugar beet. For tobacco, gel electrophoresis of the RT–PCR products from amplicons E, F, and G gives evidence that at least the 118 bp and 120 bp alternative introns are also spliced in this species to very low amounts (data not shown).
To further investigate alternative splicing, RT–PCR was performed with primers that comprise two introns each. These are represented by amplicons H, I, and J (Fig. 5a; Table 2). The expected splice products were 1468, 1304, and 716 bp in length, respectively. Figure 5c shows that these products were the major PCR products amplified from transgenic sugar beet and tobacco. No larger unspliced products could be detected for amplicons H and I. The larger amplicons amplified for J from transgenic sugar beet can be explained by alternative splicing of intron four which has been confirmed by cloning and sequencing PCR products for amplicon D (see above). Most interestingly was the observation that smaller fragments could be amplified from both transgenic species with primers for amplicons H and I. To investigate the molecular nature of these smaller fragments, these were cloned and sequenced from transgenic sugar beet. The sequence of these PCR products revealed novel large introns within the second and third exon. Two such different PCR products derived from primers corresponding to amplicon H were obtained (Fig. 6). The 724 bp and 702 bp long products were generated by splicing of a 744 bp and a 766 bp intron, respectively, in which the same alternative splice donor site at position 1584 and two alternative splice acceptor sites at positions 2327 and 2349 were used. For amplicon I three different PCR products were cloned and sequenced (Fig. 6). The 799, 793, and 785 bp long products were generated by splicing of a 505, 511, and a 519 bp intron, respectively, in which the same alternative splice donor site at position 2736 and three alternative splice acceptor sites at positions 3240, 3246 and 3254 were used.
In this respect it may be interesting to investigate if such alternative splice products may also occur in maize, the original host species from which Ac was isolated. Although this analysis should be perfomed experimentally, an initial bioinformatic analysis was performed here. For this, the sequence of the Ac transposable element colinear with the primary transcript was subjected to a bioinformatic analysis using the program SplicePredictor (Brendel and Kleffe 1998). SplicePredictor assigns a score from 1 to 15 to each splice site, where 15 means that this sequence is predicted as a highly efficient site in the host organism selected. Table 3 shows that the splice sites of all four major introns in the primary transcript of Ac have values from 11 to 15 for splicing efficiency in Zea mays and Arabidopsis thaliana. Of the alternative introns detected here (Fig. 6) only the introns with splice donor sites at position 1584 (value 15), 3885 (value 12), 3915 (value 11), and splice acceptor site at position 2327 (value 15) are predicted to be as efficiently spliced in maize as the four major introns (Table 3). The splice donor and acceptor sites of the 744 bp alternative intron in exon two (positions 1584 and 2327) both have the highest values of 15 (Table 3).
In summary, these results indicate that intron four of the Ac transposase gene is sometimes alternatively spliced in sugar beet and much less frequently in tobacco. Exon two and three harbour at least five novel introns that are spliced in sugar beet and tobacco. Most interestingly, a SplicePredictor analysis suggests that the 744 bp alternative intron in exon two may be efficiently spliced in maize.
Discussion
Sugar beet as a new host for biotechnological applications of the maize Ac/Ds transposon system
The work presented here extends the host range of the maize Ac/Ds system to sugar beet. Several lines of evidence suggest that the elements are functional in this species. First, the elements integrate at novel positions within the sugar beet genome. In total, two complete and two partial insertions were recovered (Fig. 2). In case of the cloned partial insertions it was found that the ends of the elements are separated by only 280 bp. This may be caused by two independent transposition events from two transferred T-DNAs. Alternatively, Ds may have transposed consecutively during replication of the DNA. In this case transposition of Ds may have occured after the replication fork has passed. Ds could have transposed from one replicated chromatid into unreplicated DNA leading to two linked insertions. Such a model has been proposed earlier for the Modulator element of maize (Greenblatt 1984). Such close distance transpositions are reminiscent of Ac transposition in tomato where linked clusters of insertions were found (Osborne et al. 1991). In addition to the analyses of functional transposition, RT–PCR analyses of the Ac transcript in sugar beet reveal that alternative splicing occurs only in a very low proportion of the primary transcript. Based on these observations it is proposed that sugar beet is a suitable host species for the maize Ac/Ds transposon system.
