
Abstract
This paper reports the results obtained from sulphidation tests on a low-alloyed steels 15HM (T/P12), 16M (T/P1), 18K and 10HM2 (T/P22) in the temperature range 450–550 °C for 100 h. Tests were conducted using a gas mixture of 1%H2S 99% Ar at 1 bar. The results indicate that the low-alloyed steels underwent a high degree of corrosion degradation due to the formation of an Fe1−XS scale. The scale thicknesses and chemical compositions of the scales formed on the exposed samples were analysed by means of standard techniques, including scanning electron microscopy coupled with energy X-ray dispersive spectroscopy. In addition, a phase diagram for the Fe–S system was calculated using FACTSAGE software to aid interpretation of the results. Based on the study conducted, relevant defect equations were proposed.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Development of modern technologies and enrichment of societies around the globe requires higher amounts of electricity outputs from power stations. However, this demand can be met only when much better steels are employed. A new generation of steels need to be able to withstand the harsh conditions they are subjected to during long-term exposure in an oxidising or reducing atmosphere. Currently, many pulverised coal-fired power plants from the 70’s operate in conditions where steam temperature reaches 540 °C with a steam pressure up to 140 bar producing an efficiency of 35% [1]. Many of those power units in Europe, UK, USA, Poland and China burn coal containing different amounts of sulphur in organic and inorganic forms [2]. Generally, coal is mined in four different ranks: lignite, subbituminous, bituminous and anthracite. Lignite gives the lowest energy outcome during burning, whereas anthracite is the hardest coal with greatest amount of energy stored (92.1–98%) [3]. Furthermore, lignite is the youngest coal in terms of geological lifespan; such a type of coal contains significant amounts of volatile matter like methane (CH4), hydrogen and carbon monoxide as well as incombustible gases such as carbon dioxide (CO2) and nitrogen (N2). Lignite contains moisture and low amounts of fixed coal producing relatively small amounts of ash upon burning (4.2 wt%). In contrast, anthracite possesses low amounts of volatile matter and moisture, but the highest amount of fixed carbon, producing a relatively high amount of ash upon burning (9–20 wt%) causing a reduction in burning capacity, increased handling costs and a lower combustion and boiler efficiency through the clinkering and slagging processes [3]. The formation of H2S depends on sulphur content, conditions during high-temperature exposure and volatile matter in coal [4]. Burning of coal containing sulphur often leads to the formation of sulphide scales on the high-temperature components. Such scales are much less protective than oxide scales, and metal sulphidation rates are much higher than oxidation rates [5,6,7,8]. Particular attention needs to be paid to the section where highly reducing atmospheres are formed. In pulverised coal-fired power plants, a low excess air and two-stage combustion process is used in order to minimise NOx formation [9]. The region between burners and air ports forms an aggressive reducing atmosphere rich in H2S, which invokes corrosion of the waterwall tubes [10]. Likewise, and as previously mentioned, coal containing sulphur produces harsh reducing conditions upon burning. Such an atmosphere promotes the formation of highly aggressive hydrogen sulphide (H2S) leading to accelerated sulphidation and hence fast corrosion degradation of low-alloyed steels [10]. Due to the low price, good weldability, high thermal conductivity and low thermal expansion coefficient (CTE), low-alloyed ferritic steels are widely used in pulverised coal-fired boilers for waterwall tubes and other sections working under relatively low temperatures (e.g. economizer) [11, 12]. These alloys possess a low Cr content, not exceeding 3 wt%. Ferritic steels with low Cr content in the metal matrix show susceptibility to severe high-temperature degradation due to an inability to form an outermost protective scale, such as Cr2O3, or a slightly less protective FeCr2O4 [13]. Due to the above-mentioned issues, corrosion in the presence of H2S is still a major concern. Moreover, there is currently little on-going research concerned with sulphide corrosion invoked by H2S in part due to aspects such as health and safety [14, 15]. In this matter, the present study shows the outcomes of high-temperature studies performed on low-alloyed ferritic steels 18K, 16M (T/P1), 15HM (T/P12) and 10H2M (T/P22) in the temperature range 450–500 °C for 100 h.
