
Abstract
This study addresses the effect of pre-oxidation on the corrosion resistance of the following three commercial steels: a low-alloyed ferritic 10CrMo9-10 steel; a Nb-stabilized austenitic AISI 347 steel; and a high-alloyed austenitic Sanicro 28 steel. Samples of these three steels were pre-oxidized under various conditions and then exposed to potassium chloride (KCl) for 168 h at 550 °C under dry conditions. In the case of the ferritic 10CrMo9-10 and the austenitic AISI 347 steels, despite differences in oxide scale composition and structure, their corrosion resistance could not be improved by any of the studied pre-oxidation procedures. In contrast, pre-oxidation conditions resulting in chromium-enriched oxides improved the corrosion resistance of the high-alloyed austenitic Sanicro 28 steel in terms of oxide scale growth during exposure to KCl.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Substituting renewable fuels for fossil fuels has steadily increased during the last two decades. The main reasons are the limited reservoirs of fossil fuels and because of high greenhouse gas and particle emissions, which are often coupled with coal combustion [1]. In contrast to fossil fuels, biomass is considered to be a CO2-neutral fuel with vast reservoirs, making it an appealing alternative. In the year 2011, 16% of the total global energy demand was covered by biomass-related power production [2]. In the USA, for example, the amount of biomass-based electricity is expected to increase significantly by 2050 [3]. In addition to biomass, different types of waste-derived fuels are considered to be an option when striving for environmentally more sustainable power production. Unfortunately, from a chemical point of view, renewable fuels are inhomogeneous, usually with a high concentration of water and considerable amounts of potassium, chlorine, and/or heavy metals, all of which are known to affect the durability of superheater tubes [e.g., 4,5,6]. To slow or reduce corrosion, power plants using renewables are operated at lower steam temperatures, thus reducing the power production efficiency. Therefore, more efficient ways to prevent corrosion are needed, so that power plants using biomass and waste-derived fuels can operate at higher steam temperatures.
Alloying elements such as nickel and chromium have long been used to improve steels’ capability to withstand corrosion at elevated temperatures (400–600 °C). In these iron-based alloys, chromium forms a very dense and thus protective chromium oxide (Cr2O3) on the steel surface, whereas nickel is responsible for stabilizing the austenite (FCC) crystal structure of the steel. In addition, nickel can be used as the base metal of an alloy, further improving the material durability at high temperatures under harsh conditions. Unfortunately, despite superior corrosion resistance, nickel-based alloys are used only in the most critical parts of a boiler due to their high costs. In order to make biomass and waste-derived fuels more appealing to power producers, more cost-efficient ways to protect the heat-transfer surfaces are required. Different kinds of coatings such as laser-cladded [7,8,9] and Al-based diffusion coatings [10,11,12] have been studied in laboratory scale, and the results have been promising. However, optimization regarding coating composition and parameters is still required before corrosion resistance based on applied coatings will reach its full potential. In addition, the coatings are relatively brittle, preventing the superheater tubes, for example, to be coated before installation. Before more malleable coatings are designed, various approaches to generate on-site coatings of the heat-transfer surface are of interest. One approach for material improvement could be temperature- and time-based controlled pre-oxidation of the heat-transfer materials, resulting in oxide scales thick or dense enough to withstand material degradation at higher material temperatures and for longer times than materials, which have not been pre-oxidized. The possibilities of pre-oxidation have previously been addressed in the case of alumina formers [13], Ni-based superalloys [14], and in studies addressing corrosion resistance to gaseous chlorine species [15,16,17]. Generally speaking, pre-oxidation may improve the high-temperature corrosion resistance of an alloy compared with a non-oxidized material. However, the beneficial effects of the pre-treatment strongly depend on the pre-oxidation procedure and on the material in question. Therefore, more information concerning the impact of pre-oxidation parameters on the high-temperature behavior of various steels is desirable.
