
Abstract
The KCl-induced corrosion of the FeCrAl alloy Kanthal® APMT in an O2 + N2 + H2O environment was studied at 600 °C. The samples were pre-oxidized prior to exposure in order to investigate the protective nature of alumina scales in the present environment. The microstructure and composition of the corroded surface was investigated in detail. Corrosion started at flaws in the pre-formed α-alumina scales, i.e. α-alumina was protective in itself. Consequently, KCl-induced corrosion started locally and, subsequently, spread laterally. An electrochemical mechanism is proposed here by which a transition metal chloride forms in the alloy and K2CrO4 forms at the scale/gas interface. Scale de-cohesion is attributed to the formation of a sub-scale transition metal chloride.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Renewable fuels such as biomass and waste are increasingly used for power production worldwide. Burning biomass and waste results in a more corrosive fireside environment, e.g., it contains more alkali chloride than fossil fuels [1–9]. Consequently, the corrosion of critical components in a power boiler, e.g., the superheater tubes, is a major challenge. To mitigate such corrosion problems, the maximum steam temperature is kept relatively low (typically 400–550 °C depending on the fuel). To increase the competitiveness of this power source, higher operating temperatures are needed, which creates demands for new materials that can cope with the aggressive environment. Currently, low alloyed steels and stainless FeCr and FeCrNi steels are the material of choice in these power boilers. The ability of the stainless steels to withstand high temperatures relies on the formation of a protective, Cr-rich oxide. It has previously been shown that KCl, O2 and H2O react with chromia in the, normally protective, scale on stainless steels, thus forming K2CrO4 and leaving a poorly protective iron oxide behind [10–14]. A possible strategy to avoid component failure in a power boiler would be to use another type of alloy that is not susceptible to this kind of attack. FeCrAl alloys are a suitable candidate because the reaction of KCl with Al2O3 which produces potassium aluminate (KAlO2) is much less favoured thermodynamically than the formation of K2CrO4 at the relevant temperatures. Thus, alumina-forming alloys may provide a viable alternative for high temperature applications in which materials are exposed to alkali chlorides under high p(O2) conditions at intermediate temperatures.
Previous studies have shown that small amounts of KCl in an O2 + N2 + H2O environment at 600 °C cause rapid corrosion of the FeCrAl alloys Kanthal® APMT and AF [15–17]. The aim of this study is to use pre-formed alumina scales prior to exposure to the KCl-containing environment in order to mitigate the corrosion.
Materials and Methods
The rapidly solidified powder-based alloy Kanthal® APMT with the nominal composition given in Table 1 was investigated in the present study.
The material was supplied by Sandvik Heating Technology in the form of a 2 mm thick sheet, from which samples with the dimensions 15 × 15 mm2 were prepared. The main surface was ground with 320 grit SiC paper and polished in three steps using a 9, 3 and 1 µm diamond suspension. The sample edges were only ground with 320 grit SiC paper. The polished coupons were degreased and cleaned using ultrasonic agitation in water, acetone and finally ethanol. The samples used in the corrosion exposures were first pre-oxidized in a tube furnace at 900 and 1,100 °C for 1 and 24 h in 5 % O2 + 95 % N2. The flow rate was 1,000 ml/min in the tube furnace which corresponds to 3.2 cm/s.
KCl-induced corrosion of the pre-oxidized samples was studied through isothermal exposures in a tube furnace at 600 °C. A saturated solution of KCl in water/ethanol was used to add KCl to the pre-oxidized sample surface prior and between the exposure cycles. The specimen was alternately sprayed with the solution and dried with flowing air (~35 °C) to avoid the formation of droplets. The samples with 0.2 mg/cm2 KCl were then allowed to stabilize in a desiccator before their weight was recorded on a six-decimal Sartorius balance. The exposure environment was 5 % O2 + 40 % H2O + 55 % N2 with a flow rate of 1,000 ml/min which corresponds to 3.2 cm/s. The specimen was exposed for 24-h cycles until breakaway corrosion was observed. The samples with applied KCl were put into a heated furnace (600 °C) in order to limit the heating period. The samples were removed from the furnace and cooled in static lab air in a desiccator.
A Siemens D5000 powder diffractometer was used to determine the crystalline corrosion products. The instrument was equipped with a grazing-incidence-beam attachment together with a Göbel mirror. The samples were exposed to a source of CuKα radiation (λ = 1.5418 Å) with an incident angle of 0.5–1.0°. The moving detector collected data in the range of 20° < 2θ < 70° with a step size of 0.05°.
Cross-sections with a width of roughly 500 µm were prepared using a Gatan Ilion+ Broad Ion Beam (BIB) system. The ion gun was operated at 6 kV and the sputter time was 2–3 h. The cross-sections were analysed with Scanning Electron Microscopy (SEM). An FEI Quanta 200 FEG ESEM operated in high vacuum mode was used in this study. The instrument comes equipped with a Schottky Field Emission Gun (FEG) and an Oxford Inca EDX system, which was used for chemical quantification.