Experimental Procedures
Test specimens with dimensions of 1 × 1 cm and a thickness of 4 mm were cut from unused pipe segments. The samples were cut using a diamond cut-off wheel. Following the cutting process, the samples were ground with different SiC papers (200, 600 and 1200) and then polished with diamond suspension paste (1 and 3 μm). The dimensions of the polished samples were accurately measured using a micrometre (length of sample, wall thickness, chord length). A further step in the sample preparation procedure involved ultrasonic cleaning for 15 min at 40 °C. Finally, the samples were weighed using an analytical balance (SARTORIUS CPA-225D). Nominal chemical compositions of the low-alloyed steels used in this work, according to Polish Norm: PN-75/H-84024, are provided in Table 1.
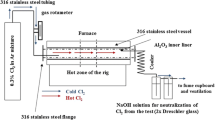
Sulphidation tests of the low-alloyed ferritic steels 18K, 16M (T/P1), 15HM (T/P12) and 10H2M (T/P22) were performed for 100 h total. The tests were comprised of 4 × 25 h cycles at 450, 500 and 550 °C. According to the illustration presented in Fig. 1, 1% H2S-99% Ar at 1 bar pressure mixture was delivered directly to the furnace.

Schematic illustration of 1% H2S-99% Ar mixture experimental test rig for high-temperature tests at 450, 500 and 550 °C for 100 h
For precise gas flow control, an accurate rotameter was used. The gas mixture was constantly controlled via an on-line camera set-up using gDMSS HD Lite software. The rotameter was placed between the gas cylinder and the furnace, and the flow rate was set to 50 Nml/min (Nml/min at 20 °C, 1 bar—standard conditions). The low-alloyed steel test samples were placed on a double deck Al2O3 (99.5%) holder and moved into the hot zone of a tube furnace (CARBOLITE). The hot zone of the furnace was controlled using a programmable controller from EUROTHERM. Prior to the high-temperature sulphidation tests, the furnace was carefully calibrated for testing of three samples at once. For health and safety reasons [14, 15], the reaction zone was contained in a 316 stainless-steel vessel capped at both ends with 316 stainless-steel flanges locked by four screws. In addition, the stainless-steel vessel was lined with an Al2O3 tube. The exiting gas mixture was passed through a NaOH solution for scrubbing and then directed to a ventilation system. Post-exposure investigations were carried out using a single-lens DSLR camera Canon EOS 70D camera coupled with a Canon MP-E 65 mm f/2,8 macro-lens and two scanning electron microscopes. Low magnification images were captured using a Hitachi 3000 tabletop microscope operated in backscatter electron mode (BSE). Higher magnifications of the cross-sectioned samples were carried out using a SCIOS FEI Dual Beam SEM. Chemical analyses of the exposed samples were conducted by means of energy X-ray dispersive spectrometry (EDS) from Bruker and EDAX. Standard mass change data were collected using a highly accurate scale, and data were collected every 25 h.