The aim of this study was to investigate whether the corrosion resistance of commercial heat-transfer materials can be improved by pre-oxidizing them under well-controlled conditions. The corrosion resistance was tested by exposing the pre-oxidized steels to potassium chloride. The study is a sequel to a paper, in which the oxidation behavior of the studied steels was described in detail [18].
Experimental Procedures
Three commercial steels (Table 1) typically used in different locations in power plants firing biomass were pre-oxidized in various ways before exposing them to 0.250 g of KCl (J.T.Baker®, Baker Analyzed grade) deposit with a particle size of 125–297 μm. The precise pre-oxidation procedure has been described in detail elsewhere [18], but Table 2 summarizes the key parameters of the pre-oxidation procedures used.
The exposures were carried out in a quartz reactor positioned inside of a tube furnace. The KCl-deposited samples laid horizontally in a sample holder with three thermocouples attached: one on each end of the sample holder and the third one in the middle. The temperature difference between the samples at both ends and the sample in the middle was measured to be less than 5 °C. The target temperature was 550 °C, to which the furnace was heated with a heating rate of 5 °C min−1. The exposure time was 168 h, and the prevailing atmosphere was synthetic air (21% O2, 79% N2) with a flow rate of 400 ml min−1. Before entering the reactor, the synthetic air was heated to 220 °C to avoid unnecessary cooling of the samples. The gas feed was on during both the heating and the cooling periods. After cooling, the samples were mounted in a chlorine-free epoxy and cut in half to produce cross-sectional samples for the analyses. Prior to the analyses, the cross-sectional samples were first ground with 500, 1200, and 2000 grit SiC papers, then sequentially polished with 3 and 1 μm diamond pastes, and finally, cleaned in petroleum ether. The polished cross sections were analyzed with a scanning electron microscope (LEO 1530 Gemini) equipped with an X-ray detector (Thermo Scientific Ultra Dry Silicon Drift Detector) and an energy-dispersive X-ray analysis system (Thermo Scientific Thermo NORAN Vantage X-ray). The microscope was operated at an accelerating voltage of 15 or 20 kV for general imaging and 20 kV for EDX analyses. The backscatter electron mode was used for the images affiliated with the EDX analyses, while both backscatter electron and secondary electron modes were applied for the general imaging of the cross sections. The oxide scale thickness was determined using automatic image analysis, which identified different regions (salt, oxide, steel) by contrast analysis, after which the images were colored to highlight the formed oxide (Fig. 1). A more detailed description of the test method and experimental setup is provided in, for example, [19]. The method enables the determination of the maximum, minimum, average, and median values of the oxide scale thickness, which can be presented as thickness distribution graphs. The shape of the distribution curve can be used to estimate the type of corrosion: A narrow distribution suggests that homogeneous corrosion occurred over the whole surface, whereas a broad and possibly tailing distribution implies that more heterogeneous corrosion took place in (Fig. 2).

Steps in coloring the image for oxide scale thickness measurement: a the original SEM image taken from the cross-sectional sample with backscattered electrons, b a partly processed image representing the epoxy in blue and the non-reacted steel in red, c the finalized image showing the formed oxide in green (Color figure online)

Graphs of oxide scale distribution as a function of thickness demonstrating a homogeneous and b non-homogeneous oxidation
The potential reaction between the protective Cr2O3 scale and the corrosive KCl was also studied with an SDT Q600 simultaneous thermogravimetric analyzer/differential scanning calorimeter (TGA/DSC) apparatus (TA Instruments). A mixture of Cr2O3 powder and KCl with a molar ratio of 1:1 was heated to 700 °C with a heating rate of 2 °C min−1 in synthetic air.
Results and Discussion
Oxide Scale Thickness after Exposure to KCl
The oxide thickness values were calculated as a mean based on measurements at 10,000 points along the part of the cross section covered with the salt deposit. The minimum, maximum, and mean values of the oxide thicknesses together with corresponding standard deviations are summarized in Table 3. A brief description of the oxides formed during the pre-oxidation is given before the results of corrosion resistance for each alloy. The pre-oxidation results are comprehensively presented in [18].