The mechanism of transposition of the maize Ac/Ds transposable element system in sugar beet
In the present study it is shown that the elements transpose from the T-DNA vector into the genome of sugar beet. The molecular events observed at the excision site of the elements in the T-DNA vector support the hairpin model for transposon excision (Kunze and Weil 2002). This model was recently experimentally supported with an in vitro assay using the hAT transposable element Hermes (Zhou et al. 2004). This model predicts that excision of the element induces a double strand break at either end adjacent to the element at the donor site, with a one base pair overhang that is followed by ligation of the top and bottom strand generating a hairpin structure. Such a hairpin structure involves an intra-helix 3′ to 5′ trans-esterification intermediate that is reminiscent of V(D)J recombination. This structure has been shown to be generated in vitro by the Hermes transposase at the donor site (Zhou et al. 2004). Upon repair, the hairpin structures are endonucleolytically cleaved randomly within the hairpin, followed by a fill in reaction or an exonucleolytic removal of the overhang, and religation of the ends. Upon this, single bp exchanges and/or one bp deletions may occur at the donor site (Kunze and Weil 2002). The observation that all donor sites obtained for Ac and Ds in sugar beet show a one bp exchange and/or a one bp deletion supports the hairpin model for Ac and Ds excision also for sugar beet (Figs. 3a, 4a).
Integration of Ac and Ds in maize and other heterologous host plants induces an eight bp duplication at the target site (Kunze and Weil 2002). Such an eight bp duplication was observed at the integration site of Ac in sugar beet (Fig. 4b). This indicates that Ac has retained its mechanism for transposition in this host. However, the integration event observed for the Ds element lacked an eight bp duplication (Fig. 3b). The occurence of a two bp deletion at the integration site is reminiscent of the molecular events that occur during excision which have been described above. It may be conceivable that Ds has integrated in a genomic position that was generated by a double strand break followed by a repair reaction that may have deleted the nucleotides. The free ends may have been the substrates for Ds integration which explains the lack of an eight bp duplication. A lack of an eight base pair duplication at the insertion site of Ds has been noted before in heterologous host plants. For example in tomato three insertion sites have been cloned in which both ends were present. While two harboured an eight bp duplication adjacent to Ds, the third one showed a 7 bp sequence duplication in the vicinity but not directly adjacent to the element (Carroll et al. 1995). Using a specifically modified Ds element that harboured reversed terminal ends to induce chromosomal rearrangements in Arabidopsis thaliana, nine inversions for which both Ds termini have been recovered were analysed moleculary. Three cases had eight bp target site duplications, two cases had only 1 bp duplications, and the other four cases did not show any target site duplication at all (Krishnaswamy et al. 2008).
Alternative splicing of the Ac transposase transcript
In the course of this work a comparative analysis of transcription and splicing of the Ac transcript in sugar beet and tobacco was performed. In addition to the four known introns spliced from the primary Ac transcript, eight additional introns were detected (Fig. 6). The RT–PCR analyses suggest that only those transcripts with alternative splicing of the fourth intron are of significantly higher abundance in sugar beet than in tobacco. On the contrary the alternative introns in exon two and exon three may be present in both species at similar proportions (Fig. 5).
The first indication that the Ac transcript is alternatively transcribed or processed in heterologous host plants came from Arabidopsis thaliana. Northern blot analysis revealed many unexpected transcripts leading to the conclusion that only a fraction of the Ac messenger is correctly processed, whereas the majority of the transcripts is misprocessed (Grevelding et al. 1992). In contrast, Northern blot analysis in transgenic tobacco and in its native host maize did not indicate that misprocessing of the Ac transcript in these plants occurs in a significant proportion (Hehl and Baker 1990; Kunze et al. 1987). Further insight into misprocessing of the Ac transcript in A. thaliana came from two other studies (Jarvis et al. 1997; Martin et al. 1997). The splicing efficiencies of introns three and four were determined to be 57 and 30%, respectively (Jarvis et al. 1997). This is in contrast to our RT–PCR experiments in sugar beet and tobacco where no unspliced intron three was detected and the detected unspliced transcripts of intron four were not very abundant and more evident in sugar beet than in tobacco (Fig. 5b). Although no precise quantification has been performed here, it is estimated, with the help of a digital imaging program, that the PCR product which represents the unspliced intron four is not more than 2.5% relative to the overall product of amplicon D in sugar beet (Fig. 5b, data not shown). In addition to the unspliced transcript containing intron four, several fragments of intermediate sizes were detected in A. thaliana suggesting alternative splicing within or in the vicinity of intron four (Jarvis et al. 1997; Martin et al. 1997). These studies revealed that those introns correspond to several of the alternative introns detected here in sugar beet (Fig. 6). The 88, 118, and 120 bp introns within intron four detected for sugar beet have also been detected in A. thaliana (Martin et al. 1997). Only one alternative intron found in A. thaliana, which harbours splice donor and acceptor sites at positions 3616 and 3770, respectively (Martin et al. 1997) has not been identified in sugar beet.