Results and Discussion
Macro-images
Macro-images of the exposed low-alloyed steels are presented in Figs. 2, 3, 4 and 5. In general, the presented images showed that the exposed samples underwent catastrophic corrosion degradation due to the formation of highly porous, non-protective sulphide scales that exfoliate from the metallic core of the substrate. The initial stages of sulphidation of the low-alloyed steels indicated that a tri-sulphur radical anion (S−3) was formed at 450 °C in the first 25 h providing a blue coloured tint to the sulphide scale. It is interesting to note that 10HM2 (T/P22) steel showed the most intense blue colour. The presence and thermodynamical stability of a tri-sulphur radical anion (S−3) at high temperatures is limited; at higher temperatures (500 °C) the low-alloyed steels indicated the formation of red sulphide scale. Exposure up to 75 h leads to the formation of a grey iron sulphide scale then further, a mixed grey–orange sulphide scale (100 h). Simultaneously, mass gain of the exposed low-alloyed steels accelerates significantly. Macro-observations of the surface changes under sulphidation of the samples are shown in Figs. 2, 3, 4 and 5. The observed results indicate that acceleration of mass gain in low-alloyed steels is related to porous, cracked sulphide scale structure where S access from the ambient atmosphere is unlimited. Such behaviour can be considered only when linear growth of the scale is considered. In general, at high temperatures when scale is thin and protective, corrosion rates decrease with time (parabolic behaviour) due to an increasing scale thickness acting as a stronger diffusion barrier. However, due to the formation of highly porous, poorly adherent or cracked non-protective layers as was observed during this study, corrosion rates remain linear. The low-alloyed steels are unable to form a thin protective scale in an aggressive atmosphere due to their low Cr content and tend to corrode according to linear kinetics. Finally, it is important to note that rate of sulphide scale growth exceeds the spallation process of the scale. Kinetic curves at every temperature show no mass gain, suggesting that sulphide scale in the present conditions developed at a high rate.

18K steel high-temperature corrosion induced by 1%H2S in Ar

16M (T/P1) steel high-temperature corrosion induced by 1%H2S in Ar

15HM (T/P12) steel high-temperature corrosion induced by 1%H2S in Ar

10H2M (T/P22) steel high-temperature corrosion induced by 1%H2S in Ar
Kinetic Data
Kinetic data obtained at 450, 500 and 550 °C are shown in Fig. 6a–c, respectively. The measured results clearly indicate that all low-alloyed steels showed very similar high-temperature sulphidation behaviour. At the highest temperature (550 °C), the mass gain was the largest. There was no apparent spallation of the formed sulphide scales, at every temperature tested even when mass gain was at the level of 30 mg/cm2. Moreover, the tested samples showed an acceleration of mass gain after 75 h of exposure at every temperature. The observed changes in mass gain originate from the formation of a more porous sulphide scale, where more H2S gas makes contact with the metallic core. It is interesting to note that steel 18K showed a continuously increasing rate at 500 °C, though mass change of the steel was slightly lower than that observed at a lower temperature 450 °C. A similar behaviour was observed by Zurek et al. [16], during oxidation of T91 ferritic martensitic steel with around 9 wt% Cr. Even so, the results shown here indicate that a sulphur-rich atmosphere induces abnormal corrosion kinetic behaviour of low-alloyed steel. Due to negligible differences in chemical composition (Cr content), the exposed steels showed similar high-temperature behaviour. In this study, all steels showed two cases in terms of high-temperature behaviour:

Kinetic curves for the samples exposed in 1% H2S gas in 99%Ar at high temperature a 450, b 500 and c 550 °C for 100 h
-
(1)
If a compact sulphide scale formed, it will be diffusion-controlled and sulphidation follows parabolic law (initial stages of sulphidation).
-
(2)
If a porous sulphide scale formed, it will be chemical reaction-controlled and then sulphidation will obey a linear law (rest of the sulphidation process).
The calculated exponents for all materials and temperatures are summarised in Table 2. In general, all n factors are between n = 1–2, but calculated values are closer to n = 1, suggesting that low-alloyed steels underwent more linear behaviour than parabolic.
The obtained results are slightly in contradiction to the previous work presented by Hauffe and Rahmel [17]. Those authors found that sulphidation of iron-based materials, i.e. low-alloy steels in wide range of temperatures and pressures, proceeds according to parabolic law only via outward diffusion of iron ions throughout ionised cationic gaps. The formation of sulphide scales and the rate of sulphidation depend mainly on macro- and microstructures. Where a scale is porous, a sulphur flux from an ambient atmosphere to the metal surface is high; hence, the scale formation is determined by chemical reaction at the substrate–sulphide scale interface. Such postulate is in contradiction to the overall theory, where linear kinetics are determined by chemical reaction in the interface between scale and the atmosphere, whereas diffusion process throughout compact adherent scale determines parabolic rate of corrosion.