In order to obtain detailed microstructural information a Scanning Transmission Electron Microscope (STEM) was used. Transmission Electron Microscopy (TEM) characterization was carried out on a Philips model CM200 equipped with a Schottky FEG operated at an accelerating voltage of 200 kV and with an X-ray Energy Dispersive Spectrometer (EDS), from EDAX which was used for analytical analyses. Bright-field High resolution-STEM imaging and EDS mapping was obtained at 200 kV with a sub-Ångström probe on an aberration-corrected JEOL 2200FS-AC STEM using a CEOS Cs-Corrector equipped with a Bruker XFlash® 6|30 silicon drift detector (SDD). STEM samples were prepared with a focus ion beam (FIB) in situ lift-out technique using a Hitachi NB5000 FIB-SEM and an FEI Strata DB 235 M FIB-SEM.
Results
Pre-oxidation (900 °C and 1,100 °C for 1 and 24 h)
Before the corrosion exposures, all samples were pre-oxidized isothermally in a tube furnace fitted with an alumina tube at 900 or 1,100 °C for 1 and 24 h in 5 % O2 + 95 % N2. The chosen pre-oxidation conditions were studied more in detail by Hellström et al. [18, 19]. A short description of the obtained alumina scales can be found below.
A well adherent alumina scale had formed after all pre-oxidation treatments, scale thickness ranging from 0.1 to 1.5 µm depending on oxidation time and temperature (Fig. 1). Both samples oxidized at 1,100 °C and the sample oxidized for 24 h at 900 °C clearly exhibited a two-layered oxide scale. The bottom scale layer was barely visible after 1 h at 900 °C (Fig. 1). A TEM investigation by Hellström et al. confirmed the presence of a two-layered α-Al2O3 scale after oxidation at 900 °C, the inward growing alumina layer was however not continuous over the entire surface [18]. Analysis using XRD showed that α-Al2O3 was present on all pre-oxidized samples. There was no evidence of metastable forms of alumina.

Corrosion Experiments (O2, N2, H2O and KCl at 600 °C)
Figure 2 shows the distribution of KCl on the sample surface before exposure, with salt crystallites and aggregates ranging in size from a few μm to ~100 μm. While the surface between the salt particles appears to be free from KCl, it was shown previously [17, 20] that small amounts of KCl were present also in these areas.

Optical image (a) and SEM-BSE micrograph (b) of the salt distribution on the alumina scale prior to corrosion exposure
Corrosion of Samples Pre-oxidized 1 h at 900 °C
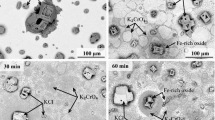
The alumina scales formed during 1 h pre-oxidation at 900 °C failed during the first corrosion exposure (Fig. 3a1), i.e., the entire surface suffered breakaway oxidation. This is illustrated in the SEM-BSE top view micrograph in Fig. 3a2, which shows a representative area of the corroded surface with a former KCl particle in the middle of the micrograph, surrounded by a cracked alumina scale. During the first corrosion exposure, iron-rich oxide had grown on top of the alumina scale, covering most of the sample surface. Analysis using XRD after the corrosion exposure showed the presence of potassium chromate (K2CrO4) and hematite (α-Fe2O3) in addition to α-Al2O3, which was present after pre-oxidation (Table 2). SEM imaging at higher magnification revealed numerous 1–2 µm-size particles on the surface (not distinguishable in Fig. 3a2). Analytical analysis using SEM/EDX confirmed the presence of K, Cr and O and it was concluded that they consisted of K2CrO4.

Optical images and SEM-BSE micrographs of top-view and cross-sections of the pre-oxidized samples a1–a4 (1 h, 900 °C) to the left and b1–b4 (1 h, 1,100 °C) to the right exposed for one corrosion exposure (O2 + H2O + N2 + KCl at 600 °C for 24 h)
The one-sided cross-section in Fig. 3a3 and a4 (a4 is an enlargement of a part of a3) shows a layered oxide scale. The alumina layer from the pre-oxidation stage contains several cracks and relatively thick layers of iron-rich oxide have formed in connection to these cracks, both above and below the alumina. The K2CrO4 particles detected in the top view can also be seen in the cross section (Fig. 3a3), and are always found on top of the cracked alumina layer, being associated with iron oxide. Below the iron oxide/alumina/iron oxide sandwich a chromium-rich oxide layer is present (Fig. 3a3). Just beneath the chromium-rich oxide layer there is an Oxidation Affected Zone (OAZ) in the alloy substrate containing small needle-like particles. EDX point analyses showed that the particles are enriched in nitrogen and aluminium and their needle-like shape is consistent with aluminium nitride (AlN), which suggests the presence of AlN in the OAZ.
