
Abstract
Understanding the molecular basis of plant performance under water-limiting conditions will help to breed crop plants with a lower water demand. We investigated the physiological and gene expression response of drought-tolerant (IR57311 and LC-93-4) and drought-sensitive (Nipponbare and Taipei 309) rice (Oryza sativa L.) cultivars to 18 days of drought stress in climate chamber experiments. Drought stressed plants grew significantly slower than the controls. Gene expression profiles were measured in leaf samples with the 20 K NSF oligonucleotide microarray. A linear model was fitted to the data to identify genes that were significantly regulated under drought stress. In all drought stressed cultivars, 245 genes were significantly repressed and 413 genes induced. Genes differing in their expression pattern under drought stress between tolerant and sensitive cultivars were identified by the genotype × environment (G × E) interaction term. More genes were significantly drought regulated in the sensitive than in the tolerant cultivars. Localizing all expressed genes on the rice genome map, we checked which genes with a significant G × E interaction co-localized with published quantitative trait loci regions for drought tolerance. These genes are more likely to be important for drought tolerance in an agricultural environment. To identify the metabolic processes with a significant G × E effect, we adapted the analysis software MapMan for rice. We found a drought stress induced shift toward senescence related degradation processes that was more pronounced in the sensitive than in the tolerant cultivars. In spite of higher growth rates and water use, more photosynthesis related genes were down-regulated in the tolerant than in the sensitive cultivars.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice is one of the world’s most important staple foods and provides 30% of the calories consumed in Asian countries. The conditions of rice cultivation vary from flooded wetland to rainfed dryland. In rainfed systems, that occupy about one-third of the growth area, drought is the major environmental factor that reduces productivity by 13–35% (Jongdee et al. 1998; Lafitte et al. 2006). However, as a consequence of centuries of breeding efforts in environments with different water availability, drought tolerance in rice cultivars ranges from very susceptible to highly tolerant. In recent years, the use of this genotypic variation for genomic research on drought tolerance mechanisms has been enhanced by the development of introgression lines from drought tolerant donor cultivars into elite cultivars and the selection of drought tolerant backcross populations (Li et al. 2005; Lafitte et al. 2006). The genotypic variation in drought tolerance together with the genetic tools available for rice, such as marker maps, sequence information, and microarrays (Matsumoto et al. 2005; Rensink and Buell 2005) and the possibility to test the agronomic relevance of a scientific discovery (Xu et al. 2006), make rice a most interesting model system for research in drought tolerance of grass crops.
Drought is a multifaceted stress condition. It comes in many forms with respect to timing and severity, ranging from long drought seasons where the water supply by rain is lower than the demand, to short periods without rain where plants rely completely on the available water in the soil (Fukai and Cooper 1995; Lafitte et al. 2006). In addition, water availability in the soil varies with respect to amount and distribution (Clark et al. 2002). Different durations of stress require different physiological adaptations: short periods of severe stress might favor a “wait and see” tolerance strategy. Long periods rather require an avoidance strategy: growth can be kept up by increased water uptake from lower soil layers by deeper roots combined with an improved water conduction capacity of the root system or a decreased water potential of the plant (Levitt 1972). Decrease in water potential often correlates with a decrease in osmotic potential through the accumulation of osmolytes (Turner and Jones 1980). In addition, compatible osmolytes are assumed to protect macromolecular structures from conformational changes at lower water potentials (Hanson et al. 1994; Zhang et al. 2000; Jongdee et al. 2002). These strategies help to keep up turgor and thus growth and water uptake under reduced water availability. Correlations between the capacity for osmotic adjustment and the performance under early-season drought have been shown, e.g. for wheat cultivars (Blum et al. 1999; Zhang et al. 1999) whereas direct evidence for a role of osmotic adjustment in drought tolerance of rice is still limited (Mitra 2001; Jongdee et al. 2002; Hazen et al. 2005). Another important drought resistance strategy is the optimization of CO2 gain through stomatal aperture while minimizing water loss (Price et al. 2002). Efficient regulation of transpiration can result in higher water use efficiency, thus resulting in increased biomass at the end of a drought period. Higher seedling vigor and maintenance of a high leaf water potential correlate with a better drought recovery and thus a better performance of plants after early season drought (Jongdee et al. 2002; Kamoshita et al. 2004; Siopongco et al. 2006). Studies of the response of rice plants to long-term water stress may thus result in the identification of drought tolerance mechanisms relevant for the development of drought-adapted crops that will ultimately give more,crop per drop’.
Here we used gene expression profiling in combination with a detailed physiological analysis of drought sensitive and tolerant rice cultivars to identify plant reactions that may contribute to long-term drought tolerance. In addition, we used published quantitative trait loci (QTL) data to evaluate the significance of candidate genes identified in our profiling experiments (Wayne and McIntyre 2002; Hazen et al. 2005). Our approach was to compare the drought response of unrelated tolerant and sensitive cultivars to identify common responses of tolerant in comparison to sensitive cultivars. We thus aim to identify traits which are of general importance for drought tolerance in rice and, using orthology as well as synteny approaches, other cereal crops as well.
Material and methods
Plant material, cultivation and drought stress treatment
Seeds of rice (Oryza sativa L.) cultivars Nipponbare (IRGC accession 12731) (NB), Taipei 309 (IRGC accession 42576) (TP), and IR57311-95-2-3 (IRGC accession 17509 (INGER)) (IR) were obtained from the International Rice Research Institute (IRRI, Manila, Philippines), seeds for the rice cultivar LC-93-4 (LC) were obtained from the Institute of Biotechnology (Hanoi, Vietnam). The seeds from NB, TP and IR that were used in the experiments were derived from plants grown at the Max-Planck-Instiute of Molecular Plant Physiology.
Rice plants were grown under water sufficient and water limiting conditions in three independent experiments (#1–3 in a controlled climate chamber) together with 17 additional cultivars from a Vietnamese tolerance breeding programm. The design was a split-plot design with five blocks per drought or control treatment. Each treatment and cultivar was represented by five replicate pots with one plant per pot. Pots were randomized within the blocks. Block position was rotated daily. In two additional experiments (#4 and 5), five plants per cultivar and treatment were grown in a Latin square design.
Seeds were pre-germinated in tap water at 28°C for ten days. Plantlets were transferred to a climate chamber with 12 h day length at a photon flux density of 600 μE m−2 s−1 (Lamps: Iwasaki Eye MT 400 DL/BH E40, DHL Licht, Wülfrath, Germany); temperature was 26°C in the light and 22°C at night, with a relative humidity of 75% in the light and 70% at night. Plantlets were grown in 10 cm diameter pots (TO 10 D, Kauseck, Mittenwalde, Germany), filled with 540 g sand mixed with 8 g of Lewatit HD 50 (Lanxess, Langenfeld, Germany), an ion-exchange resin loaded with nutrient ions and 0.4 g Fetrilon Combi (Compo, Münster, Germany) (Köhl 1996). Soil layer was 7.5 cm deep. Pots were positioned in polypropylene boxes filled with water to the level of the substrate surface. Pot surfaces were covered with black, pinpricked polythene film (Aquafol, Reinmann, Emsdetten, Germany) to prevent growth of algae. Twenty-six days after sowing, water was removed from half of the boxes and plants were left to dry for four days, until the soil water content had reached the average permanent wilting point (PWP) for 50% of all plants in the experiment (105 plants from 21 cultivars). Thereafter, the soil water content was kept constant to the fixed average PWP value over a period of 14 days by daily weighing each pot at the end of the light period and adding the amount of water lost during the last 24 h. Water use efficiency was calculated from these data as the average daily evapotranspiration during the drought treatment divided by the dry biomass of the plant at harvest. The daily evapotranspiration was determined from the pot mass after addition of water minus pot mass after 24 h evapotranspiration (prior to addition of water). Data were not corrected for the daily water loss from pots without plants, which was about 8 g water per day and pot.
During this moderate drought stress period, stressed and control plants were characterized by repeated measurements of leaf length, tiller number and scoring (1 growth normal, leaves green, 3 some leaves discolored, 5 most leaves discolored, 7 most leaves dry, 9 complete plant dying) based on the stress damage score of the IRRI (Mitchell et al. 1998) in experiments #1–3. Water potentials were measured in the experiments #4 and 5. Pre-dawn water potential was measured after 18 days of stress using a Scholander pressure bomb (Plant water status console 3000 series, Soil Moisture Equipment Corporation, Santa Barbara, CA, USA). Pots were removed from the climate chamber 30 min before the end of the dark period and kept dark until the measurement. Mid-day leaf water potential was measured in the middle of the light period, 24–26 days after the beginning of stress treatment.