As this study is concerned with low-alloyed steels with distinct additions of Mn, Mo and other elements, it should be noted that the rate of sulphidation of such materials is similar to the sulphidation of pure metal (Fe). The sulphidation process in this work was not influenced by hydrogen because the concentration of defects in the sulphides of Fe was too high to be affected by hydrogen dissolution in the growing scale [18]. Furthermore, based on nominal chemical composition of the low-alloyed steels as shown in Table 1, it was observed that addition of 2.5 wt% Cr to 10H2M (T/P22) steel showed a lack of improvement in terms of corrosion resistance in the sulphidising environment. The observed results are in good agreement with previous work [18]. Those researchers found that corrosion resistance of iron-based materials depends on Cr content to the extent that corrosion resistance in terms of Cr content could be divided into main two thresholds: when Cr concentration is lower than 4 wt%, and when Cr concentration is higher than 4 wt% but lower than 40 wt%.
Surface Microstructures (SEM) and Surface Analyses (EDS)
The SEM microstructures and EDS chemical composition of the low-alloyed steels after 50 and 100 h of exposure in 1%H2S-99% Ar gas mixture at 1 bar total pressure at 450, 500 and 550 °C were performed and investigated. In general, the SEM observations (not shown in this work) revealed the formation of a scale consisting of large, thick crystals which are plate-like and grow perpendicular to the substrate surface after 50 and 100 h. EDS analyses carried out from the surface after 50 h in the temperature range of 450–550 °C indicated some differences in chemical composition of the scales. The steels with no or very little Cr (18K, 16M) showed the development of a sulphide scale consisting of 65 wt% Fe and 35% S. Despite a 0.25–0.35 wt% content of Mo in bulk 16M (T/P1) material, after sulphidation molybdenum was not detected. The 15HM (T/P12) steel with around 1 wt% Cr and 0.55 wt% Mo showed the detection of as little as 0.11 wt% Cr on the surface; however, no Mo was found. Finally, after 50 h of exposure at 450 °C, the steel with the highest Cr and Mo content in the substrate indicated surface enrichment in Mo (15 wt%) and a low concentration of Cr (0.15 wt%), indicating higher affinity of S to Mo rather than to Cr.
The EDS analyses performed on the surface of the samples exposed for 100 h sulphidation on indicated some changes. Initially, the exposed steels developed crystals with as high as 65 wt% and 35 wt% of Fe and S, respectively, after 50 h of sulphidation. After 100 h of sulphidation atmosphere, the concentration of Fe increased further, up to 93 wt% conversely the concentration of S decreased to 7 wt%. These findings clearly indicate that outward diffusion of Fe ions showed a higher rate of inward diffusion compared to S in an ambient atmosphere. The findings correlate with kinetic data as well, where a change in the kinetic mechanism was observed Mass change data of the low-alloyed steels shown in Fig. 6a–c significantly increased after 75 h of exposure due to higher diffusion rates of the elements with a higher atomic mass (Fe ma = 55.845 u) compared to S atoms (S ma = 32.065 u). Nevertheless, the results indicate that despite different chemical compositions, addition of a low amount of alloying elements showed no kinetic behaviour effect; therefore, the steel with 2.5 wt% Cr behaves in the same way as the steel with no alloying additions (18K). Similar behaviour was observed in the steels with an addition of 1 and 2.5 wt% Cr, respectively, where concentrations of Fe and S showed around 50–50 wt%. After 100 h of sulphidation, the concentration of S was reduced to around 1.5 wt% and Fe to around 98 wt%. It is important to note that a high concentration of Mo after 50 h of exposure decreased to a negligible level after 100 h in 10H2M (T/P22) steel. In general, it can be concluded that despite the different chemical compositions of the exposed steels used in the sulphidation tests at 450–550 °C, the formed scales showed similar morphologies and similar corrosion mechanisms; therefore, it can be suggested that the ferritic steels with low Cr content (up to 2.5 wt%) present similar behaviour. A content of 2.5 wt% Cr seems to be meaningless against a sulphidation atmosphere because in such harsh conditions the addition of Cr shows no effect in comparison to 18K steel with lack of Cr.