Corrosion of Samples Pre-oxidized 1 h at 1,100 °C
Pre-oxidation for 1 h at 1,100 °C also failed to be protective during the first corrosion exposure, with more than half of the sample surface exhibiting breakaway oxidation (Fig. 3b1). The SEM-BSE top view micrograph in Fig. 3b2, shows a relatively lightly corroded area (corresponding to the grey area in Fig. 3b1) consisting of an alumina scale containing several cracks and fissures. SEM/EDX point analysis showed that the bright areas associated with the cracks are dominated by iron oxide. Similar to the samples pre-oxidised at 900 °C for 1 h (see above), K2CrO4 particles were detected on the surface using SEM/EDX. In addition to the α-Al2O3 formed during pre-oxidation, XRD analysis detected α-Fe2O3 and potassium chromate (Fig. 4).

XRD diffractogram of pre-oxidized APMT (1 h, 1,100 °C) exposed for one corrosion exposure (O2 + H2O + N2 + KCl at 600 °C for 24 h). The symbols indicate: K2CrO4 (filled diamond), α-Fe2O3 (filled circle), α-Al2O3 (filled triangle), and substrate (filled square)
The one-sided cross-section in Fig. 3b3 was prepared from an area corresponding to the top view image in Fig. 3b2, which shows a cracked alumina layer with a thickness of about 0.4 μm. In contrast to the previous case (900 °C, 1 h pre-oxidation), the iron-rich oxide is mainly found beneath the alumina layer (compare Fig. 3a3, b3). A chromium-rich oxide is present underneath the iron-rich oxide and there is an OAZ containing needle like aluminium nitride precipitates in the substrate. There is less nitride formation compared to the sample pre-oxidized for 1 h at 900 °C, which indicates that the alumina scale was more protective in this case (compare Fig. 3a3, b3). The image of the cross-section in Fig. 3b4 shows the early stage of a corrosion attack, at a location where the alumina scale has cracked. A wedge-shaped layer consisting of iron-rich oxide (SEM/EDX) has formed underneath the protective alumina scale, accompanied by nitride formation in the alloy substrate.
Corrosion of Samples Pre-oxidized 24 h at 900 °C
The samples pre-oxidized 24 h at 900 °C were much less corroded after the first corrosion exposure compared to both 1 h pre-oxidations (compare Figs. 5, 3). Nevertheless, the alumina scale had failed at a few locations. The failures were located both on the polished surface (1 µm diamond suspension) and close to the ground edges (320 SiC paper). The second corrosion exposure resulted in lateral growth of the areas that suffered localised attack during the first corrosion exposure. The areas exhibiting localized corrosion were similar to the ones described above (1 h pre-oxidations). Thus, cracks can be seen in the alumina layer, and are associated with iron-rich oxide and K2CrO4 particles (Fig. 6). It can be noted that K2CrO4 was also observed at a distance from the cracks in the alumina scale. The presence of α-Fe2O3 and K2CrO4 was corroborated using XRD (Table 2).

Optical images of pre-oxidized APMT (24 h, 900 °C) exposed for one and two corrosion exposures (O2 + H2O + N2 + KCl at 600 °C for 24 h). The SEM-BSE top view micrograph (left) and SEM-SE image on the right-hand side shows the corrosion front with a visible crack tip after two corrosion exposures

SEM-BSE micrograph of pre-oxidized APMT (24 h, 900 °C) exposed for two corrosion exposures (O2 + H2O + N2 + KCl at 600 °C for 24 h) showing the cracked alumina scale. Iron oxide and K2CrO4 have formed in the cracks
The top view SEM-BSE image of an area at the corrosion front in Fig. 5 (lower left), far away from the iron-rich oxide, shows the end of a crack in the alumina scale. Iron-rich oxide was not detected in imaging from top view. In order to investigate the early stages of alumina scale failure, a TEM specimen was prepared at the crack tip normal to the direction of the crack (see dotted line). An image of the TEM sample before the final thinning is shown in Fig. 5 (lower right), and reveals a wedge-shaped oxide underneath the alumina layer. SEM/EDX analysis showed that the oxide wedge is iron-rich. Figure 5 shows that the alumina/iron oxide scale is partly detached from the alloy. The point where the alumina scale is detached from the alloy coincides with the tapered end of the iron rich oxide wedge and is considered to correspond to the corrosion front. A comparison of the top view and cross section images in Fig. 5 reveals that the crack tip, seen in top-view, does not coincide with the corrosion front as captured by the cross-section image. Figure 7 shows a STEM micrograph of the corrosion front, complemented by STEM/EDS elemental maps. The analysed TEM foil had a thickness of about 100 nm and carbon and tungsten were deposited for protection of the surface during sample preparation. The interface between the carbon/tungsten deposit and the alumina scale is illustrated with the dotted line in Fig. 5. During final sample thinning, part of the iron-rich oxide wedge was milled away by gallium ions, thus enlarging the void seen in Fig. 5. The void was also affected by re-deposition. Therefore, the STEM/EDS analysis was performed at the far left in the micrograph (boxed area in Fig. 5).