Irrespective of the pre-oxidation procedure, an iron oxide scale consisting of Fe2O3 and Fe3O4 formed on all 10CrMo-9 steel samples [18]. The oxide scale thickness increased as functions of pre-oxidation time and temperature, and the morphology changed from initial oxidation along the surface grain boundaries (200 °C) to a more homogeneous, whisker-containing oxide (500 °C), and finally, to a thick porous oxide (700 °C). The thickness of the oxide after pre-oxidation varied from 17 nm (5 h at 200 °C) to above 20 μm (24 h at 700 °C).
In the case with the ferritic 10CrMo-9 steel, all pre-oxidation procedures resulted in thinner oxide scales compared to the non-treated reference when exposed to KCl. However, the oxide scale thicknesses did not alter systematically as a function of pre-oxidation temperature or time, but appeared to be constant at around 150 microns. After pre-oxidation, all samples had a chromium-rich region below the formed iron oxide scale [18], which might have slowed further oxidation, resulting in thinner oxides compared to the reference sample. A few samples had clearly thinner oxide scales (< 70 μm), but the present study did not give answers to this observation.
Pre-oxidation of the Nb-stabilized austenitic AISI347 steel at 200 °C and 500 °C resulted in a duplex oxide scale with an iron-rich outer and a chromium-rich inner layer [18]. In addition, niobium oxide nodules formed at 500 °C in an uncontrolled manner through the surface oxide. At 700 °C, the niobium oxide particles were completely covered with an oxide scale consisting of almost pure chromium oxide. However, the porous morphology and the particle size (above one micron) of the chromium oxide scale suggested that it could not protect the steel in the presence of corrosive species such as KCl. The thickness of the oxide after pre-oxidation varied between 7 nm (5 h at 200 °C) and 629 nm (24 h at 700 °C).
The oxide scales formed on the austenitic stainless AISI 347 steel samples in the presence of KCl were clearly thinner than those formed on the ferritic steel. However, the pre-oxidation procedure did not appear to have a notable influence on the thicknesses of the oxides: All the formed oxide scales have more or less the same thickness, which is of the same magnitude as the one measured from the reference sample. The thinner oxides indicate better corrosion resistance, which can be attributed to the chromium content of the steel, but no clear beneficial effect of a certain pre-oxidation procedure could be observed in terms of oxide scale thickness.
The oxide scales formed 200 and 500 °C on the austenitic Sanicro 28 steel samples had a duplex structure with an iron-rich outer layer and a chromium-rich inner layer [18]. A non-porous and well-adhered oxide scale of almost pure chromium formed at 700 °C. The thickness of the oxide after pre-oxidation varied between 3 nm (5 h at 200 °C) and 184 nm (24 h at 700 °C).
The high-alloyed austenitic Sanicro 28 steel samples can be divided into two categories on the basis of the measured oxide scale thicknesses formed in the presence of KCl: group 1 with oxide thicknesses below 40 μm and group 2 with oxide thicknesses above 60 μm. The depth profiles of the oxides after pre-oxidation indicated chromium enrichment in all the oxides in group 1 [18]. In the case of the two samples pre-oxidized at 700 °C, a virtually pure chromium oxide scale had formed. These two samples also exhibited the best resistance against KCl-induced corrosion. Based on the measured oxide scale thickness values, it appears to be possible to improve the corrosion resistance of the high-alloyed austenitic Sanicro 28 steel through oxide manipulation by affecting the scale composition and/or porosity.
Oxide Scale Composition and Structure
The oxide scales formed on all low-alloyed 10CrMo9-10 steel samples were multilayered and consisted of almost pure iron oxide with traces of chromium (Fig. 3). The chemical composition and the structure of the oxide remained unaltered despite the pre-oxidation procedure. As the oxide consisted almost solely of iron oxides (Fe2O3 and Fe3O4) after the pre-oxidation [18], the protective properties of the scale were very limited.