Cross Sections
Cross-sectioned images of the low-alloyed steels 18K, 16M (T/P1), 15HM (T/P12) and 10H2M (T/P22) are shown in Fig. 7.

Cross-sectioned images performed by means of SEM in BSE mode after 100 h exposures at 450, 500 and 550 °C for 18K, 16M (T/P1), 15HM (T/P12) and 10H2M (T/P22) steels
The images were captured using SEM in BSE mode. The low-alloyed steels exposed in a highly reducing atmosphere showed very poor corrosion resistance and a thick scale formed where plate and needle-like structures rich in Fe–S phase developed. A calculated thickness of the sulphide scale is shown in Table 3.
The thickness measurements were carried out from the top of the sulphide scale to the substrate interface. Higher magnification showing the sulphide scale–substrate interface in 15HM (T/P12) and 10H2M exposed at 550 °C is presented in Fig. 8a, b, respectively, where EDS analyses are provided as well. The findings clearly show that there are much differences between two steels. In both, similar chemical composition was found after the sulphidation test. Figure 9 shows EDS X-ray mapping of the inner part of the scale developed upon exposure of 15HM (T/P12) material at 500 °C for 100 h in a rich sulphur environment. The results indicate the formation of a thick sulphur scale, rich in sulphur with very little concertation of Cr. The presented SEM images in Fig. 8 showed a thick sulphide scale of two low-alloyed steels with 1 and 2.5 wt% of Cr, respectively. The scale structure developed under sulphidation strongly affects the ability of the scale to withstand strains under high temperature. Based on EDS analyses (not shown here) from cross-sectioned samples performed after exposure for 100 h at 450, 500 and 550 °C, it was found that the outermost part of the sulphide scale consisted of a high concentration of S reaching 70 wt% and 30 wt% Fe. The middle part of the formed scale possessed high concentrations of S equivalent to 35 wt% and Fe concentrations reaching 65 wt% indicating the formation of a non-stoichiometric Fe1−XS phase. Furthermore, as was stated previously, there is no difference in terms of corrosion resistance between the steel with the lowest and with the highest concentration of Cr.

Cross-sectioned images presenting the sulphide scale—substrate interface in: a 15HM (T/P12) at 500 °C b 10H2 M (T/P22) steels at 550 °C for 100 h exposure to sulphide atmosphere

Cross-sectioned EDS X-ray mapping performed on a low-alloyed steel exposed in rich sulphide atmosphere: 15HM (T/P12) steel at 500 °C for 100 h
According to previous work [18], when the Cr concentration is lower than 4 wt% (as in this work) the sulphidation rate has no influence of chemical composition of an alloy; the rate is the same as for pure iron. In such situations, Cr does not build up into the crystal structure of the scale which instead consists of an Fe1−xS phase only. The sulphide scale growth depends on outward diffusion of metal from the metal matrix. In the case where the Cr content is higher than 4 wt%, the formed scale consists of two different phases, where the outermost layer consists of FeS. However, the layer that forms in the interface between the sulphide scale and the substrate consists of spinel-type phase: Fe(Fe2−xCrx)S4.
The exact chemical composition based on EDS results shows a sulphide scale (all steels) consisting of a non-stoichiometric Fe1−XS phase. The middle part of the sulphide scale showed the formation of high number of voids; it is believed that the voids are formed due to the condensation of vacancies created within the sulphide scale during the transfer of metal atoms from the metal lattice into the sulphide lattice [19, 20]. Underneath the developed Fe1−XS scale, the layer consisted of 80 wt% Fe and up to 20 wt% S suggesting the formation of Fe2S band rich in Fe with an addition of S. The band was doped with Cr showing a concentration of 2–4 wt% (only 15HM and 10H2M steels). In order to rationalise the formation of phases found in the exposed low-alloyed steels, sulphide corrosion may be explained based on the results derived from two steels 15HM (T/P12) and 10H2M (T/P22) with the highest Cr content.