Elemental maps of the boxed area in the STEM-HAADF micrograph of pre-oxidized APMT (24 h, 900 °C), exposed two corrosion exposures (O2 + H2O + N2 + KCl at 600 °C for 24 h)
Figure 7 depicts a High Angle Annular Dark Field (HAADF)-STEM micrograph of the area where the alumina scale detached from the alloy substrate (corresponding to the corrosion front). To the left of the corrosion front, the alumina scale is well adhered to the alloy substrate, i.e., no porosity was detected in the alumina/alloy interface. The boxed area in Fig. 7 was subjected to STEM/EDS mapping. The corresponding elemental maps for iron and chromium indicate that a Fe, Cr oxide is present just to the right of the corrosion front, to the far right in the boxed area. This corresponds to the remains of the iron-rich oxide wedge in Fig. 5. Gallium was detected together with the Fe,Cr oxide to the far right in the boxed area, showing that this area had been affected by sample preparation. The remaining part of the boxed area was not affected by the re-deposition. Potassium was enriched in the interface between the alumina scale and alloy close to the corrosion front while chlorine was detected throughout the whole alumina/alloy interface (Fig. 7). In addition STEM/EDS analyses (not shown) showed that the alloy grain boundary in Fig. 7 was enriched in Mo while the alloy grains were depleted in Mo. It should be noted that Mo is homogeneously dispersed in the alloy matrix in the as-delivered state. No evidence of nitride formation was found in the TEM foil beneath the oxide scale.
Corrosion of Samples Pre-oxidized 24 h at 1,100 °C
The samples subjected to 24 h of pre-oxidation at 1,100 °C showed no signs of corrosion after two consecutive corrosion exposures (not shown). However, the third corrosion exposure caused the alumina scale to fail locally, the corrosion morphology was similar to the one described for the other pre-oxidized samples (Fig. 8). In this case, the corrosion started at the sample edges. This is in contrast to the samples pre-oxidized for 24 h at 900 °C where corrosion sometimes started on the polished surface, far away from the edges. After initiation, the corroded areas grew laterally over the surface (Fig. 8). Similar to the other pre-oxidized samples, iron-rich oxide and K2CrO4 were detected in association with the cracks in the alumina layer and on areas where the alumina scale had spalled (see Table 2; Fig. 8). It may be noted that K2CrO4 particles were detected, not only in association with cracks in the alumia layer but also on top of the intact alumina surface, far away from cracks. It is believed that the chromate had formed through Reaction (1) (see “Discussion” section). It is suggested that the chromia needed for the reaction to occur originated from the transient oxide from the pre-oxidation.

Optical images of pre-oxidized APMT (24 h, 1,100 °C) exposed for three and four corrosion exposures (O2 + H2O + N2 + KCl at 600 °C for 24 h). The SEM-SE top-view micrograph (lower left, four corrosion exposures) of the periphery of a corroded area shows the cracked and partly spalled alumina layer. The cross section was obtained at a crack tip in the alumina layer as indicated by the dotted line
A TEM specimen was prepared from the area at a crack tip, see dotted line in Fig. 8 (top view image, lower left). Figure 8 shows an image of the TEM sample before thinning (lower right), in which the cracked alumina layer with a thick oxide scale can be seen underneath. The STEM micrograph in Fig. 9 was complemented by the STEM/EDS line scan from A to B and by point analyses.

STEM-HAADF micrograph of pre-oxidized APMT for (24 h, 1,100 °C), after four consecutive corrosion exposures (O2 + H2O + N2 + KCl at 600 °C for 24 h). The micrograph shows the area at the crack tip in Fig. 8. The EDS line profile from A to B probes the bottom part of the original alumina scale, the oxide underneath it and the corresponding IOZ
The STEM-HAADF image of the cross-section in Fig. 9 reveals that the crack seen in the top view image (Fig. 8) does not reach all the way through the alumina scale. It may be noted that is difficult to prepare a TEM foil specimen exactly at the crack tip because the TEM foil is only about 100 nm thick. The crack is believed to run through the entire alumina layer at some location close to the region represented by the obtained TEM foil. The area sampled by the TEM foil shows a relatively advanced stage of corrosion attack, with a rather thick (~1.5 µm) oxide present underneath the alumina layer. It can be seen that the oxide scale is partly cracked and contains several large voids. At the bottom of the scale an Internal Oxidation Zone (IOZ) seems present (see below).
In the vicinity of the voids on the left-hand side in Fig. 9, there are signs of re-deposition from sample preparation (mostly Ga). Consequently, the EDS analysis was conducted on the right-hand side of the micrograph. The RE particle indicated in the alumina layer contained low amounts of Ta and Zr together with alumina. The EDS line-scan (A to B in the micrograph) probed the bottom part of the alumina layer, the oxide underneath it and the OAZ in the alloy. The top 4/5 of the oxide below the alumina layer was dominated by iron oxide but also exhibited Al- and Cr-rich bands containing small amounts of K, Si and Cl. Below the iron oxide there was a ~200 nm thick layer which consisted of chromia and alumina, and where the alumina component was closest to the alloy. The XRD (Table 2; Fig. 4) analyses did not result in any diffraction from Cr2O3 and it is believed to be due to the relative thin layers that are present in the scale. EDS point analysis indicated that the area immediately below the chromia/alumina layer had been partly oxidized. This ~200 nm thick layer is interpreted as an IOZ.