After a total of 19 days of drought stress, plants from experiments #1–3 were harvested 5 h after the beginning of the light period. Samples for expression profiling and osmolality determination were harvested from the middle section of the blades of fully expanded green leaves and immediately frozen in liquid nitrogen. The middle sections of leaves were selected to avoid taking material from the elongation zone at the base of the leaf blade or senescent tissue at the top of the leaves, especially in stressed plants. Fresh (FW) and dry weight (DW) of the remaining leaf blades, and of total shoots and roots were determined. Actual shoot water content was determined as (FW − DW)/DW and expressed as g water per g dry weight. Saturation water content was determined after 24 h resaturation in tap water (Turner 1981; Lafitte 2002). From each plant, a leaf was cut, weighed to determine the fresh weight and positioned with the cut end in tap water in a closed vessel. After 24 h incubation at 4°C, saturation weight (SW) was determined. Leaf samples were then dried for 48 h at 60°C to determine dry weight (DW). Relative water content was calculated as (FW − DW)/(FW − SW). For leaf osmolality measurements, frozen leaf material was homogenized and mixed with 1 μl ddH20 per mg sample. After centrifugation, osmolality of the supernatant was measured in a vapour pressure osmometer (Vapro 5520, Vescor, Logan, USA). The readings were corrected to the water content of the orginal samples.
The distribution of log-transformed dry weights of shoot and root, untransformed shoot:root ratios, actual and relative water content and water potentials was checked by proc uniform (SAS 9.2, SAS-Institute, Cary, NC, USA) and values beyond means ± 3 standard deviations were removed from the data set. Analysis of variance was performed using proc GLM on the terms condition, cultivar and the condition × cultivar interaction, means were compared by the Ryan-Gabriel-Welch-Test.
Genotyping
For each cultivar, DNA was isolated from leaf material from three independed plants using the CTAB extraction method (Doyle and Doyle 1990). DNA concentration was measured photometrically (NanoDrop ND-1000 UV-Vis spectrophotometer, NanoDrop Technologies, Wilmington, DE, USA). Seven subspecies-specific sequence tagged site (SS-STS) markers were selected from (Chin et al. 2007), primer sequences are given in Supplementary Table 1. Polymerase chain reaction was carried out in 50 μl reaction volume with 100 ng DNA, 0.1 nmol forward and reverse primer, 25 ng dNTP mix, 57.5 nmol Mg2+) at an annealing temperature of 53°C and an elongation temperature of 72°C for 40 cycles. Reaction products were separated on a 3% agarose gel.
Gene expression profiling
Transcript profiles were measured on NSF rice 20 K oligonucleotide microarrays (http://www.ricearray.org), on which 50–70 mer oligonucleotides representing 20,230 randomly chosen genes from the rice genome are spotted. The profiling was performed with a modified version of a previously developed quality controlled method (Degenkolbe et al. 2005). PolyA + -RNA was extracted with magnetic beads (Dynabeads oligo (dT)25, Dynal, Oslo, Norway). After DNase treatment, concentration and quality of extracted mRNA was measured photometrically and with a Bioanalyzer (Agilent Technologies, Santa Clara, CA). Contamination with DNA was checked by quantitative PCR (see below) using an intron specific primer pair (LOC_Os01g01840) and a primer pair specifically amplifying intergenic DNA. The sequences of all primers used in this study and the identifiers of all corresponding genes are given in Supplemental Table S1. To minimize biological variance, mRNA from four plants originating from the same experiment, condition and cultivar was pooled. cDNA was synthesized using SuperScript III (Invitrogen, Carlsbad, CA, USA) and purified by precipitation, using Bioline Sure Clean (Bioline, Luckenwalde, Germany). Yield of cDNA synthesis was determined photometrically. The quality of cDNA synthesis was evaluated by qRT-PCR using primer pairs specific to the 3′ and 5′ end of the cDNA of the housekeeping gene actin 1 (Supplemental Table S1), and cDNA synthesis was repeated for samples with a difference of Ct3’ and Ct5’ of more than 1.5 cycles. Samples were directly labeled with the fluorescent dyes Alexa Fluor 532 and 647 (Qiagen, Hilden, Germany) following the manufacturers instructions. Labelling efficiency was measured photometrically (NanoDrop ND-1000 UV-Vis spectrophotometer, NanoDrop Technologies, Wilmington, DE, USA). Labelled cDNA was hybridized to the arrays in the hybridization station Hybarray12 (Perkin Elmer, Wellesley, MA, USA).
After hybridization and washing, microarrays were scanned with a FLA-8000 laser scanner (Fuji, Tokyo, Japan). The software GeneSpotter 2.4.3 (Microdiscovery, Berlin, Germany) was used to fit the grid position of the spots and to calculate spot intensities. Spots for which intensity was affected by dust or dirt were flagged manually.
Hybridization design
A total of 28 samples, each representing mRNA from four pooled individual plant extractions, were hybridized to 14 arrays. The hybridization design was optimized for the estimation of the effects of condition and the condition × cultivar interaction taking a variance minimization approach (Landgrebe et al. 2006). In a two-step procedure, a smaller design for one sensitive and one tolerant cultivar was enlarged to encompass all four cultivars by integrating eight additional arrays. The optimization was programmed using R (R Development Core Team, 2007) and carried out on the 16-node Beowulf Linux-Cluster at the University of Potsdam. R-scripts are available upon request and from ‘http://bioinformatics.mpimp-golm.mpg.de/projects/own/d2cma/’. Allocation of the three biological replicates of each of the combinations of treatment and cultivar was completed such that a balanced distribution with respect to the labeling was achieved. Also, each combination of condition and cultivar from all experiments was used at least once (Supplemental Fig. 1).
Background correction, normalization and testing
Microarray signal intensities were background corrected and normalized before statistical analysis. Background correction was done based on the expression signal of 218 spots representing a hygromycin resistance gene that is not in the genome of the investigated cultivars. Any spot with an expression below the mean plus threefold standard deviation of the intensity of the hygromycin gene spots of the respective array and dye was labelled as below background. This applied to 40–70% of the spots on all arrays, in good agreement with results obtained for published images from 20 K NSF rice arrays (http://www.ricearray.org). Spots identified as below background were given the weight of zero in normalization and excluded from further analysis.
Normalization and statistical testing was performed using the R package limma (R 2.3.1, limma version 2.7.3; Smyth 2005). The normalization methods Median, Loess, Robustspline, and Printtiploess for within array normalization and Vsn, Scale and Quantile for between array normalization were compared. The methods Robustspline for within array normalization and Quantile for between array normalization yielded the smallest differences between arrays with respect to the position of the median, the variation and the shape of the distribution curve between arrays and were used to normalize the data. The p-value distribution for the effects of array, dye (red or green), condition (control or drought), tolerance group (tolerant or sensitive) and the interaction term condition × tolerance group was calculated in SAS 9.2 using proc glm and proc uniform. In spite of the almost identical distribution of the normalized data, we found that both array and dye had a significant (F-Test, p < 0.1) effect for more than 25% of the genes (Supplemental Table S2). Thus, the linear model fitted in the R package limma (version 2.7.3) to model the systematic variation in the data included the main effects dye, condition (E-effect), tolerance group (G-effect), and the G × E interaction. Afterwards, for the comparisons of interest, moderated t-statistics that use an empirical Bayes method were calculated. Differentially expressed genes were identified using the decideTests function (method global, fdr corrected p-value < 0.05; Benjamini and Hochberg 1995) in the R package limma. The interaction factor I was calculated based on normalized logarithmic expression values E as I = (E(dT) − E(cT)) − (E(dS) − E(cS)), with c indicating control, d indicating drought treatment, T = tolerant cultivars, S = sensitive cultivars).
Gene mapping and MapMan annotation
Genomic positions of rice genes were determined by aligning the un-spliced genes to the rice genome using BLAST (Altschul et al. 1990). Genomic as well as gene sequence information was obtained from the TIGR Rice Genome Annotation resource (http://rice.tigr.org). To establish a mapping of the genes represented on the NSF rice array to the MapMan bins (Thimm et al. 2004), translated sequences of the transcripts were aligned (BLASTX version 2.2.12) against the TAIR Arabidopsis peptide database version 6. The best blast hit was extracted. Genes whose array annotation and annotation of the best blast hit were identical were put in the MapMan bin of the best blast hit. Additionally, the GO (gene ontology) Term was used to sort genes into MapMan bins in cases of differences in the annotations of the gene and the best blast hit, poor blast E-values or for large gene families. A Wilcoxon rank sum test implemented in MapMan was used to extract bins whose gene members exhibited a significantly different regulation compared to all other bins (fdr corrected p-value < 0.1). Additionally, the Fisher exact test of the software PageMan (Usadel et al. 2006) was used to test for significant overrepresentation of significantly induced or repressed genes within the MapMan bins.