Some of the introns within exon two and three detected in sugar beet and tobacco (Fig. 5c) have also been detected in other transgenic host plants. The 505 bp long intron in exon three has been detected in A. thaliana expressing a modified Ac cDNA (Jarvis et al. 1997). The 744 and 766 bp introns in exon two have also been detected in Ac transgenic barley (Friedrich 2007). In addition, two even larger introns were detected in the second exon of Ac in barley, all starting from the same donor site. Also within the third exon, alternative introns, all starting at the same donor site, were detected in barley, two of which are distinctly smaller than those found in sugar beet. Most interestingly, a comparative PCR analysis between transgenic barley and a maize line harbouring Ac suggests that these introns may also be spliced from the Ac transcript in maize (Friedrich 2007).
Consequences of alternative splicing for the Ac transposase
Without the isolation and analysis of complete cDNAs, a discussion of how the observed splicing events may affect transposase function is purely speculative. Therefore, discussion is restricted to events that have functional implications for transposition. Except of those, most of the alternative splice products lead to frame shift mutations with premature termination codons that may be substrates for nonsense-mediated mRNA decay (Rebbapragada and Lykke-Andersen 2009). Besides, the consequences of transcripts harbouring cryptic introns within intron four have been discussed before (Jarvis et al. 1997; Martin et al. 1997). Because in sugar beet the proportion of misspliced intron four is very low and the missplicing has no effect on any of the functional domains of the transposase, it may be hard to conceive that this has any negative effect on transposition frequency in sugar beet.
The most interesting alternatively spliced Ac transcript may occur if, in addition to the four major introns, the 744 bp intron within exon two is spliced. This is tempting to speculate because this intron and the four major introns are predicted to be efficiently spliced in maize (Table 3). In contrast to the majority of the other alternatively spliced products, this mRNA does not change the native transposase reading frame, but removes the DNA binding domain and two nuclear localization signals, and retains the dimerization domain (Essers et al. 2000; Feldmar and Kunze 1991). Such a transposase, if in fact translated, may be able to interfer with the normal function of Ac by forming non-functional heterodimers. Such a transcript may not have been isolated yet from maize because its expression could be regulated or depend on Ac dosage. In this respect it seems interesting that in nuclear extracts from maize a second protein of 70 kDa cross reacts with an Ac specific antibody in addition to the native Ac transposase of 110 kDa (Fusswinkel et al. 1991). If the 744 bp intron together with the four major introns are removed from the primary transcript the encoded protein is predicted to be a 63 kDa protein. The data shown here may stimulate molecular analysis of novel functions encoded by alternatively spliced maize Ac transposase transcripts.