A typical oxide scale at the low-alloyed 10CrMo9-10 steel, featuring a multilayered Fe2O3 structure. The sample was exposed to KCl at 550 °C for 168 h
Formation of a multilayered product on a low-alloyed steel resulting from KCl-induced high-temperature corrosion has been reported earlier [20, 21] and is most likely related to iron chloride (FeCl2) formation. As KCl reacts with iron, chlorine is released, either as molecular chlorine according to Eq. 1 [24] or as a chloride ion [23]. Chlorine reacts with iron, forming a solid iron chloride, which then easily vaporizes due to its high vapor pressure at 500 °C and above (Eqs. 2, 3) [22]. As the gaseous FeCl2 diffuses toward a higher oxygen partial pressure, it will oxidize rapidly, leaving a porous oxide behind (Eq. 4). Due to a virtually exhaustless supply of chlorine, the reaction described above will repeat itself, resulting in a multilayered structure like that presented in Fig. 3. Based on the thickness distribution curves (not shown here), the oxidation of the low-alloyed steel was homogeneous despite the pre-oxidation procedure. Homogeneous oxidation indicates a fast surface transport mechanism of the oxidative/corrosive species, which in the present case for the low-alloyed steel, could take place due to the presence of a eutectic FeCl2-KCl melt [25].
As a summary, based on the appearance of the formed oxides, it can be stated that none of the tested pre-oxidation conditions could improve the corrosion resistance of the studied 10CrMo9-10 steel.
Compared to the low-alloyed 10CrMo9-10 steel, the chromium content of the austenitic stainless AISI 347 steel is high enough to form and sustain a protective chromium-based oxide [26]. Although minor differences in the oxide structure were detected, the general composition of the formed oxide scale was the same: The oxide contained principally iron together with chromium (Fig. 4). Below the oxide, a nickel-enriched region was detected, evidencing the diffusion of iron and chromium from the steel into the formed oxide. The nickel-enriched region was very porous and indicated localized corrosion deeper into the steel, most likely along the grain boundaries (Fig. 5). The porosity originated from material degradation, in other words, from the selective outward diffusion of iron and chromium. Corrosion along the grain boundaries was also observed in the shape of the thickness distribution curves, which displayed a typical asymmetry (Fig. 6). In addition, traces of potassium chromate (K2CrO4) were found on top of some of the oxide scales. K2CrO4 is a solid intermediate, which is known to be rapidly formed in the reaction between KCl and the protective chromium-rich oxide, impairing the corrosion resistance of the alloy through chromium depletion [27]. In the case of a pure chromium oxide scale, the reaction would proceed according to Eq. 5 [28]. However, it should be borne in mind that the change in the Gibbs free energy of the discussed reaction is positive (363 kJ at 600 °C), suggesting it to be non-spontaneous.

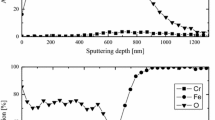
Elemental maps of a typical oxide formed on the austenitic stainless AISI 347 steel. The sample was exposed to KCl at 550 °C for 168 h

Local corrosion and the formed porous region on the steel/oxide interface of the austenitic stainless AISI 347 steel, exposed to KCl at 550 °C for 168 h

Thickness distribution curve of the austenitic stainless AISI 347 steel, exposed to KCl at 550 °C for 168 h. Prior to the exposure, the sample was pre-oxidized at 700 °C for 5 h
Before the exposure to KCl, all oxides of the pre-oxidized AISI 347 samples formed below 700 °C consisted of a duplex oxide with an iron-rich outer layer and a chromium-rich inner layer. In addition, niobium oxide granules had pierced the surface oxide, providing pathways for corrosive species to diffuse relatively rapidly to the oxide/metal interface. Unlike with the other pre-oxidation temperatures, the pre-oxidation at 700 °C resulted in a pure chromium oxide [18]. However, the porosity of the formed oxide was most likely too high to withstand corrosion. Based on this, the high chromium content of a surface oxide alone is not sufficient to improve the protective properties of the oxide, but the structure of the oxide also plays a role. In summary, it appears also that in the case with the austenitic stainless AISI 347 steel, the tested pre-oxidation conditions could not improve its corrosion resistance.