The lateral crack between the alumina and the iron-rich oxide layer (boxed area in Fig. 9) was investigated further with STEM/EDS elemental mapping (Fig. 10). The analysis showed that the crack was enriched in Cr, K and Cl. It may be noted that potassium and chlorine (corresponding to KCl) were also present in a grain boundary in the alumina layer (to the right in Fig. 10).

EDS Elemental maps of the boxed area in the STEM-HAADF micrograph in Fig. 9 of pre-oxidized APMT (24 h, 1,100 °C), exposed two corrosion exposures (O2 + H2O + N2 + KCl at 600 °C for 24 h). A alumina grain boundary is indicated in the STEM-BF image
A TEM sample was prepared from an area far away from the corrosion front on the sample shown on the upper right in Fig. 8 (after 4 corrosion cycles). In this area top-view imaging revealed no cracks in the alumina scale. Figure 11 shows a STEM-BF image of the cross-section with an adherent alumina layer. No corrosive species e.g. Cl or K were detected using EDS point analyses and elemental mapping (not shown) either in the alumina scale/alloy interface or in the oxide grain boundaries. On top of the alumina scale there was a layer consisting of K, Si, O and Al. Because no crystalline corrosion products were found other than alumina, this layer is suggested to consist of potassium aluminium silicate glass. Previously, crystalline KAlSiO4 was identified on Kanthal® APMT after pre-oxidizing at 700 °C for 24 h in O2 and then exposed to an O2 + H2O environment in the presence of KCl at 600 °C [17].

STEM-BF micrograph of pre-oxidized APMT (24 h, 1,100 °C), exposed four corrosion exposures (O2 + H2O + N2 + KCl at 600 °C for 24 h). The TEM specimen was obtained far away from the corrosion front
The observation that the KCl-induced corrosion of the pre-oxidized samples always began at the sample edges prompted an investigation of the alumina scale formed by pre-oxidation for 24 h at 1,100 °C. While SEM/EDX top-view imaging of the polished surface showed a very smooth alumina scale, the ground sample edges revealed small (~5–10 µm in size) iron-rich oxide nodules (Fig. 12a). It was estimated that about 200 of these iron-rich oxide nodules were present at the sample edges after pre-oxidation. Figure 12b and c shows top-view and cross-section SEM images of one of the iron-rich oxide nodules. The top-view image shows the scratches from grinding. The cross-section shows that a continuous alumina layer was present underneath the iron-rich oxide although it was thinner than the alumina layer formed next to the nodule. Figure 12d illustrates the situation at the edge of a sample before the corrosion exposure, showing a KCl particle in the vicinity of an iron-rich oxide nodule. KCl was also detected on top of some of the iron-rich protrusions before exposure.

SEM-BSE images (a, d) and SEM-SE (b, c) of top view and cross section of one grounded edge (320 SiC paper) of pre-oxidized APMT (24 h, 1,100 °C) showing iron-rich oxide nodules. d shows an image of the pre-oxidized edge with added KCl before exposure
Discussion
Previous studies have shown that small amounts of KCl in an O2 + N2 + H2O environment at 600 °C causes rapid corrosion of the FeCrAl alloys Kanthal® APMT and AF [15–17]. In other words in the presence of KCl, the normally protective and alumina-rich external oxide was replaced with a rapidly growing iron-rich scale, and the two FeCrAl alloys behaved similar to FeCrNi alloys in the same environment [10]. It was reported that the top part of the iron-rich scale contained potassium chromate while transition metal chloride i.e. FeCl2 was detected at the alloy/scale interface. In addition, AlN and Al2O3 particles were found in an OAZ in the alloy substrate. The failure of the alloys to form a protective alumina layer can be explained primarily as chromate formation according to Reaction (1):
It has been argued that the Cr depletion of the external oxide and the metal substrate resulting from (1) made the formation of an external alumina scale more difficult, considering the important role played by Cr in the formation of protective alumina layers [21]. It has been suggested that the resulting iron-rich scale was porous, allowing N2 molecules to penetrate to the alloy. The ensuing precipitation of aluminium nitride in the alloy made the formation of an alumina scale more difficult. Accordingly, it has been reported that after some time, when the KCl source was exhausted, alloy APMT formed a chromia layer underneath the iron-rich scale, rather than an alumina layer [15].
This study investigated the ability of a pre-formed external alumina scale to prevent KCl-induced corrosion. The scale cross sections in Fig. 1 and the XRD analysis showed that all four pre-oxidations (1 and 24 h at 900 and 1,100 °C in O2 + N2) generated continuous α-alumina layers. The oxidation behaviour is in line with literature reports on the ability of alloy APMT and other FeCrAl alloys to form highly protective α-alumina scales in the presence of O2 at 900 °C and higher [22–24].