Comparison to quantitative trait loci (QTL)
The genome position of the genes represented on the NSF array was compared to the position of drought stress QTL published for rice in the Gramene Database (http://www.gramene.org). Genes were considered to map to QTL regions when the midpoint of the mapping coordinates of the start and end positions of the corresponding gene fell within the QTL region boundaries.
To test whether candidate genes detected from the expression studies were significantly overrepresented in known drought QTL regions, we used the Fisher exact statistical test applied to the 2 × 2 contingency table containing the Yes/No counts for “Is Candidate Gene” and “Maps to QTL”, respectively.
Quantitative RT-PCR (qRT-PCR)
Leaf material of experiment #4 was used to validate the microarray expression data on a subset of 45 genes that showed significant condition × tolerance group interaction and mapped within QTL. For each combination of cultivar and treatment, three plants were sampled. RNA isolation and cDNA synthesis were performed as for microarray expression profiling, but mRNA samples from single plants were used for cDNA synthesis. qRT-PCR was performed with the ABI Prism 7900HT (Applied Biosystems, Foster City, CA, USA) in 5 μl reaction volume (0.5 μl cDNA, 2 μl primer mix (0.5 μM each), 2.5 μl SYBR Green Master Mix (Eurogentec, Seraing, Belgium). Primers for qRT-PCR were designed using the software PrimerExpress (Version 2.0, Applied Biosystems) and all primer sequences together with the gene identifiers are given in Supplemental Table S1. Quality of the primers was checked with the webtool NetPrimer (PREMIER Biosoft International). To ensure specific amplification in japonica as well as indica cultivars. Primer sequences were blasted on the Gramene Database Webpage and on the Beijing Genomics Institute database (http://rice.genomics.org.cn). Correct size of the amplified region for each primer pair was checked by agarose gel electrophoresis.
Data were normalized based on the expression data of the housekeeping genes actin 1 and cyclophilin. Normalized expression of the genes of interest was calculated by dividing the average relative expression (primer efficiency P to the power of cycle number Ct) of the two housekeeping genes (H1 and H2) by the relative expression of the gene of interest (GOI): ((PH1^CtH1 + PH2^CtH2)/2)/PGOI^CtGOI. Primer efficiency was calculated using LinRegPCR (Ramakers et al. 2003).
A linear model that included the factors condition, tolerance group and the condition × tolerance group interaction was fitted and an ANOVA was performed to identify genes with a significant effect of tolerance group or condition × tolerance group interaction on gene expression.
Results
Characterization of cultivars by their physiological response to long-term drought stress
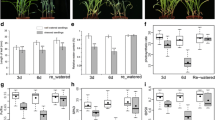
Drought treatment significantly reduced total biomass (root plus shoot dry weight) of the four rice (Oryza sativa L.) cultivars Nipponbare (NB), Taipei 309 (TP), IR57311 (IR) and LC-93-4 (LC) by up to 79% (Fig. 1). Shoot:root ratio increased significantly under drought stress but showed no significant cultivar effect (Table 1). Dry weight of both shoots and roots was significantly higher in the cultivars LC and IR than in NB and TP (Table 1) under both control and drought conditions. Likewise, LC and IR scored better in a visual scoring test (Supplemental Table S3). More than 75% of the LC and IR plants were scored 3 or better under drought stress, whereas 75% of NB plants and 50% of TP plants scored 5 or worse. At harvest, control and drought treated plants of all cultivars were still in the vegetative tillering phase (BBCH 20–29). Thus, LC and IR were judged as tolerant and NB and TP as sensitive to long-term drought in the juvenile stage of the plant.

Total dry weight of the rice cultivars Nipponbare (NB), Taipei (TP), LC-93-4 (LC) or IR57311 (IR) after 18 days of growth under control or drought treatment. Mean values from three experiments with five plants per cultivar, experiment and treatment condition. Biomass of cultivars was compared within a condition, different letters indicate that log-transformed means differ significantly (α = 0.05). F for condition = 357, p < 0.001; F for condition × cultivar = 0.46, p = 0.708
In all cultivars, drought treatment significantly reduced the water content of the shoot at the end of the drought period by about 45% compared to control conditions (Table 1). Interestingly, the water content of the leaf blade was much lower than the total shoot water content and changed less in response to drought (Table 2). Under drought stress, shoot water seemed to be mainly depleted from the tissues of the leaf sheath (data not shown). Shoot water content was higher in the tolerant cultivars IR and LC than in the sensitive cultivars NB and TP. For the cultivar LC, this difference to the sensitive cultivars was significant under control and drought conditions. At harvest, mean water potential of the leaf blades (Table 2) ranged between −0.12 and −0.34 MPa pre-dawn and between −0.96 and −2.42 MPa at mid-day. Pre-dawn and mid-day leaf water potentials were significantly lower under drought than under control conditions. At mid-day, leaf water potential was higher in the tolerant cultivars LC and IR than in the sensitive cultivars NB and TP. Likewise, osmotic potential was significantly lower under drought than under control conditions, with the interesting exception of the tolerant cultivar LC that showed a very negative osmotic potential already under control conditions and no osmotic adjustment under drought stress.
During the first 4 days after plants were removed from the water, cultivars did not differ in the amount of water used per day (Table 3). After day four, when the daily water loss was depleted at the end of the light period, the total amount of water used per gram final plant dry weight was significantly lower in the tolerant cultivars IR and LC than in the sensitive cultivars NB and TP (Table 3). However, both tolerant cultivars depleted the soil water content to significantly lower values than the sensitive cultivars, indicating that the tolerant cultivars were not saving water but were rather using the available water more efficiently. Both cultivar groups differed clearly in their response to drought stress, whereas the cultivars within a group showed similar responses and where thus analyzed together as members of a ‘tolerance group’.
Genotyping of the cultivars
For all four cultivars, genotyping with subspecies specific STS markers was performed for six locations (Fig. 2). TP and NB showed, as expected, the length of the PCR product predicted for ssp. japonica, IR the amplicon length for ssp. indica. The fourth cultivar, LC, for which published pedigree information is missing, showed japonica specific lengths of the PCR products.

Gel picture of PCR amplicons for subspecies-specific STS markers S01022, S03020, S03136, S04128, S07011, S070103 with expected fragment sizes for japonica (J) and indica (I) cultivars. Cultivars Nipponbare (NB), Taipei (TP), LC-93-4 (LC) and IR57311 (IR)
Drought and drought × tolerance group effects on gene expression
To reduce the effect of biological variation between parallel plants on the within treatment variance, we pooled samples from four plants per experiment and cultivar. We used material from three independent experiments to allow stringent statistical data analysis. Genes with expression levels that were significantly affected by drought were identified by fitting a GLM. The final GLM included the main effects dye, condition (E-effect), tolerance group (G-effect), and the G × E interaction effect on the normalized expression level as response variables. Both tolerance groups contained two cultivars each. Genes were identified as significantly affected by tolerance group or condition, when the t-test on the normalized expression values had a fdr corrected p-value below 0.05 and the induction or repression factor was at least 1.5.
The number of genes that were significantly differentially expressed between sensitive and tolerant cultivars increased twofold to 225 genes under drought stress compared to 123 genes under control conditions. (Fig. 3a). Fifty genes were differentially expressed under both conditions.

Venn diagrams for groups of genes with significantly different expression levels. Numbers indicate the number of genes in a given group. Numbers in the circle overlap indicate the number of genes common to both compared groups, numbers outside the overlap indicate the number of genes exclusive to the group defined by the criteria given above the circle. a Comparison of significantly differently expressed genes between tolerant (T) and sensitive (S) cultivars under control condition (cT − cS) and under drought stress conditions (dT − dS). b Comparison of significantly drought-repressed genes in the tolerant group (dT − cT) and the sensitive group (dS − cS). c. Comparison of significantly drought-induced genes, abbreviations as in a. d Comparison of genes that were significantly drought induced or repressed in all cultivars taken together (d − c) with genes that showed a significant condition x tolerance group interaction effect
To identify genes that were generally affected by drought stress in our experiments, we compared mean gene expression values of plants from all four cultivars grown under drought conditions with those of plants from all cultivars grown under control conditions. Drought stress significantly induced 413 genes and repressed 245 genes. Among the genes most highly influenced under drought conditions were genes coding for metallothionein like protein (induction factor 35.2), and late embryogenesis abundant protein (induction factor 23.2). Five genes encoding cytochrome P450 family proteins and three genes encoding serine/threonine protein kinases were found to be highly drought induced as well. The genes that were most strongly repressed by drought stress were mostly coding for unknown or hypothetical proteins, among the known gene products were a putative EF hand and SANT/MYB domain containing protein (Supplemental Table S4).