In the case of Sanicro 28, although minor differences in the oxide structure were detected, the general composition of the formed oxide scale was the same, independent of the pre-treatment parameters: The oxide contained iron and chromium and in some cases, nickel (Fig. 7). The formed oxides were porous and in most cases, detached from the unreacted steel. In most of the samples, a nickel-enriched region was detected below the formed oxide, evidencing the diffusion of iron and chromium from the steel into the formed oxide. In addition, traces of metal chlorides (MClx) were found in the steel/oxide interface of some of the samples, indicating that chloride-involving interactions had taken place below the surface oxide, which was no longer protective. Since the focus of this study lies in the protective properties of the oxide rather than on the details of oxide formation, the origins for the minor differences in the oxide structure were not studied further.

Elemental maps of a typical oxide formed on the high-alloyed austenitic Sanicro 28 steel. The sample was exposed to KCl at 550 °C for 168 h
The asymmetric shape of the oxide scale thickness distribution curves suggested localized oxidation, in other words, grain boundary corrosion. Although grain boundaries have been reported to be responsible for increased corrosion resistance through fast chromium supply to the surface oxide [29], the grain boundaries will become depleted in chromium after longer exposure times in aggressive environments, leaving the grain boundaries enriched with nickel (Fig. 8). In their initial stage, the grain boundaries are usually chromium-rich due to the presence of chromium carbides (Cr7C3, Cr23C6), which have beneficial effects on steel-strengthening mechanisms [30]. However, chromium carbides are known to react with alkali chlorides [31, 32], which could explain the selective material degradation of the studied Sanicro 28 steel. The nominal carbon content of the Sanicro 28 steel is below 0.020 wt%, which might be too low to stabilize the carbides. Therefore, the importance of chromium carbides in the chromium depletion along the grain boundaries remained unresolved in the present study.

Elemental maps of the high-alloyed austenitic Sanicro 28 steel, demonstrating grain boundary corrosion. The sample was pre-oxidized at 500 °C for 5 h and then exposed to KCl at 550 °C for 168 h
As an exception from the other samples, pre-oxidation of Sanicro 28 steel at 700 °C resulted in a pure, dense, and relatively thin (100–200 nm) chromium oxide, measured with XPS [18]. After a 168-h exposure to KCl at 550 °C, the thickness of the formed oxide was below one micron, i.e., below the detection limit of the utilized SEM. This suggested that a very protective oxide was successfully formed on the steel surface. Based on thermodynamic calculations, the reaction between KCl and pure Cr2O3 according to Eq. 5 is not favored and hence it is non-spontaneous [33]. This was further verified by studying a mixture of powdered KCl and pure Cr2O3, during heating up to 700 °C (Fig. 9). No mass or temperature change was observed during the heating, indicating no reaction between the two studied species. The reactivity of chromia-forming materials has been reported several times [e.g., 34], and the reactivity originates most likely from the fact that instead of consisting of solely chromium, the protective oxide is a mixed spinel with a nominal composition of either Ni(Fe,Cr)2O4 or (Fe,Cr)3O4. The spinel structure often deviates from stoichiometry and contains point defects such as cation vacancies [35], increasing the reactivity of the mixed spinel compared to pure chromium oxide.

Relative mass (black) and the DTA curve (red) of a Cr2O3/KCl mixture (molar ratio 1:1) as a function of temperature in synthetic air with a heating rate of 2 °C min−1 to 700 °C (Color figure online)
Interestingly, the pre-oxidation procedure, which generated a pure and a thick enough chromia scale, remarkably improved the ability of Sanicro 28 steel to withstand KCl-induced corrosion.