While this study confirms that a pre-formed alumina scale can provide protection in a KCl + O2 + H2O environment at 600 °C, it also shows that the preformed scales do not survive exposure to repeated KCl corrosion cycles. In view of the differences observed between the pre-formed scales (see above) their differences in effectiveness in protecting the alloy does not come as a surprise. The relative protectiveness of the preformed scales was: (900 °C 1 h) < (1,100 °C 1 h) < (900 °C 24 h) < (1,100 °C 24 h), see Figs. 3 and 8. The present study investigated the mode of failure of the pre-formed alumina scale in the presence of KCl and the subsequent corrosion process.
Break-up of the Pre-formed Alumina Scale
As shown in Results, corrosion and the associated break-up of the alumina scale were initially highly localized and much of the surface was seemingly unaffected. Repeated KCl corrosion exposures caused corrosion to spread over the sample surface (Figs. 3, 5 and 8). The lateral movement of the “corrosion front” is associated with the spreading of cracks in the alumina scale (Figs. 3, 8). The absence of signs of corrosion e.g.,de-cohesion of the scale/substrate interface, penetration of Cl in the areas where the alumina scale is not cracked (Fig. 11), indicates that the preformed alumina scale is inherently protective in this environment. Thus, this study implies that KCl-induced corrosion is initiated at flaws in the alumina layer, because an intact alumina layer is impermeable to attack. It is inherently difficult to investigate how corrosion starts using “post-mortem” investigations. However, it is suggested that the periphery of the corroded areas corresponds to early stages of corrosion. More specifically, this applies to the end of the laterally growing cracks in the alumina scale where the alumina layer has not yet separated from the alloy substrate. It is argued that investigation of those areas can help elucidate, not only how the “corrosion front” moves but also the initiation of corrosion (Figs. 5, 6, 7, 8, 9, and 10).
A typical case is shown in Figs. 5, 6 and 7 (24 h pre-oxidation at 900 °C, one and two corrosion cycles). The cracks in the alumina scale are associated with potassium chromate and iron-rich oxide at the scale/gas interface (Fig. 6). Below the alumina, a layered iron-rich oxide had formed as a result of corrosion. It may be noted that the crack in the alumina scale seen in the top-view (Fig. 5 lower left) corresponds to de-cohesion of the alumina/alloy interface (Figs. 5, 7). Significantly, chlorine was detected at the alumina/alloy interface in front of the crack tip, in a region where the Al2O3 layer remained seemingly intact and in contact with the alloy substrate (Fig. 7). The same image shows that chlorine penetrated far ahead of potassium, which indicates that transition metal chlorides (FeCl2 and/or CrCl2) were present at the alumina/alloy interface. Chlorination with HCl(g) (generated by Reaction (1)) is not considered to be the source of subscale chloride in this case because the HCl generated by Reaction (1) will form at the gas/scale interface and will be transported away from the sample by the flowing gas.
The influence of alkali chlorides and other chlorine-containing species on the high temperature corrosion of different metals/alloys has been investigated by several researchers [13, 14, 25–29]. The strong acceleration of corrosion by HCl, KCl and NaCl has been attributed to alloy chlorination, e.g. by the “chlorine cycle” mechanism proposed by Grabke et al. [30] and more recently by an electrochemical mechanism [15, 16, 20, 31].
In the present case, it is proposed that the formation of sub-scale transition metal chloride is part of an electrochemical reaction involving the metal substrate, KCl, Cr2O3 and O2 (written for FeCl2 formation):
∆G°873K = −141.458 kJ [32], corresponding to a standard cell potential E° of +0.73 V.
According to observations, the products of Reaction (2) accumulate at the scale/gas interface (K2CrO4) and at the scale/alloy interface (FeCl2) (see Figs. 3a3, 6, and 7). It is argued that, although all the reactants are present at their respective positions, Reaction (2) cannot run if the α-alumina scale is intact because the alumina scale constitutes an effective barrier to the diffusion of ions (Fe2+, Cl−). Once the scale cracks, the situation is fundamentally changed. It is proposed that, when a crack forms during the exposure, KCl immediately enters it by means of surface diffusion, forming an adsorbed layer on the freshly exposed alumina in the crack. This is illustrated in Fig. 13, steps I and II. This is supported by the observation of KCl in a newly formed crack, close to the scale/alloy interface after exposure (Fig. 7). It is proposed that KCl in the crack provides the medium for the ionic current i.e., transport of chloride ions, which is necessary for Reaction (2) to run. Moreover, it is proposed that the corresponding electronic current is carried by alumina (Fig. 13, step III).

Schematic illustration of the initiation of the corrosion attack coupled to Reaction (2)
Thus, in the presence of a KCl “salt bridge” across the scale, the anodic formation of FeCl2 at the scale/alloy interface becomes coupled to the formation of potassium chromate at the scale/gas interface (step IV in Fig. 13). Close to the scale/alloy interface, the anodic formation of FeCl2 (Reaction (2), Fig. 7) causes charged chloride ion vacancies to form in the adsorbed KCl. Simultaneously, at the scale/gas interface, the cathodic reaction consumes K+ ions, thus generating an excess of chloride ions in KCl.