When low-alloyed steels with up to 3 wt% Cr are exposed to a rich sulphur atmosphere, Fe–S like phases are likely to develop under sulphidising conditions. The FeS phase possesses some degree of non-stoichiometry in the crystal lattice according to work performed by Gesmundo et al. [21], Mrowec [22] and McCammon [23]; thus, the real formula should involve such non-stoichiometry and the phase should be written as Fe1−XS, where x = 0–0.2 [24]. The phase also possesses p-type semiconductor properties, where holes are the majority carriers and electrons are the minority carriers. Defects in Fe1−XS develop due to the absorption of sulphur from the gaseous phase onto the Fe1−XS crystal; furthermore, diffusion of iron cations from the metal matrix occur via chemisorption with S2− anions. The chemisorption reaction forms the new lattice structure of the F1−XS phase. The adsorbed S2− ions on the surface involve an electron-withdrawal mechanism resulting in the formation of electron holes (h·). The formation of defects in the Fe1−XS phase can be rationalised by the following balanced equation:
The equilibrium constant can be calculated for Eqs. 1–4 according to the following:
where V—vacancies, M—metal, X—gas reagent, h·—hole. Similar to a standard chemical reaction, in this case an electroneutrality condition needs to be met:
Based on the theoretical consideration performed via Eqs. 1–4, it can be deduced that the number of ionic and electron defects increases with increase in sulphur pressure. The formation of an Fe1−XS scale depends on the concentration of S in the atmosphere as stated previously. In this work, the atmosphere was rich in S, 1% H2S in inert 99% Ar under 1 bar total pressure. At high temperatures such as 450–550 °C, when low-alloyed steels with very low alloying element additions are exposed, several reactions may take place. Firstly, at high temperatures, decomposition of H2S takes place according to the following reaction shown below:
Furthermore, inward diffusion and adsorption of sulphur on the low-alloyed steel surface and the formation of an Fe1−XS phase needs to be considered.
Hence, for non-stoichiometric Fe1−XS phase defects equations can be considered based on Eqs. 1–4. It is well known that in sulphur-rich atmospheres iron can form two sulphides: Fe1−XS and FeS2. According to Gibbs free energy of formation presented via the Ellingham diagram for sulphide formation in Fig. 10, the formation of Fe1−XS is more likely to occur than FeS2 due to a more negative value of Gibbs free energy formation and depends on pS2 value [25].

Ellingham diagram for the selected metal sulphides
Moreover, FeS is more stable than FeS2. Hence, the formation of an FeS2 phase probably occurred when Fe1−XS was developed via the reaction presented below:
The steels with the highest concentration of Cr showed enrichment in Cr; 2.46 and 3.78 wt% Cr was found in 15HM (T/P12) and 10H2M (T/P22), respectively. The enrichment zone was localised between the Fe1−XS scale and the steel substrate. However, concentrations of chromium in the enriched band presented in Figs. 8 and 9 (Cr EDS X-Ray map) were too low to form a Cr2S3 compound. Based on the simple calculations, to form an exclusively Cr2S3 layer, at least 49 wt% S and 51 wt% Cr is needed; therefore, the amount of Cr found in the band diffused outwardly from the steel metal matrix was far too low. It is well known, similar to other environments, that a higher Cr concentration improves corrosion resistance in atmospheres where a high content of sulphur is met. It was found that the influence of Cr in Fe–Cr steels can be divided into three different segments. When the concentration of Cr reaches 4 wt%, corrosion degradation proceeds with a similar swiftness than that of pure Fe material and a sulphide scale develops due to the outward diffusion of Fe. When the Cr concentration increases up to 12 wt%, the sulphide scale possesses a two-layered structure, where the inner layer consists of an Fe(Fe2−xCrx)S4 phase and the outer layer consists of an Fe1−XS compound. Finally, when the concentration of Cr in a steel is as high as 40 wt% the sulphide scale offers complete protection due to the formation of a continuous Cr2S3 phase scale [26]. In this work, the steels possessed up to 2.5 wt% Cr, the steel with the highest Cr content (10H2M), produced a band where the concentration of Cr reached up to 4 wt%. This concentration corresponds with segment 1, where the ability of Fe(Fe2−xCrx)S4 spinel to form is impossible. The formation of an Fe(Fe2−xCrx)S4 spinel can be formed only when a steel possesses as high as 12 wt% Cr in the metal matrix; then, the formation of spinel can be described by the following reaction:
The formation of Cr3S4 and Cr2S3 phases in this work can be omitted based on the mentioned dependency that these phases develop under sulphidation conditions when exposed steels possess a high enough concentration of Cr [27]. Likewise in Fe–Cr alloys exposed in oxidising atmospheres, when the concentration of Cr in Fe-based material is relatively low, the formation of a FeCr2O4 spinel-type phase is impossible to form. However, when Cr concentration reaches 25 wt% it is expected to develop a Cr2O3 protective scale exclusively [28]. To confirm the findings in the performed studies, the stability diagram for FeS elements at high temperatures is shown in Fig. 11.