Conclusions
This study addressed the effect of pre-oxidation on the corrosion resistance of three commercial steels (a low-alloyed ferritic 10CrMo9-10 steel, a Nb-stabilized austenitic AISI 347 steel, and a high-alloyed austenitic Sanicro 28 steel). The goal was to induce dense corrosion-resistant scales at steel surfaces. Samples of the three steels were pre-oxidized under various conditions and then exposed to potassium chloride (KCl) for 168 h at 550 °C under dry conditions.
Pre-oxidation conditions resulting in chromium-enriched oxides improved the corrosion resistance of the high-alloyed austenitic Sanicro 28 steel in terms of oxide scale growth during the exposure to KCl. In the case with the ferritic 10CrMo9-10 and the austenitic AISI 347 steels, despite the differences in oxide composition and structure, their corrosion resistance could not be improved by any of the studied pre-oxidation procedure.
The study suggests that an optimized pre-oxidation procedure enhances the corrosion resistance of commercial steels without any other material manipulation such as the addition of reactive elements. The most beneficial set of pre-oxidation parameters depends on the steel in question as well as on the conditions prevailing in the application. Since the superheater tubes have bends, the pre-oxidation should be carried out when the heat-transfer parts are in their final form. This might be easier to do before the installation of the parts, although the protective oxides might get damaged during the installation, whereupon the improved corrosion resistance is lost. The other option would be on-site pre-oxidation after the installation of the heat-transfer parts, although this might not be economically reasonable. Furthermore, certain industrial processes such as biomass combustion expose the material to physical wear (erosion) in addition to the chemical corrosion. Therefore, the effect of erosion on possible spalling of the pre-oxidized surfaces should be addressed in the coming studies.
References
B. Yousaf, G. Liu, Q. Abbas, R. Wang, M. U. Ali, H. Ullah, R. Liu, and C. Zhou, Applied Energy 208, 142 (2017).
Renewables 2011—Global Status Report, REN21, Secretariat Paris, France, ISSN 143792140X (2011).
U.S. Department of Energy, Energy Information Administration, Office of Integrated and International Energy Analysis, Annual Energy Outlook 2018 with Projections to 2050, Washington, DC, February 2018, https://www.eia.gov/outlooks/aeo/pdf/AEO2018.pdf.
L. L. Baxter, T. R. Miles, T. R. Miles Jr., B. R. Jenkins, T. Milne, D. Dayton, R. W. Bryers, and L. L. Oden, Fuel Processing Technology 54, 47 (1998).
P. Vainikka, D. Bankiewicz, A. Frantsi, J. Silvennoinen, J. Hannula, P. Yrjas, and M. Hupa, Fuel 90, 2055 (2011).
Y. Kawahara, Oxidation of Metals 85, 127 (2016).
X.-Z. Li, Z.-D. Liu, H.-C. Li, Y.-T. Wang, and B. Li, Surface and Coatings Technology 232, 627 (2013).
Z. Pala, M. Bai, F. Lukac, and T. Hussain, Oxidation of Metals 88, 749 (2017).
L. Reddy, P. Shipway, C. Davis, and T. Hussain, Oxidation of Metals 87, 825 (2017).
A. Agüero, I. Baraibar, V. González, R. Muelas, and D. Plana, Oxidation of Metals 85, 263 (2016).
S. Kiamehr, T. N. Lomholt, K. V. Dahl, T. L. Christiansen, and M. A. J. Somers, Materials and Corrosion 68, 82 (2017).
J. Metsäjoki, E. Huttunen-Saarivirta, and T. Lepistö, Fuel 133, 173 (2014).
N. Israelsson, J. Engkvist, K. Hellström, M. Halvarsson, J.-E. Svensson, and L.-G. Johansson, Oxidation of Metals 83, 29 (2015).
S.C. Okoro, M. Kvisgaard, M. Montgomery, F. J. Frandsen, and K. Pantleon, Surface Engineering 33, 428 (2017).