This scenario is based on physical properties, i.e., conductivities, reported for alkali chlorides and α-alumina. Alkali chlorides are good ionic conductors at 600 °C, since electrical current is carried solely by ions [33]. The diffusion of chloride ions in KCl has been reported to occur by the jump of an ion into a neighbouring anion vacancy, the activation energy for a single anion vacancy jump being 0.95 eV [34]. The size of the activation energy was recently confirmed by Density Functional Theory (DFT) calculations performed at Chalmers University of Technology.
At 600 °C, the diffusion coefficient for the chlorine ion vacancy diffusion in KCl has been reported to be 4 × 10−11 cm2/s [34]. The time needed for a chloride ion to diffuse across the scale through the KCl film can be estimated from the diffusion coefficient and using the equation for the mean square displacement in three dimensions. The diffusion time becomes 167 s if the random diffusion distance is assumed to be 2 µm. Thus, in the present scenario, chloride transport is suggested to occur by jump diffusion in KCl, driven by the gradient in the vacancy concentration and by the potential difference set up between anode and cathode.
The electrical conductivity of α-alumina has been reported to be dominated by electronic conduction, since α-alumina is a very poor conductor of ions [35]. Thus, at 600 °C, single-crystal α-alumina is an extrinsic semiconductor with E a = 0.4 eV, conduction being dominated by impurity states in the crystal [35]. Based on [33, 35] the ionic conductivity of single-crystal KCl at 600 °C is estimated to be 1010–1012 larger than the ionic conductivity of single-crystal alumina. Thermally grown alumina scales on high temperature alloys differ from the material investigated in [35], because alumina scales are both polycrystalline and relatively impure. The electric conductivity and diffusion properties of alumina scales formed on FeCrAl alloys have been reported by e.g. Badini and Laurella, and Öijerholm et al. [23, 36]. Öijerholm et al. [36] investigated electrical conductivity and ionic transport in α-Al2O3 scales formed at 900 and 1,000 °C on the FeCrAl alloy Kanthal® AF (the measurements were performed at 800–1,000 °C). The results of that research essentially confirm the conclusions drawn from the single-crystal sapphire study cited above. These findings were that the alumina scales were electronic conductors that behaved as heavily doped, wide-band-gap semiconductors, and that they were very poor ionic conductors. Thus, the ability of the intact alumina scale to conduct electrons and its very poor ionic conductivity explains why the presence of small amounts of KCl on the walls of a crack through the alumina scale can trigger a corrosion attack by establishing electrolytic contact between the scale surface and regions close to the alloy/scale interface.
Transition metal chloride formation is associated with a large expansion in volume. It is proposed that this is the primary cause behind the de-cohesion of the alumina scale/alloy interface and the cracking of the alumina scale. The process of scale de-cohesion and cracking is expected to continue for as long as KCl is present. In the region behind the crack tip, the alloy substrate becomes exposed to the gas environment, resulting in the rapid growth of an iron-rich oxide scale below the alumina layer (Fig. 9). The growth of the iron oxide layer also produces an expansion in volume that further contributes to the cracking of the alumina scale. The observation that the alloy is unable to re-form an alumina layer behind the crack tip, where it is exposed to O2, is attributed to the relatively low temperature (600 °C) and the presence of KCl. The sequence of events described above that lead to scale failure is schematically illustrated in Fig. 13. Electrochemical reactions similar to (2) have previously been suggested to explain the formation of sub-scale transition metal chloride after the exposure of FeCrAl alloys to oxidising environments with KCl [15, 16]. However, in those cases KCl was not detected below the alumina scale. Consequently, the transport of chloride ions was suggested to occur, not in KCl, but in the oxide grain boundaries in the scale.
It is argued that the initiation of the KCl-induced corrosion of the FeCrAl alloy with a pre-formed alumina scale is similar to the process sketched above that explains scale de-cohesion and alumina crack spreading. Thus, it is suggested that the electrochemical Reaction (2) has a seminal role and that corrosion starts at a crack or some other type of flaw in the alumina scale that allows KCl to penetrate the scale. This puts the scale/gas and alloy/scale interfaces into electrochemical contact, allowing the reaction to proceed and form transition metal chloride at the alumina/alloy interface and K2CrO4 on the scale surface. It is considered significant that corrosion mainly starts at the sample edges (see Fig. 8). The thermal mismatch between alumina scale and alloy substrate gives rise to mechanical stress and strain, e.g., during cooling after pre-oxidation and between corrosion cycles, that can damage the scale. Thus, the tendency for corrosion to start at the sample edges may be explained by the tendency of thermal-mismatch-generated flaws in the alumina scale to be concentrated to those regions because of geometrical constraints.
Another option is that the distribution of initial attack is related to the state of the surface of the alloy. For instance, while the front and back side of the sample (15 × 15 mm) were polished before exposure (using 1 μm diamond suspension), the rims of the sample (2 × 15 mm) were ground using 320 mesh SiC paper. Consequently, the morphology of the pre-formed alumina scale differed on the two types of surface, and iron-rich oxide nodules appeared on the ground rims that were absent on the polished surface (Fig. 12). In the cross section of an iron-rich nodule shown in Fig. 12c, the iron oxide is separated from the alloy by an alumina layer which is significantly thinner than the alumina scale in general. It can be speculated that the alumina scale had been damaged at the oxide nodules and that the corrosion started there which would explain the tendency for corrosion to start at the edges. With the information at hand, the initiation of alumina scale failure and corrosion in the presence of KCl may be explained by either cracks caused by thermal mismatch stresses or by the iron oxide nodules at the edges.