The stability diagram for Fe–S element at high temperatures
Calculations were performed using FACTSAGE software. The black spots shown in Fig. 11 indicate the chemical composition derived from EDS results obtained after exposure. The results gained from the calculated Fe–S phase diagram strongly correlate with the data obtained under the test conditions studied; the area with black spots shows 65.48 wt% Fe and 34.52 wt% S corresponding to a pyrrhotite phase; iron sulphide mineral with a non-stoichiometric formula [29]. The region where the Fe1−XS phase is present co-exists with a FeS2 phase. Finally, in the Fe–S phase diagram the formation of an Fe2S phase has been indicated via black spots, the phase is often omitted in phase diagrams; however, it is extremely important to adopt the phase because the formation of Fe2S is often observed under sulphidising conditions.
In order to consider further, development of sulphide scale in rich sulphur atmosphere, Mrowec and Weber [27] found that sulphide scale formation in H2S atmosphere develops due to simultaneous iron and sulphur diffusion within the sulphide scale. A porous external layer of the scale develops due to the outward diffusion of iron and the internal compact layer of the scale forms due to inward diffusion of sulphur. In contrast, according to work carried out by Haycock [30] a significant rate of inward diffusion of sulphur ions during sulphide scale development results from blockage of outward diffusion of iron ions in FeS due to dissolving hydrogen in proton state. As mentioned in this work several times, iron sulphide (FeS) presents high defect structure (Fe1−xS) on the cationic side due to electron holes and empty lattice nodes. In such situations, sulphide scale growth should proceed due to outward diffusion of iron ions and electrons, not by inward diffusion of sulphur ions.
Conclusions
This paper reported the results of a study carried out to investigate the effect of 1%H2S in 99% Ar under 1 bar pressure on the corrosion resistance of the low-alloyed steels 18K, 16M (T/P1), 15HM (T/P12) and 10H2M (T/P22) that are often used in coal power plants. Based on the performed research, it can be stated that low-alloyed steels in rich sulphur atmospheres showed no protection due to the formation of porous, thick and non-protective sulphide scales. Based on EDS analyses performed after exposures at 450, 500 and 550 °C, the exposed low-alloyed steel indicated the formation of an Fe1−xS phase across the scale. Furthermore, addition of up to 2.5 wt% Cr showed no influence on corrosion resistance.
References
R. G. Holcomb, Steam Turbine Materials and Corrosion, (National Energy Technology Laboratory, Albany, OR, 97321, USA 2008).
W. H. Calkins, Fuel 73, 475 (1994).
R. Stefanenko, Coal Mining Technology: Theory and Practice. Society for Mining Metallurgy (1983).
G. Y. Lai, High-Temperature Corrosion and Materials Applications, Chapter 10—Coal Fired Boilers, ASM International, Materials Park, Ohio, USA (2007), pp. 259–314.