To identify those genes that may be relevant for the differences in drought stress tolerance between cultivars, we compared the responses of the two tolerant and the two sensitive cultivars. Strikingly, the number of genes that were significantly up- or down-regulated under drought stress was much higher in the sensitive than in the tolerant cultivars (Fig. 3). The number of genes exclusively drought-repressed in the sensitive cultivars was twice the number of genes exclusively repressed in the tolerant cultivars (Fig. 3b). For the induced genes, almost six times as many were exclusively induced in the sensitive compared to the tolerant cultivars (Fig. 3c). To find genes that differed in their treatment-depended expression between cultivars of contrasting tolerance (genes with G × E effect), we singled out those genes that showed a significant t-test for the condition × tolerance group term contrast and an interaction factor (compare Materials and Methods) higher than 1.5. A significant G × E effect was found for 236 genes. Within this group, 78 genes were also significantly affected by drought when all cultivars were compared (Fig. 3d). Among the genes with a significant condition × tolerance group effect, almost three times as many were drought regulated in both sensitive cultivars than in both tolerant cultivars. This difference resulted from a much higher number of drought induced genes in the drought sensitive than in the drought tolerant cultivars.
Functional testing of selected gene lists
To identify the parts of metabolism mostly affected by drought stress in rice and responding differently in sensitive compared to tolerant cultivars, we used the published gene ontology annotation (http://rice.tigr.org) to sort the genes into metabolic groups. The physiological role of the products of those genes that were significantly induced or repressed in all cultivars under drought stress compared to control conditions was visualized with the software MapMan (Thimm et al. 2004) (Fig. 4).

Induction or repression (log-scale) of genes under drought treatment in all four cultivars encoding enzymes involved in metabolism grouped in functional bins according to MapMan. Red indicates down-regulated, blue up-regulated genes
For assignment of rice transcripts to MapMan bins, the already established Arabidopsis bin classification was used as a basis. In total, the translated sequences of 11,208 rice transcripts were compared (BLASTX; Altschul et al. 1990) to the TAIR Arabidopsis peptide database version 6. For the majority of transcripts (83%), an Arabidopsis hit with a blast E-value of less than 10E−10 was found, for 39% the E-value was even lower than 10E−100. Sixteen percent of the best blast hits had a rather poor similarity, with an E-value higher than 10E−10. The proportion of blasted sequences, for which no hit in the Arabidopsis peptide database was found, was very low (0.6%). For transcripts with a blast result with an E-value lower than 10E−10, the MapMan bin classification of the best Arabidopsis sequence homolog was used to assign those rice genes to a MapMan bin. Afterwards, classification was curated manually with the help of the rice annotation and gene ontology data, if available. Bin 35 “not assigned” that contains all genes with unknown function and restricted gene ontology information contained a higher percentage (47.5%) of the genes in the rice classification than in the Arabidopsis classification (38.5%). The second highest number of genes was classified to the bin “protein” (bin 29), followed by “RNA” (bin 27). Overall, distribution of known and expressed genes from the 20 K NSF array to bins is similar to the distribution for Arabidopsis MapMan bin classifications (Supplemental Table S5).
To identify those bins that were significantly affected by drought stress, we used two approaches. In the first approach, we calculated the induction factor of all genes in a bin and compared the average induction factor of a bin to that of all other bins by Wilcoxon rank sum test. In a second approach, we counted the genes whose expression was significantly influenced by the condition and the condition x tolerance interaction and used the Fisher exact test to determine whether induced or repressed genes are overrepresented in a bin compared to all other bins (Table 4). The average change in gene expression under drought stress compared to control conditions for all four cultivars is depicted in a MapMan graph (Fig. 4) to give an overview of the general regulation pattern of genes encoding enzymes involved in major biochemical pathways.
Under drought stress, we found a highly significant down-regulation of genes that code for proteins involved in the photosynthetic light reactions, especially those of photosystem II, both at the level of average induction factors as well as the number of repressed genes. Gene repression was furthermore found for isoprenoid metabolism and a number of protein synthesis bins, especially amino acid activation and synthesis of ribosomal proteins (Table 4). Concordantly, genes for amino acid and lipid degradation were up-regulated.
To identify the metabolic pathways that differed in drought-induced changes between tolerant and sensitive cultivars, we used the parameters induction factor calculated separately for drought tolerant and drought sensitive cultivars, and interaction factor. The absolute interaction factor is high when the compared cultivars show opposite responses, and close to zero when the compared cultivars show concordant responses. Bins were compared for both induction and interaction factors by Wilcoxon rank sum test. Again, the percentages of drought-induced or repressed genes or genes with a significant interaction effect within each bin were compared to the general distribution by Fisher exact test (Table 4). We identified a number of pathways, in which gene expression was differentially affected by drought in sensitive and tolerant cultivars. In the sensitive cultivars, genes for protein synthesis were strongly down-regulated, especially those genes coding for ribosomal proteins of plastids. The effect was not a consequence of the down-regulation of a few genes, but rather a response of all genes in the bin. Concordantly, genes for protein degradation were strongly up-regulated in the sensitive cultivars. The induction factor for cysteine proteases and ubiquitin E3 ligases were significantly higher than average for the sensitive cultivars whereas the tolerant cultivars showed no drought effect on the gene expression of these pathways. In the next steps after protein degradation, namely amino acid degradation and the metabolisation of the carbon bodies by the TCA cycle, up-regulated genes were overrepresented in the sensitive, but not in the tolerant cultivars. Likewise, genes for the lipid degradation pathway, which feeds into the TCA cycle as well, were significantly induced in the sensitive, but not in the tolerant cultivars. The overall picture is that the high number of genes differentially expressed under drought stress in drought sensitive and drought-tolerant cultivars indicates a shift of metabolism towards degradation pathways in sensitive cultivars.
Drought stress strongly down-regulated photosynthesis genes in both sensitive and tolerant cultivars. However, for the polypeptide subunits of photosystem I and photosystem II, the number of down-regulated genes was, surprisingly, higher in the tolerant than in the sensitive cultivars, in spite of the higher growth rate of the tolerant cultivars (Table 4). Drought repressed gene expression specifically in tolerant cultivars for photosystem II protein D2 and a photosystem II 44 kDa protein, two chlorophyll a/b binding proteins, the photosystem I reaction center subunits III and IX, ribulose bisphosphate carboxylase small subunit C and the alpha and beta chains of cytochrome b559 (Supplemental Table S4).
The genes of the cytochrome P450 bin (Table 4, bin 26.10), which contained one of the most highly drought induced genes, were generally up-regulated under drought stress. The induction factor of the entire gene family was significant in the tolerant but not in the sensitive cultivars, which makes this bin another candidate for pathways contributing to drought-tolerance. Five cytochrome P450 genes exhibited a significant G × E interaction effect on their expression level. Two cytochrome P450 cyp86A2 genes were induced under drought in the tolerant but not in the sensitive cultivars resulting in a significant positive G × E interaction. In contrast, cytochrome P450 76C2 was 10-fold induced in the sensitive, but just two-fold in the tolerant cultivars and thus showed a negative G × E interaction.
Among the other bins that contained the most highly drought induced genes, the bin with the LEA proteins (bin 33.2) was represented with too few genes on the slide to allow a general statement. The bin with the metallothionein genes (bin 15.2) contained a significantly higher number of up-regulated genes in the sensitive than in tolerant cultivars.
Thus, the analysis of drought effects on gene expression yielded two candidate bins that may contribute to improved performance of tolerant cultivars, namely the bins containing photosynthesis and cytochrome P450 genes.
Mapping of candidate genes to drought tolerance QTL and confirmation by quantitative RT-PCR (qRT-PCR)
To identify genes that localize to genomic regions contributing to drought tolerance under field conditions, we mapped our candidate genes to drought tolerance QTL available in the Gramene Database. Location of the QTL was estimated with the help of the flanking markers and QTL longer than 5 million bases were excluded.
Of the 236 genes with a significant G × E interaction, 108 (45.8%) fell into a published QTL (Table 5 and Fig. 5). Likewise, 44.5 % of the genes that were significantly affected by drought (E effect) fell into a QTL. Among the genes that had no significant effect of G × E or E, 42.8% fell into published QTL. The hypothesis that there is an overrepresentation of genes with significant effect in QTL is thus to be rejected with an error of p = 0.2. Genes that were drought affected in our climate chamber experiments were thus only slightly and not statistically significantly overrepresented in drought related QTL. However, as many of these QTL have been identified in field trials, the location of a candidate gene within a QTL increases the likelihood that the gene is relevant for drought tolerance under field conditions. We thus used the location within a QTL as an additional filter to narrow down the list of candidate genes gained from our climate chamber experiments.