References
Aarts MGM, Dirkse WG, Stiekema WJ, Pereira A (1993) Transposon tagging of a male sterility gene in Arabidopsis. Nature 363:715–717
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Babwah V, Waddell S (2002) Trans-activation of the maize transposable element, Ds, in Brassica napus. Theor Appl Genet 104:1141–1149
Baker B, Schell J, Lörz H, Fedoroff NV (1986) Transposition of the maize controlling element “Activator” in tobacco. Proc Natl Acad Sci USA 83:4844–4848
Bosemark NO (1993) Genetics and breeding. In: Cooke DA, Scott RK (eds) The sugar beet crop: science into practice. Chapman and Hall, London, pp 67–119
Brendel V, Kleffe J (1998) Prediction of locally optimal splice sites in plant pre-mRNA with applications to gene identification in Arabidopsis thaliana genomic DNA. Nucleic Acids Res 26:4748–4757
Calvi BR, Hong TJ, Findley SD, Gelbart WM (1991) Evidence for a common evolutionary origin of inverted repeat transposons in Drosophila and plants: hobo, Activator, and Tam3. Cell 66:465–471
Carroll BJ, Klimyuk VI, Thomas CM, Bishop GJ, Harrison K, Scofield SR, Jones JD (1995) Germinal transpositions of the maize element Dissociation from T-DNA loci in tomato. Genetics 139:407–420
Deblaere R, Bytebier B, De Greve H, Deboeck F, Schell J, Van Montagu M, Leemans J (1985) Efficient octopine Ti plasmid-derived vectors for Agrobacterium-mediated gene transfer to plants. Nucleic Acids Res 13:4777–4788
Dinesh-Kumar SP, Whitham S, Choi D, Hehl R, Corr C, Baker B (1995) Transposon tagging of tobacco mosaic virus resistance gene N: its possible role in the TMV-N-mediated signal transduction pathway. Proc Natl Acad Sci USA 92:4175–4180
Douglas SE (1995) DNA strider. An inexpensive sequence analysis package for the Macintosh. Mol Biotechnol 3:37–45
Elliott MC, Weston GD (1993) Biology and physiology of the sugar-beet plant. In: Cooke DA, Scott RK (eds) The sugar beet crop: science into practice. Chapman and Hall, London, pp 37–66
Essers L, Adolphs RH, Kunze R (2000) A highly conserved domain of the maize Activator transposase is involved in dimerization. Plant Cell 12:211–224
Fedoroff N, Wessler S, Shure M (1983) Isolation of the transposable maize controlling elements Ac and Ds. Cell 35:235–242
Feldmar S, Kunze R (1991) The ORFa protein, the putative transposase of maize transposable element Ac, has a basic DNA binding domain. EMBO J 10:4003–4010
Friedrich CK (2007) Untersuchungen zur Expression der Ac-Transposase aus Mais (Zea mays L.) in Gerste (Hordeum vulgare L.). Dissertation, Universität Hamburg, Germany
Fusswinkel H, Schein S, Courage U, Starlinger P, Kunze R (1991) Detection and abundance of mRNA and protein encoded by transposable element activator (Ac) in maize. Mol Gen Genet 225:186–192
Greenblatt IM (1984) A chromosomal replication pattern deduced from pericarp phenotypes resulting from movements of the transposable element Modulator in maize. Genetics 108:471–485
Grevelding C, Becker D, Kunze R, von Menges A, Fantes V, Schell J, Masterson R (1992) High rates of Ac/Ds germinal transposition in Arabidopsis suitable for gene isolation by insertional mutagenesis. Proc Natl Acad Sci USA 89:6085–6089
Hall TD (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows. Nucl Acids Symp Ser 41:95–98
Hehl R (1994) Transposon tagging in heterologous host plants. Trends Genet 11:385–386
Hehl R, Baker B (1989) Induced transposition of Ds by a stable Ac in crosses of transgenic tobacco plants. Mol Gen Genet 217:53–59
Hehl R, Baker B (1990) Properties of the maize transposable element Activator in transgenic tobacco plants: a versatile inter-species genetic tool. Plant Cell 2:709–721
Hehl R, Nacken WKF, Krause A, Saedler H, Sommer H (1991) Structural analysis of Tam3, a transposable element from Antirrhinum majus, reveals homologies to the Ac element from maize. Plant Mol Biol 16:369–371
Honma MA, Baker BJ, Waddell CS (1993) High-frequency transposition of Ds-ALS in Arabidopsis. Proc Natl Acad Sci USA 90:6242–6246