F. Gesmundo, D. J. Young and S. K. Roy, High Temperature Materials and Processes 8, 149–190 (1989 ).
S. Mrowec, Materials and Corrosion 31, 371 (1980).
K. Nakagawa, M. Kitagawa, Y. Tumitam and S. Ooki, Journal De Physique IV 3, 787 (1993).
S. Mrowec, Oxidation of Metals 44, 177 (1995).
A.L. Moretti, C.S. Jones, Advanced Emissions Control Technologies for Coal-Fired Power Plants, Babcock & Wilcox Power Generation Group, Inc. Barberton, Ohio, U.S.A, Technical Paper BR-1886 (2012).
H. Shirai, M. Ikeda and H. Aramaki, Fuel 114, 114 (2013).
M. J. Peet, H. S. Hasan and H. K. D. H. Bhadeshia, International Journal of Heat and Mass Transfer 54, 2602–2608 (2011).
D. Laverde, T. Gomez-Acebo and F. Castro, Corrosion Science 46, 613–631 (2004).
R. Viswanathan, J. Sarver and J. M. Tanzosh, Journal of Materials Engineering and Performance 15, 255 (2006).
B. Doujaiji and J. A. Al-Tawfiq, Annals of Saudi Medicine 30, (1), 76 (2010).
C.-H. Selene and J. Chou, Hydrogen Sulfide: Human Health Aspects, Concise International Chemical Assessment Document 53, (World Health Organization (WHO), Geneva, 2003).
J. Ehlers, E.J. Smaardijk, A.K. Tyagi, M. Thiele, W.J. Quadakkers, Effect of Steel Composition on the Bell Shape Temperature Dependence of Oxidation in Water Vapour Containing Environments Proceedings 14th Int. Corr. Congress, 26.9–1.10.1999, Cape Town, South Africa Proceedings, Publ. by CorrISA, Kelvin, South Africa, Paper No. 336.
K. Hauffe and A. Rahmel, Zeitschrift für Physikalische Chemie 199, 152 (1952).
S. Mrowec, Oxidation of Metals 44, (1/2), 177–209 (1995).
S. Jansson, W. Hubner, G. Ostberg and M. D. Pourbaix, British Corrosion Journal 4, 21 (1969).
M. G. Cox, B. McEnaney and V. D. Scott, Philosophical Magazine 31, 331 (1975).
F. Gesmundo, D. J. Young and S. K. Roy, High Temperature Materials and Processes 8, 149 (1989).
S. Mrowec, Oxidation of Metals 44, 177 (1995).
C. A. McCammon and L.-G. Liu, Physics and Chemistry of Minerals 10, 106 (1984).
E. J. Fasiska, Physica Status Solidi 10, 169 (1972).
A. N. Klein, K. P. Furlan, R. M. Schroeder, G. Hammes, C. Binder, J. B. R. Neto, S. H. Probst and J. D. B. de Mello, Powder Technology 271, 193 (2015).
A. S. Khanna, Introduction to High Temperature Corrosion, (ASM International, USA, 2000).
S. Mrowec, T. Weber, Korozja gazowa Metali/Gasous corrosion of Metals Wyd. Slask, in Polish (1975).
U. Krupp, V. B. Trindade, P. Schmidt, H.-J. Christ, U. Buschmann and W. Wiechert, Materials and Corrosion 57, 263 (2007).
D. J. Vaughan and J. R. Craig, Mineral Chemistry of Metal Sulfides, (Cambridge University Press, Cambridge, 1978).
E. Haycock, Journal of The Electrochemical Society 106, 764 (1959).
Acknowledgements
Authors like to acknowledge the financial support of EDF Poland. The study was a part of the project “Fundamental research of high temperature resistance of boiler steels”. Grant number: R&D/ZN/KWA/04072014/099.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dudziak, T., Jura, K., Dudek, P. et al. Sulphidation of Low-Alloyed Steels Used in Power Industry. Oxid Met 92, 379–399 (2019). https://doi.org/10.1007/s11085-019-09929-7
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-019-09929-7