Published QTL related to drought tolerance in rice with a size below 5 Mb and location of genes with a significant E effect (circles) or a significant G × E effect (squares). Red symbols indicate repression of gene expression or interaction factor <−1.5, blue symbol induction of gene expression or interaction factor >1.5
We chose 45 of the 108 QTL located genes with a significant G × E effect, based on the p-values, for an additional analysis by qRT-PCR (Table 6), using material from an independent experiment (#4). Due to the smaller number of plants sampled (three instead of 12), the test power was lower than in the statistical analysis of the array data. In the qRT-PCR analysis, 22 genes showed a significant G × E interaction at the p = 0.1 level. For 32 genes, the p-value was lower than 0.25 (Table 6). Among the genes with a significant G × E effect in both array and qRT-PCR analyses was a putative LEA protein, a MYB transcription factor and an ethylene responsive transcription factor, but also a number of genes with unknown function that would not have been identified as candidates in a search focused on functional categories.
Discussion
Physiological response of rice to drought stress
The aim of this study was to identify mechanisms with a general relevance for drought tolerance in rice by comparing cultivars that differ in tolerance to long-term drought stress. We focused on long-term stress, as we were most interested in mechanisms that contribute to performance of rice in an agronomic environment under upland growth conditions where drought stress often persists for a considerable time of the plant’s life cycle. For the varieties used in our study, life cycle is three to four months. A stress treatment of more than two weeks in the juvenile phase, which is an especially drought-sensitive growth stage (Banoc et al. 2000; Kamoshita et al. 2004), can thus be considered long-term. Seedling vigor, the ability to keep a high biomass alive during drought stress, has been shown to be essential for recovery and final yield in field and greenhouse experiments (Kamoshita et al. 2004). Mechanisms identified to keep the plant vital during drought stress in the juvenile stage are thus relevant for performance in a drought-prone environment.
The response of plants to stress will depend not only on the duration, but also on the degree of stress imposed. We used the parameters leaf water potential, growth reduction and drought score to characterize the degree of stress and to allow comparison with results from other experiments. The water potentials observed under drought stress in our experiments were comparable or higher (less negative) to those found in drought stress experiments under field conditions (Turner et al. 1986; Jongdee et al. 2002; Kamoshita et al. 2004). The reduction of shoot biomass by about 75% was more severe than in moderate drought stress trials that resulted in 25–50% yield loss (Babu et al. 2003; Fischer et al. 2003; Lanceras et al. 2004), but less severe than in terminal drought stress trials (Lafitte et al. 2006). Based on the drought score, the stress imposed in our experiments yielded less or similar damage than the stress treatment in field trials (Babu et al. 2003). Thus, the stress imposed can be classified as moderate to strong long-term drought stress comparable to stress under field trial conditions.
The relevant parameter for a stress tolerant crop is yield: varieties that produce more grain under stress than sensitive cultivars are considered tolerant (Fischer et al. 2003). The parameter yield cannot be determined in a short-term test like ours. We therefore used so-called secondary traits to estimate tolerance. The parameter absolute biomass at the end of the drought stress was chosen as it is associated with superior recovery ability after stress release (Fukai and Cooper, 1995; Kamoshita et al. 2004). The parameter drought score, which is based on leaf survival, was used as it correlates to yield and shows the best heritability of those secondary traits that can be scored in the vegetative stage (Fischer et al. 2003). Furthermore, we found a higher reproducibility of a tolerance classification based on these parameters compared to other parameters (e.g. PAM measurements, height, tiller numbers; data not shown).
Based on the secondary traits absolute biomass and drought score, 21 cultivars, including 17 Vietnamese cultivars from a breeding program for drought stress resistance, were characterized for drought tolerance in our experimental system. The two sensitive cultivars (NB and TP) and the two tolerant cultivars (LC and IR) were chosen as they showed the most stable response over three independent experiments. The characterization of drought tolerance was done in an experimental system with a low soil depth, in which water was supplied from above. This system mimics an upland field with a shallow soil layer and insufficient water supply by rain or irrigation. The effect of differences in rooting depth on the tolerance assessment, which is often linked to superior performance under drought conditions (Kamoshita et al. 2000; Wade et al. 2000), was reduced in the experimental system. Indeed, shoot:root ratios under drought stress did not differ significantly between cultivars. In spite of that, both tolerant cultivars depleted the soil water more than the sensitive cultivars. At the same time, the higher (less negative) mid-day water potentials in the tolerant cultivars suggest a lower degree of stress compared to the sensitive cultivars. This is confirmed by the higher harvest biomass and significantly higher water use efficiency in the tolerant compared to the sensitive cultivars. Thus, the tolerant cultivars were able to use more of the available water and use it more efficiently for dry matter production. Maintenance of a high transpiration rate during periods of severe drought correlates with a superior recovery of young plants when drought is released (Wade et al. 2000). Within a group of closely related double-haploid rice lines, not only high transpiration rates during drought stress were linked to drought tolerance, but also high water use efficiency (Siopongco et al. 2006). The adaptive mechanisms of LC and IR, that both show high water uptake and water use efficiency, are thus relevant for the selection of improved cultivars within the ‘more crop per drop’ strategy.
Drought effects on gene expression
Transcript profiles of leaf samples from control and drought stressed plants were generated to identify genes and pathways that may contribute to the higher tolerance and water use efficiency of LC and IR compared to NB and TP. The sequence data from one of these cultivars, Nipponbare (NB) are the basis of the gene models from the TIGR Rice Annotation, that were used to design the NSF oligonucleotide microarray. This array contains about 50% of the rice predicted genes models. As the oligonucleotides on the array are short (50–70 bases) and only a single oligonucleotide has been spotted per gene, sequence differences between the cultivars could result in a stronger hybridization of labelled cDNA from the japonica cultivars compared to the indica cultivar IR. Obviously, also the expression of genes in the indica cultivars that are not present in the japonica genome could not be detected with the arrays used in our study. We did not optimize the design of the experiments and data evaluation to identify constitutive differences in gene expression between tolerant and sensitive cultivars, although they could also be a source of increased stress tolerance.
We focused on genes that differed in their response to drought stress between two tolerant cultivars on the one hand and two sensitive cultivars on the other hand. In statistical terms, this means that we searched for genes showing a significant interaction effect between condition and tolerance group. To validate our method, we checked, whether genes that had previously been described as drought induced in rice or other monocots can be found among those that showed a significant effect of condition on expression in our experiments. Among the genes that were significantly drought induced, we indeed found metallothioneins and late embryogenesis abundant proteins that had previously been found to be induced in young rice plants under long-term drought stress (Reddy et al. 2002; Hazen et al. 2005; Markandeya et al. 2005, 2007) and in barley and Arabidopsis thaliana (Ozturk et al. 2002; Seki et al. 2002; Talame et al. 2007) under drought stress. Also, cytochrome P450 family proteins and serine/threonine protein kinases that were prominent among genes in EST libraries from drought-stressed rice plants (Reddy et al. 2002) showed a significant effect of condition in our study.
To facilitate a functional interpretation of the changes in gene expression of rice in response to drought stress, we used the published sequence of Oryza sativa cv. Nipponbare (Matsumoto et al. 2005) for a homology search to the Arabidopsis genome and sort the genes that we found expressed on the NSF array into functional categories, using the established MapMan bins. We used two statistical methods to identify those bins in which gene expression was strongly affected by drought. In the first approach, the mean induction factor for all genes in a bin was calculated and compared to the mean induction factors of all other bins. In the second approach, the percentage of genes with significantly changed expression in a bin was compared to the overall percentage of genes with significantly altered expression. Both approaches can lead to completely different but biologically meaningful results. If half of the genes in a bin are strongly repressed and the other half is strongly induced, the average induction factor will not be significantly different from zero. However, the percentage of differentially expressed genes will be 100% and therefore significantly different from the overall percentage of regulated genes. Such a pattern might be expected if expression of genes within a large family switches from a set of genes coding for nontolerant isoenzymes to stress tolerant isoenzymes. On the other hand, most of the genes in a family could be induced just below the set threshold and only a few above it. In this case, the percentage of significantly induced genes would not be different from the general mean, but the average induction factor for the bin could be significantly higher than the average over all other bins. As both situations, switch to different genes of a family and weak but concordant induction of many genes in a functional group, could be important for the identification of functional categories relevant for drought stress responses, we used both approaches.
Like other authors (Munne-Bosch and Alegre 2004; Hazen et al. 2005), we found strong evidence that drought stress causes a transition of metabolism from protein synthesis to degradation in rice. Amino acid activation and synthesis of ribosomal proteins were down-regulated, and amino acid and protein degradation, especially by the ubiquitin pathway, were up-regulated. Together with the general down-regulation of protein synthesis, genes coding for proteins of the photosynthetic light reactions were repressed as well, especially those of photosystem II. This corresponds to the visible bleaching of drought-stressed leaves and a decrease in photosynthetic activity (Do and Zuther, unpublished data). Photosystem II activity and its main regulatory mechanisms are severely affected by drought (Pieters and El Souki 2005). Down-regulation of photosynthesis genes under drought stress has been observed before in rice and barley under moderate long-term drought-stress in the field (Ozturk et al. 2002; Hazen et al. 2005) and under controlled conditions (Talame et al. 2007).