Jacobs G, Dechyeva D, Menzel G, Dombrowski C, Schmidt T (2004) Molecular characterization of Vulmar1, a complete mariner transposon of sugar beet and diversity of mariner- and En/Spm-like sequences in the genus Beta. Genome 47:1192–1201
Jarvis P, Belzile F, Dean C, Page T (1997) Inefficient and incorrect processing of the Ac transposase transcript in iae1 and wild-type Arabidopsis thaliana. Plant J 11:921–931
Kloos DU, Oltmanns H, Dock C, Stahl D, Hehl R (2002) Isolation and molecular analysis of six taproot expressed genes from sugar beet. J Exp Bot 53:1533–1534
Knapp S, Coupland G, Uhrig H, Starlinger P, Salamini F (1988) Transposition of the maize transposable element Ac in Solanum tuberosum. Mol Gen Genet 213:285–290
Krishnaswamy L, Zhang J, Peterson T (2008) Reversed end Ds element: a novel tool for chromosome engineering in Arabidopsis. Plant Mol Biol 68:399–411
Kunze R, Weil CF (2002) The haT and CACTA superfamilies of plant transposons. In: Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) Mobile DNA II. ASM Press, Herndon
Kunze R, Stochaj U, Laufs J, Starlinger P (1987) Transcription of transposable element Activator (Ac) of Zea mays L. EMBO J 6:1555–1564
Kuykendall D, Shao J, Trimmer K (2008) Coe1 in Beta vulgaris L. has a Tnp2-domain DNA transposase gene within putative LTRs and other retroelement-like features. Int J Plant Genomics 2008:360874
Laufs J, Wirtz U, Kammann M, Matzeit V, Schaefer S, Schell J, Czernilofsky AP, Baker B, Gronenborn B (1990) Wheat dwarf virus Ac/Ds vectors: expression and excision of transposable elements introduced into various cereals by a viral replicon. Proc Natl Acad Sci U S A 87:7752–7756
Lawson EJ, Scofield SR, Sjodin C, Jones JD, Dean C (1994) Modification of the 5’ unteranslated leader region of the maize Activator element leads to increased activity in Arabidopsis. Mol Gen Genet 245:608–615
Lippincott JA, Lippincott BB (1969) Tumor growth complementation among strains of Agrobacterium. J Bacteriol 99:496–502
Liu H, Wang Q, Yu M, Zhang Y, Wu Y, Zhang H (2008) Transgenic salt-tolerant sugar beet (Beta vulgaris L.) constitutively expressing an Arabidopsis thaliana vacuolar Na/H antiporter gene, AtNHX3, accumulates more soluble sugar but less salt in storage roots. Plant Cell Environ 31:1325–1334
Martin DJ, Firek S, Moreau E, Draper J (1997) Alternative processing of the maize Ac transcript in Arabidopsis. Plant J 11:933–943
Menzel G, Dechyeva D, Keller H, Lange C, Himmelbauer H, Schmidt T (2006) Mobilization and evolutionary history of miniature inverted-repeat transposable elements (MITEs) in Beta vulgaris L. Chromosome Res 14:831–844
Moore I, Galweiler L, Grosskopf D, Schell J, Palme K (1998) A transcription activation system for regulated gene expression in transgenic plants. Proc Natl Acad Sci USA 95:376–381
Murai N, Li ZJ, Kawagoe Y, Hayashimoto A (1991) Transposition of the maize Activator element in transgenic rice plants. Nucleic Acids Res 19:617–622
Mutasa-Gottgens E, Qi A, Mathews A, Thomas S, Phillips A, Hedden P (2009) Modification of gibberellin signalling (metabolism & signal transduction) in sugar beet: analysis of potential targets for crop improvement. Transgenic Res 18:301–308
Olszewski NE, Martin FB, Ausubel FM (1988) Specialized binary vector for plant transformation: expression of the Arabidopsis thaliana AHAS gene in Nicotiana tabacum. Nucleic Acids Res 16:10765–10782
Oltmanns H, Kloos DU, Briess W, Pflugmacher M, Stahl DJ, Hehl R (2006) Taproot promoters cause tissue specific gene expression within the storage root of sugar beet. Planta 224:485–495
Osborne BI, Baker B (1995) Movers and shakers: maize transposons as tools for analyzing other plant genomes. Curr Opin Cell Biol 7:406–413
Osborne BI, Corr CA, Prince JP, Hehl R, Tanksley SD, McCormick A, Baker B (1991) Ac transposition from a T-DNA can generate linked and unlinked clusters of insertions in the tomato genome. Genetics 129:833–844
Osborne BI, Wirtz U, Baker B (1995) A system for insertional mutagenesis and chromosomal rearrangement using the Ds transposon and Cre-lox. Plant J 7:687–701