P. S. Sidky, and M. G. Hocking, Corrosion Science 29, 735 (1989).
H. Asteman, and M. Spiegel, Corrosion Science 49, 3626 (2007).
T. J. Pan, H. T. Ma, and Y. S. Li, Corrosion Engineering, Science and Technology 46, 499 (2011).
J. Lehmusto, D. Lindberg, P. Yrjas, and L. Hupa, Oxidation of Metals 89, 251 (2018).
B.-J. Skrifvars, R. Backman, M. Hupa, K. Salmenoja, and E. Vakkilainen, Corrosion Science 50, 1274 (2008).
J. Lehmusto, P. Yrjas, B.-J. Skrifvars, and M. Hupa, Fuel Processing Technology 104, 253 (2012).
E. Larsson, H. Gruber, K. Hellström, T. Jonsson, J. Liske, and J.-E. Svensson, Oxidation of Metals 87, 779 (2017).
H. J. Grabke, E. Reese, and M. Spiegel, Corrosion Science 37, 1023 (1995).
N. Israelsson, K. A. Unocic, K. Hellström, J.-E. Svensson, and L.-G. Johansson, Oxidation of Metals 84, 269 (2015).
S. C. Cha, and M. Spiegel, Materials and Corrosion 57, 159 (2006).
T. Jonsson, N. Folkeson, J.-E. Svensson, L.-G. Johansson, and M. Halvarsson, Corrosion Science 53, 2233 (2011).
N. Birks, G. H. Meier, and F. S. Pettit, in Introduction to the High-Temperature Oxidation of Metals 2nd Edition, (Cambridge University Press, Cambridge and New York, 2006).
J. Lehmusto, M. Bergelin, J. Sui, J. Juhanoja, B.-J. Skrifvars, and P. Yrjas, Corrosion Science 125, 1 (2017).
Y. S. Li, M. Sánchez-Pastén, and M. Spiegel, Materials Science Forum 461–464, 1047 (2004).
C. Pettersson, L.-G. Johansson, and J.-E. Svensson, Oxidation of Metals 70, 241 (2008).
R. Pillai, H. Ackermann, H. Hattendorf, and S. Richter, Corrosion Science 75, 28 (2013).
H. Fujikawa, and N. Maruyama, Materials Science and Engineering: A 120–121, 301 (1989).
M. A. Uusitalo, R. Backman, L.-M. Berger, P. M. J. Vuoristo, and T. A. Mäntylä, High Temperature Materials and Processes 21, 307 (2002).
J. Lehmusto, D. Lindberg, P. Yrjas, B.-J. Skrifvars, and M. Hupa, Oxidation of Metals 77, 129 (2012).
S. Kiamehr, K. V. Dahl, M. Montgomery, and M. A. J. Somers, Materials and Corrosion 66, 1414 (2015).
J. Töpfer, S. Aggarwal, and R. Dieckmann, Solid State Ionics 81, 251 (1995).
Acknowledgements
Open access funding provided by Abo Akademi University. This work has been carried out within the Academy of Finland project “Novel Approaches to Study Corrosion Mechanisms in High-temperature Industrial Processes” (Decision No. 296435). This work has been partly carried out within CLIFF (2014–2017) as part of the activities of Abo Akademi University. Other research partners are VTT Technical Research Centre of Finland Ltd, Lappeenranta University of Technology, Aalto University, and Tampere University of Technology. Support from the National Technology Agency of Finland (Tekes), Andritz Oy, Valmet Technologies Oy, Sumitomo SHI FW, UPM-Kymmene Oyj, Clyde Bergemann GmbH, International Paper Inc., and Top Analytica Oy Ab is gratefully acknowledged. The authors would like to thank Mr. Linus Silvander for operating the SEM apparatus and Miss Jaana Paananen for carrying out the coloring of the panoramic images.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lehmusto, J., Yrjas, P. & Hupa, L. Pre-oxidation as a Means to Increase Corrosion Resistance of Commercial Superheater Steels. Oxid Met 91, 311–326 (2019). https://doi.org/10.1007/s11085-019-09898-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-019-09898-x