Differential response of tolerant and sensitive cultivars to drought stress
To identify genes that may be relevant for the differential drought tolerance of rice cultivars, we looked for genes that showed differences in expression between the tolerance groups identified by our physiological measurements. This search strategy implies that genes contributing to tolerance show different expression patterns in the tolerant compared to the sensitive cultivars.
To find such genes, we identified those that showed a significant t-test for the condition × tolerance group term and an interaction factor higher than 1.5. To identify the source of the interaction, we compared the expression in sensitive cultivars under control (cS) and under drought conditions (dS), and in tolerant cultivars under control and drought conditions (cT, dT). The number of genes that were significantly drought-induced was much higher in the group of sensitive than in the group of tolerant cultivars. (Hazen et al. 2005) also found large differences between cultivars in the number of drought affected genes. In controlled environment experiments, moderate and severe drought stress induced a higher number of genes in IR62266, which is considered to be tolerant under these conditions, than in CT9993, which is considered to be sensitive to drought (Hazen et al. 2005).
Intuitively, one might expect more changes in the tolerant cultivars, which should carry those genes that contribute to increased tolerance. In fact, this pattern has recently been observed in Arabidopsis accessions differing in freezing tolerance (Hannah et al. 2006). However, the sensitive genotypes could show more changes if the imposed degree of stress evoked additional, damage related responses that were not yet induced in the tolerant genotypes. This pattern has been observed in salt- stressed rice, where salt stress changed expression of many more genes in the sensitive than in the tolerant cultivars (Walia et al. 2005, 2007). These differences were attributed to the higher Na+ accumulation in the sensitive cultivars that required more adjustments of metabolism. For these damage related genes, the tolerant cultivars should show low expression levels under both control and stress conditions, whereas the sensitive cultivars should show increased expression under stress. The resulting interaction factor [(dT−cT)−(dS−cS)] for these genes would then be negative.
Alternatively, genes that contribute to drought tolerance could be constitutively highly expressed in the tolerant group. If these genes are not (or very lowly) expressed in the sensitive cultivars, they will not be reliably identified with our search strategy. If these tolerance genes are drought-induced in sensitive cultivars, a negative interaction factor will result. A negative interaction factor can thus result from both stress-damage induced gene expression and stress-induced expression of tolerance genes that are constitutively expressed in tolerant cultivars. In the former case, expression levels will be low in the tolerant cultivars, in the latter case high.
Most genes with a negative interaction factor code for enzymes involved in degradation pathways, namely of lipids and proteins, especially cysteine proteases. For these genes, expression levels were generally low under control conditions for all cultivars and increased in the sensitive cultivars under drought stress. This expression pattern indicates that genes are most likely associated to damage-related responses. A similar response has been found in Fabaceae, where the activity of proteolytic enzymes increases more under drought stress in sensitive than in tolerant species (Roy-Macauley et al. 1992). In addition to lipid and protein degradation, downstream catabolic pathways of degradation products were induced in sensitive cultivars. This expression pattern was found for genes coding for enzymes of amino acid degradation pathways and of the TCA cycle that may contribute to metabolizing products of lipid degradation and fumarate produced by the urea cycle during amino acid degradation. This suggests that up-regulation of many of these genes is related to stress induced damage in the sensitive group rather than a tolerance conveying response. This is emphasized by findings in wheat (Gregersen and Holm 2007) that genes coding for enzymes involved in protein degradation as well as fatty acid and carbohydrate breakdown are induced during leaf senescence. Induction of cysteine proteases and lipid degrading enzymes were reported as part of programmed cell death in senescing leaves (Lim et al. 2007). The same authors report a down-regulation of anabolic pathways, especially of protein synthesis, rRNA and tRNA during senescence. We also found a down-regulation of many genes coding for components of the protein synthesis pathway, especially ribosomal proteins, under drought stress in the sensitive cultivars. The tolerant cultivars were much less affected, as indicated by the significantly positive interaction term. The majority of genes that were induced by drought stress in sensitive but not tolerant cultivars are thus related to senescence rather than to stress tolerance mechanisms. This interpretation is in accordance with the visual phenotype of the plants: sensitive cultivars showed yellowing and partial leaf death under drought stress, whereas the leaves of tolerant cultivars remained green. Recently, a remarkable increase of drought tolerance has been shown in plants, in which drought-induced leaf senescence was suppressed by the overexpression of isopentenyltransferase under the promoter of a senescence associated receptor protein kinase (Rivero et al. 2007). This stresses that the difference in the expression of senescence related genes between sensitive and tolerant cultivars is more than a side effect and may actually actively contribute to drought sensitivity.
As a case study for constitutively expressed tolerance genes, we compared gene expression in LC, which had a constitutively low leaf water potential, to the other cultivars by contrast analysis. Only 17 genes were significantly higher expressed in LC than in the other cultivars and showed a significant induction under drought in the latter. As especially the first comparison has a high type II error risk, the number of genes that show this expression pattern may be considerably higher. With the exception of an amino acid transporter, none of these 17 genes was involved in the synthesis or transport of known compatible solutes, although genes for trehalose, inositol and proline metabolism and 36 amino acid transporters were represented on the chip and expressed in the leaf tissues of the cultivars.
In contrast, there are some genes and gene groups, for which tolerant cultivars show more change and these are the interesting candidates for tolerance related processes. One candidate process whose regulation may contribute to drought tolerance is photosynthesis. Amounts of thylakoid membrane proteins were reduced (data not shown) and genes coding for PSI and PSII subunits were down-regulated by drought stress in all cultivars. The reduction of gene expression suggests that the observed decrease of photosynthetic capacity was not only due to drought induced damage of the photosynthetic apparatus, but may be a regulatory response. The number of significantly down-regulated photosynthesis-related genes is indeed higher in the tolerant than in the sensitive group, indicating a role for this regulation in drought tolerance. It is nevertheless unexpected, as the tolerant cultivars produced more biomass (Fig. 1) and had a higher photosynthetic capacity (data not shown) under drought conditions than the sensitive cultivars. A down-regulation of photosynthetic genes in the tolerant cultivars may therefore indicate an adaptive response to prevent photodamage during times of reduced CO2 availability in the mesophyll when stomata are closed due to water shortage. Reduction of photosynthesis is by no means drought specific, but is observed under heat, salt and chilling stress as well (Sayed 2003; Yan et al. 2006). In fact, photosynthesis-related genes are found to be massively repressed in Arabidopsis after a shift to low growth temperature (Hannah et al. 2005) and the magnitude of this repression is positively correlated with the freezing tolerance of different accessions (Hannah et al. 2006). However, drought stress seems to specifically act on proteins of the light harvesting complex of photosystem II (Sayed 2003). In agreement with this, additional photosynthesis measurements on the four cultivars (results not shown) revealed specific changes in the photosynthetic electron transport chain in response to drought. Thus, investigating the regulation of photosynthesis under drought stress may yield important insights into drought tolerance mechanisms.
Within the second candidate group, the cytochrom P450 genes, two cyp86A2 genes were induced under drought in the tolerant but not in the sensitive cultivars. In Arabidopsis, CYP86A2 catalyze the oxidation of fatty acids and are involved in the biosynthesis of extracellular lipids and cuticule development (Xiao et al. 2004). CYP86A2 transcripts are increased under various stress conditions including drought (Duan and Schuler 2005) and co-expressed among others with genes encoding enzymes involved in the TCA cycle, fatty acid elongation, wax and cutin metabolism (Ehlting 2006; Ehlting et al. 2008). In rice, epicuticular wax content is low but genetic variation of the amount exists (O’Toole and Cruz 1983). Induction of cuticula biosynthesis under drought could thus reduce non-stomatal water loss in the tolerant cultivars and thereby contribute to the observed increased water use efficiency. In Arabidopsis, cyp86A2 is furthermore coexpressed with genes coding for chlorophyll biosynthesis and photosystems, which suggests a link to the second process that has been identified as relevant for rice drought tolerance (Ehlting 2006).
Cytochrom P450 76C2, which is more highly induced in the sensitive than in the tolerant cultivars, is known to be induced during hypersensitive and developmental cell death, senescence and also under drought stress (Godiard et al. 1998; Narusaka et al. 2004), stressing the significance of both differences in P450 protein regulation and senescence associated processes for the drought-tolerance of rice.