Pohlman RF, Fedoroff NV, Messing J (1984) The nucleotide sequence of the maize controlling element Activator. Cell 37:635–643
Rebbapragada I, Lykke-Andersen J (2009) Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol 21:394–402
Reeves PA, He Y, Schmitz RJ, Amasino RM, Panella LW, Richards CM (2007) Evolutionary conservation of the FLOWERING LOCUS C-mediated vernalization response: evidence from the sugar beet (Beta vulgaris). Genetics 176:295–307
Reintanz B (1997) Funktionelle Charakterisierung der Kaliumkanal a-Untereinheit AtKC1 aus Arabidopsis thaliana. Dissertation, Universität Köln
Rotthues A, Kappler J, Lichtfuss A, Kloos DU, Stahl DJ, Hehl R (2008) Post-harvest regulated gene expression and splicing efficiency in storage roots of sugar beet (Beta vulgaris L.). Planta 227:1321–1332
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory Press, New York
Schmidt R, Willmitzer L (1989) The maize autonomous element Activator (Ac) shows a minimal germinal excision frequency of 0.2%–0.5% in transgenic Arabidopsis thaliana plants. Mol Gen Genet 220:17–24
Scholz S, Lörz H, Lütticke S (2001) Transposition of the maize transposable element Ac in barley (Hordeum vulgare L.). Mol Gen Genet 264:653–661
Scofield SR, Harrison K, Nurrish SJ, Jones JDG (1992) Promoter fusions to the Activator transposase gene cause distinct patterns of Dissociation excision in tobacco cotyledons. Plant Cell 4:573–582
Stahl DJ, Kloos DU, Hehl R (2004) A sugar beet chlorophyll a/b binding protein promoter void of G-box like elements confers strong and leaf specific reporter gene expression in transgenic sugar beet. BMC Biotechnol 4:31
Swinburne J, Balcells L, Scofield SR, Jones JDG, Coupland G (1992) Elevated levels of Activator transposase mRNA are associated with high frequencies of Dissociation excision in Arabidopsis. Plant Cell 4:583–595
Van Sluys MA, Tempe J, Fedoroff N (1987) Studies on the introduction and mobility of the maize Activator element in Arabidopsis thaliana and Daucus carota. EMBO J 6:3881–3889
Whitham S, Dinesh-Kumar SP, Choi D, Hehl R, Corr C, Baker B (1994) The product of the tobacco mosaic virus resistance gene N: similarity to Toll and the Interleukin-1 receptor. Cell 78:1101–1115
Wilson K, Long D, Swinburne J, Coupland G (1996) A Dissociation insertion causes a semidominant mutation that increases expression of TINY, an Arabidopsis Gene related to APETALA2. Plant Cell 8:659–671
Wirtz U, Osborne B, Baker B (1997) Ds excision from extrachromosomal geminivirus vector DNA is coupled to vector DNA replication in maize. Plant J 11:125–135
Yang CH, Ellis JG, Michelmore RW (1993) Infrequent transposition of Ac in lettuce, Lactuca sativa. Plant Mol Biol 22:793–805
Yoder JI, Goldsborough AP (1994) Transformation systems for generating marker-free transgenic plants. Bio/Technology 12:263–267
Yoder JI, Palys J, Alpert K, Lassner M (1988) Ac transposition in transgenic tomato plants. Mol Gen Genet 213:291–296
Zhou L, Mitra R, Atkinson PW, Hickman AB, Dyda F, Craig NL (2004) Transposition of hAT elements links transposable elements and V(D)J recombination. Nature 432:995–1001
Acknowledgments
This work was supported by the Federal Ministry of Education and Research (BMBF 0313264G and 0315210F). We would like to thank Elke Faurie for excellent technical assistance and Heike Lehmann (PLANTA GmbH) for advice regarding sugar beet hairy root transformations. We would also like to thank Juliane Dohm and Heinz Himmelbauer (Centre de Regulacio Genomica, Barcelona, Spain) for BLAST analyses with the unpublished genomic sequence of sugar beet and Norbert Käufer for critical reading of the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lisson, R., Hellert, J., Ringleb, M. et al. Alternative splicing of the maize Ac transposase transcript in transgenic sugar beet (Beta vulgaris L.). Plant Mol Biol 74, 19–32 (2010). https://doi.org/10.1007/s11103-010-9651-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-010-9651-2