Candidate selection by comparison with known QTL
In contrast to the drought-induced genes, many of which are functionally annotated, the genes with the highest repression factors were mostly of unknown or putative function. These genes could be as relevant for drought-tolerance as the highly induced genes, however, they are obviously much more difficult to interpret and much more time consuming to study functionally. To narrow down the list of genes with a significant G × E interaction to those that could be relevant in an agronomical environment, we compared their positions with published QTL, a strategy that has been successfully used before (Wayne and McIntyre 2002; Hazen et al. 2005). Indeed, four of the six cytochrome P450 genes that showed a significant G × E effect and the most highly induced gene encoding a late embryogenesis abundant protein are located in QTL. Among the five metallothionein-like protein genes represented on the chip, four, including the most highly drought induced gene, co-locate with drought QTL. Thus, the approach may yield interesting candidates for further functional studies, e.g. through transgenic approaches. The candidate list could be further narrowed down by checking candidate gene expression in DH or RIL lines characterized for their contrasting drought tolerance in the QTL region of interest. This strategy could also include genes of unknown function and thus open up the chance to discover truly unknown genes that are relevant for drought stress tolerance. The feasibility of confirming the expression pattern of such genes identified by array experiments in independent plant material has been shown in our study using qRT-PCR. In spite of the false positive risk in the array study and the high type II error in the qRT-PCR study, a significant interaction was confirmed for half of the genes. Furthermore, interaction coefficients calculated from microarray data and qRT-PCR correlated closely (data not shown). In further studies (Degenkolbe et al., manuscript in preparation), we tested the relevance of these candidate genes in an association-type approach by measuring their expression in a range of more than 20 rice cultivars of varying drought tolerance from different genetic backgrounds. Furthermore, this approach will unravel potential associations between the candidate gene and the parameters used for tolerance determination (MacNair 1993).
Abbreviations
- GLM:
-
General linear model
- GOI:
-
Gene of interest
- NSF:
-
National science foundation
- PWP:
-
Permanent wilting point
- qRT-PCR:
-
Quantitative real time polymerase chain reaction
- QTL:
-
Quantitative trait loci
- SS-STS:
-
Subspecies-specific sequence tagged site
References
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Babu RC, Nguyen BD, Chamarerk V et al (2003) Genetic analysis of drought resistance in rice by molecular markers: association between secondary traits and field performance. Crop Sci 43:1457–1469
Banoc DM, Yamauchi A, Kamoshita A et al (2000) Dry matter production and root system development of rice cultivars under fluctuating soil moisture. Plant Prod Sci 3:197–207
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300
Blum A, Zhang J, Nguyen HT (1999) Consistent differences among wheat cultivars in osmotic adjustment and their relationship to plant production. Field Crops Res 64:287–291. doi:10.1016/S0378-4290(99)00064-7
Chin J-H, Kim J-H, Jiang W et al (2007) Identification of subspecies-specific STS markers and their association with segregation distortion in rice (Oryza sativa L.). J Crop Sci Biotech 10:175–184
Clark LJ, Cope RE, Whalley WR et al (2002) Root penetration of strong soil in rainfed lowland rice: comparison of laboratory screens with field performance. Field Crops Res 76:189–198. doi:10.1016/S0378-4290(02)00039-4
Degenkolbe T, Hannah MA, Freund S et al (2005) A quality-controlled microarray method for gene expression profiling. Anal Biochem 346:217–224
Duan H, Schuler MA (2005) Differential expression and evolution of the Arabidopsis CYP86A subfamily. Plant Physiol 137:1067–1081. doi:10.1104/pp.104.055715
Ehlting J (2006) CYPedia. http://www-ibmp.u-strasbg.fr/~CYPedia/
Ehlting J, Sauveplane V, Olry A et al (2008) An extensive (co-)expression analysis tool for the cytochrome P450 superfamily in Arabidopsis thaliana. BMC Plant Biol 8:47. doi:10.1186/1471-2229-8-47
Fischer KS, Lafitte R, Fukai S et al (eds) (2003) Breeding rice for drought-prone environments. International Rice Research Institute, Los Banos (Philippines)
Fukai S, Cooper M (1995) Development of drought-resistant cultivars using physiomorphological traits in rice. Field Crops Res 40:67–86. doi:10.1016/0378-4290(94)00096-U
Godiard L, Sauviac L, Dalbin N et al (1998) CYP76C2, an Arabidopsis thaliana cytochrome P450 gene expressed during hypersensitive and developmental cell death. FEBS Lett 438:245–249. doi:10.1016/S0014-5793(98)01309-X
Gregersen PL, Holm PB (2007) Transcriptome analysis of senescence in the flag leaf of wheat (Triticum aestivum L.). Plant Biotechnol J 5:192–206. doi:10.1111/j.1467-7652.2006.00232.x
Hannah MA, Heyer AG, Hincha DK (2005) A global survey of gene regulation during cold acclimation in Arabidopsis thaliana. PLoS Genet 1:e26. doi:10.1371/journal.pgen.0010026
Hannah MA, Wiese D, Freund S et al (2006) Natural genetic variation of freezing tolerance in Arabidopsis. Plant Physiol 142:98–112. doi:10.1104/pp.106.081141
Hanson AD, Rathinasabapathi B, Rivoal J et al (1994) Osmoprotective compounds in the Plumbaginaceae: a natural experiment in metabolic engineering of stress tolerance. Proc Natl Acad Sci USA 91:306–310. doi:10.1073/pnas.91.1.306
Hazen SP, Pathan MS, Sanchez A et al (2005) Expression profiling of rice segregating for drought tolerance QTLs using a rice genome array. Funct Integr Genomics 5:104–116. doi:10.1007/s10142-004-0126-x
Jongdee B, Fukai S, Cooper M (1998) Genotypic variation for grain yield of rice under water-deficit conditions. In: Michalk D, Pratley J (eds) Proceedings of 9th Australian Agronomy Conference, Wagga Wagga, pp 403–406
Jongdee B, Fukai S, Cooper M (2002) Leaf water potential and osmotic adjustment as physiological traits to improve drought tolerance in rice. Field Crops Res 76:153–163. doi:10.1016/S0378-4290(02)00036-9
Kamoshita A, Wade LJ, Yamauchi A (2000) Genotypic variation in response of rainfed lowland rice to drought and rewatering: III. Water extraction during the drought period. Plant Prod Sci 3:189–196
Kamoshita A, Rodriguez R, Yamauchi A et al (2004) Genotypic variation in response of rainfed lowland rice to prolonged drought and rewatering. Plant Prod Sci 7:406–420. doi:10.1626/pps.7.406
Köhl K (1996) Population-specific traits and their implication for the evolution of a drought-adapted ecotype in Armeria maritima. Bot Acta 109:206–215
Lafitte R (2002) Relationship between leaf relative water content during reproductive stage water deficit and grain formation in rice. Field Crops Res 76:165–174. doi:10.1016/S0378-4290(02)00037-0
Lafitte HR, Li ZK, Vijayakumar CHM et al (2006) Improvement of rice drought tolerance through backcross breeding: evaluation of donors and selection in drought nurseries. Field Crops Res 97:77–86. doi:10.1016/j.fcr.2005.08.017
Lanceras JC, Pantuwan G, Jongdee B et al (2004) Quantitative trait loci associated with drought tolerance at reproductive stage in rice. Plant Physiol 135:384–399. doi:10.1104/pp.103.035527
Landgrebe J, Bretz F, Brunner E (2006) Efficient design and analysis of two colour factorial microarray experiments. Comput Stat Data Anal 50:499–517. doi:10.1016/j.csda.2004.08.014
Levitt J (1972) Responses of plants to environmental stresses. Academic Press, New York
Li ZK, Fu BY, Gao YM et al (2005) Genome-wide introgression lines and their use in genetic and molecular dissection of complex phenotypes in rice (Oryza sativa L.). Plant Mol Biol 59:33–52. doi:10.1007/s11103-005-8519-3
Lim PO, Kim HJ, Gil Nam H (2007) Leaf senescence. Annu Rev Plant Biol 58:115–136. doi:10.1146/annurev.arplant.57.032905.105316
MacNair M (1993) The genetics of metal tolerance in vascular plants. New Phytol 124:541–559. doi:10.1111/j.1469-8137.1993.tb03846.x
Markandeya G, Babu PR, Lachagari VBR et al (2005) Functional genomics of drought stress response in rice: transcript mapping of annotated unigenes of an indica rice (Oryza sativa L. cv. Nagina 22). Curr Sci 89:496–514
Markandeya G, Babu PR, Reddy Lachagari VB et al (2007) Identification of stress-responsive genes in an indica rice (Oryza sativa L.) using ESTs generated from drought-stressed seedlings. J Exp Bot 58:253–265
Matsumoto T, Wu JZ, Kanamori H et al (2005) The map-based sequence of the rice genome. Nature 436:793–800. doi:10.1038/nature03895
Mitchell JH, Siamhan D, Wamala MH et al (1998) The use of seedling leaf death score for evaluation of drought resistance of rice. Field Crops Res 55:129–139. doi:10.1016/S0378-4290(97)00074-9
Mitra J (2001) Genetics and genetic improvement of drought resistance in crop plants. Curr Sci 80:758–763
Munne-Bosch S, Alegre L (2004) Die and let live: leaf senescence contributes to plant survival under drought stress. Funct Plant Biol 31:203–216. doi:10.1071/FP03236
Narusaka Y, Narusaka M, Seki M et al (2004) Crosstalk in the responses to abiotic and biotic stresses in Arabidopsis: analysis of gene expression in cytochrome P450 gene superfamily by cDNA microarray. Plant Mol Biol 44:327–342. doi:10.1007/s11103-004-0685-1
O’Toole J, Cruz RT (1983) Genotypic variation in epicuticular wax of rice. Crop Sci 23:392–394
Ozturk ZN, Talame V, Deyholos M et al (2002) Monitoring large-scale changes in transcript abundance in drought- and salt-stressed barley. Plant Mol Biol 48:551–573. doi:10.1023/A:1014875215580
Pieters AJ, El Souki S (2005) Effects of drought during grain filling on PSII activity in rice. J Plant Physiol 162:903–911. doi:10.1016/j.jplph.2004.11.001
Price A, Cairns J, Horton P et al (2002) Linking drought-resistance mechanisms to drought avoidance in upland rice using a QTL approach: progress and new opportunities to integrate stomatal and mesophyll responses. J Exp Bot 53:989–1004. doi:10.1093/jexbot/53.371.989
Ramakers C, Ruijter J, Lekanne Deprez R et al (2003) Assumption-free analysis of quantiative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66. doi:10.1016/S0304-3940(02)01423-4
Reddy AR, Ramakrishna W, Sekhar AC et al (2002) Novel genes are enriched in normalized cDNA libraries from drought-stressed seedlings of rice (Oryza sativa L. subsp. indica cv. Nagina 22). Genome 45:204–211. doi:10.1139/g01-114
Rensink WA, Buell CR (2005) Microarray expression profiling resources for plant genomics. Trends Plant Sci 10:603–609. doi:10.1016/j.tplants.2005.10.003
Rivero RM, Kojima M, Gepstein A et al (2007) Delayed leaf senescence induces extreme drought tolerance in a flowering plant. Proc Natl Acad Sci USA 104:19631–19636. doi:10.1073/pnas.0709453104
Roy-Macauley H, Zuily-Fodil Y, Kidric M et al (1992) Effect of drought stress on proteolytic activities in Phaseolus and Vigna leaves from sensitive and resistant plants. Physiol Plant 85:90–96. doi:10.1111/j.1399-3054.1992.tb05268.x
Sayed OH (2003) Chlorophyll fluorescence as a tool in cereal crop research. Photosynthetica 41:321–330. doi:10.1023/B:PHOT.0000015454.36367.e2
Seki M, Narusaka M, Ishida J et al (2002) Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and hing-salinity stresses using a full-lenght cDNA microarray. Plant J 31:279–292. doi:10.1046/j.1365-313X.2002.01359.x
Siopongco JDLC, Yamatichi A, Salekdeh H et al (2006) Growth and water use response of doubled-haploid rice lines to drought and rewatering during the vegetative stage. Plant Prod Sci 9:141–151. doi:10.1626/pps.9.141
Smyth G (2005) Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W (eds) Bioinformatics and computational biology solutions using R and bioconductor. Springer, New York, pp 397–420
Talame V, Ozturk NZ, Bohnert HJ et al (2007) Barley transcript profiles under dehydration shock and drought stress treatments: a comparative analysis. J Exp Bot 58:229–240. doi:10.1093/jxb/erl163
Thimm O, Blaesing O, Gibon Y et al (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37:914–939. doi:10.1111/j.1365-313X.2004.02016.x
Turner NC (1981) Techniques and experimental approaches for the measurement of plant water status. Plant Soil 58:339–366. doi:10.1007/BF02180062
Turner NC, Jones MM (1980) Turgor maintenance by osmotic adjustment: a review and evaluation. In: Turner NC, Kramer PJ (eds) Adaptations of plants to water and high temperature stress. Wiley, New York, pp 87–103
Turner NC, O’Toole JC, Cruz RT et al (1986) Responses of seven diverse rice cultivars to water deficits I. Stress development, canopy temperature, leaf rolling and growth. Field Crops Res 13:257–271. doi:10.1016/0378-4290(86)90027-4
Usadel B, Nagel A, Thimm O et al (2005) Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of coresponding genes, and comparison with known responses. Plant Physiol 138:1195–1204. doi:10.1104/pp.105.060459
Usadel B, Nagel A, Steinhauser D et al (2006) PageMan: an interactive onotology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics 7:535. doi:10.1186/1471-2105-7-535
Wade LJ, Kamoshita A, Yamauchi A et al (2000) Genotypic variation in response of rainfed lowland rice to drought and rewatering: I. Growth and water use. Plant Prod Sci 3:173–179
Walia H, Wilson C, Condamine P et al (2005) Comparative transcriptional profiling of two contrasting rice genotypes under salinity stress during the vegetative growth stage. Plant Physiol 139:822–835. doi:10.1104/pp.105.065961
Walia H, Wilson C, Zeng L et al (2007) Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Mol Biol 63:609–623. doi:10.1007/s11103-006-9112-0
Wayne M, McIntyre L (2002) Combining mapping and arraying: an approach to candidate gene identification. Proc Natl Acad Sci USA 99:14903–14906. doi:10.1073/pnas.222549199
Xiao F, Goodwin SM, Xiao Y et al (2004) Arabidopsis CYP86A2 represses Pseudomonas syringae type III genes and is required for cuticle development. EMBO J 2004:2903–2913. doi:10.1038/sj.emboj.7600290
Xu K, Xu X, Fukao T et al (2006) Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 442:705–708. doi:10.1038/nature04920
Yan S-P, Zhang Q-Y, Tang Z-C et al (2006) Comparative proteomic analysis provides new insights into chilling stress responses in rice. Mol Cell Proteomics 5:484–496. doi:10.1074/mcp.M500251-MCP200
Zhang J, Nguyen HT, Blum A (1999) Genetic analysis of osmotic adjustment in crop plants. J Exp Bot 50:291–302. doi:10.1093/jexbot/50.332.291
Zhang J, Klueva NY, Wang Z et al (2000) Genetic engineering for abiotic stress resistance in crop plants. In Vitro Cell Dev Biol Plant 36:108–114. doi:10.1007/s11627-000-0022-6
Acknowledgements
We gratefully acknowledge helpful suggestions and support from Dr. Lothar Willmitzer, fruitful discussion especially on the Limma package with Dr. Matthew Hannah and on the MapMan/PageMan software with Dr. Björn Usadel, all from the MPIMP. We thank Dr. Le Tran Binh from the Institute of Biotechnology, Hanoi, Vietnam and Ruaraidh Sackville Hamilton and Gary Atlin from the IRRI, Manila, Phillipines, for providing rice germplasm and Camilla Caldana (MPIMP) for primers of the housekeeping genes for qRT-PCR. We thank Dr. J.E. Chin (IRRI) and Dr. Rhonda Meyer (University of Potsdam) for helpful suggestions on genotyping. Financial support for this project was provided by the Max-Planck-Society and the German Ministry for Education and Research (BMBF) through grant BMBF 0312854.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Expression profile data are available from the NCBI GEO repository at accession number GSE7766.
Electronic supplementary material
Table S4
TIGR locus identifier and annotation of all genes with a significant effect of condition (drought, probability p d-c) or condition × tolerance (probability p inter) on gene expression. OligoID = Identifier of the clone on the NSF Array, Loc_Id location identifier, coefficient for the normalized expression contrasts of d ( all samples from drought) and c (all samples from control), S (all samples from sensitive cultivars) and T (all samples from tolerant cultivars) and the respective subgroup (dS = samples from drought treated, sensitive cultivars), inter = interaction. Significance for the contrasts is -1 if p < 0.01 and coefficient < -1.5, 1 if p > 0.01 and coefficient > 1.5, 0 for all other cases. MapManBin = number of the MapMan bin, MapMan Bin Name = Name of the MapMan bin. Annotation TIGR Version 4xx = Annotation based on TIGR Rice Genome Annotation Release (http://www.tigr.org/tdb/e2k1/osa1/). QTL ID = Gramene identifier of the QTL in which the gene is located (XLS 555 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Degenkolbe, T., Do, P.T., Zuther, E. et al. Expression profiling of rice cultivars differing in their tolerance to long-term drought stress. Plant Mol Biol 69, 133–153 (2009). https://doi.org/10.1007/s11103-008-9412-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-008-9412-7