The results of this research show that repeated KCl corrosion cycles pose an increasing challenge to scale integrity; for instance, the “best” pre-formed oxide (1,100 °C, 24 h) started to break down during the third KCl consecutive corrosion cycle. This effect is suggested to be due, either to the renewal of KCl in each cycle or to the associated thermal cycling.
Nitridation
The STEM/EDS line profile in Fig. 9 shows that the oxide underneath the failed alumina layer essentially consisted of iron oxide, most likely hematite. In the middle of this layer, the iron oxide was mixed with considerable amounts of chromium and aluminium oxide together with small amounts of chlorine, potassium and silicon. It is argued that this region corresponds to the alloy immediately below the scale/alloy interface, before the failure of the alumina scale. This implies that the iron oxide layer had grown both outward and inward. Below the iron-rich oxide is a layer of relatively pure chromia. The lower part of this chromia layer, at the new scale/alloy interface, is enriched in alumina. It may be noted that the oxide underneath the failed alumina scale is very similar to the scale formed when the same alloy has been exposed to this environment without pre-oxidation [15].
After scale failure, the alloy suffered nitridation, especially in the case of the samples pre-oxidized for 1 h (Fig. 3). Once again, the corrosion behaviour was similar to that reported for the same alloy without pre-oxidation [15]. The needle-like nitride precipitates were analysed in detail in [15] and it was concluded that they consisted of AlN. Nitridation of the investigated alloy was not detected in the absence of KCl [17]. Nitridation implies that the oxide scale is permeable to N2 molecules. It is argued that the corrosion attack connected to the formation of K2CrO4 and to alloy chlorination (Reaction (2)) creates pathways/cracks through which N2 molecules to penetrate the oxide scale. N2, subsequently, dissociates at the metal surface and dissolves into the alloy. Nitridation is expected to make it more difficult for the alloy to form a new protective external alumina scale because it depletes the alloy substrate in aluminium. It may be noted that in the present study, nitride particles were not observed in the cross sections prepared from regions close to the alumina crack tips (Fig. 7). This is not unexpected in an area where the alumina scale has recently failed, because the processes of N2 diffusion, dissolution and nitride nucleation are not instantaneous.
Conclusions
-
Pre-formed α-alumina scales were unable to protect the material against corrosion when subjected to repeated exposure to KCl in an N2 + O2 + H2O environment at 600 °C.
-
Corrosion started at flaws in the pre-formed alumina scales, because the alumina is protective in itself. Consequently, KCl-induced corrosion started locally and, subsequently, spread over the surface.
-
Corrosion can be explained by an electrochemical mechanism by which transition metal chloride forms at the scale/alloy interface and K2CrO4 forms at the scale/gas interface.
-
The electrochemical process is conditioned by the presence of a flaw in the scale that allows KCl to penetrate the alumina scale, putting the scale/gas and scale/alloy interfaces into electrochemical contact. KCl acts as an electrolyte that transports chloride ions from the scale/gas interface to the alloy.
-
The formation of transition metal chloride is associated with a large volume expansion and causes de-cohesion of the scale/alloy interface and scale cracking.
-
The alloy substrate becomes exposed to the gas environment after de-cohesion due to the formation of the transition metal chloride, and this results in the rapid growth of iron-rich oxide below the alumina layer. The associated expansion in volume contributes to alumina scale cracking.
-
The formation of AlN causes a depletion of aluminium in the alloy and reduces the alloy’s ability to form a protective alumina scale.
References
H. P. Michelsen, F. Frandsen, K. Dam-Johansen, and O. H. Larsen, Fuel Processing Technology 54, 95 (1998).
B. Sander, Biomass and Bioenergy 12, 177 (1997).
F. J. Frandsen et al, in Deposition and Corrosion in Straw- and Coal-Straw Co-fired Utility Boilers—Dansih Experiences, eds. R. P. Gupta, T. F. Wall, and L. Baxter (Kluwer Academic/Plenum Publishers, New York, 1999).
L. L. Baxter, T. R. Miles, T. R. Miles Jr, B. M. Jenkins, T. Milne, D. Dayton, R. W. Bryers, and L. L. Oden, Fuel Processing Technology 54, 47 (1998).
R. L. Bain, R. P. Overend, and K. R. Craig, Fuel Processing Technology 54, 1 (1998).
B. M. Jenkins, L. L. Baxter, T. R. Miles Jr., and T. R. Miles, Fuel Processing Technology 54, 17 (1998).
H. Kassman, J. Pettersson, B.-M. Steenari, and L.-E. Åmand, Fuel Processing Technology 105, 170 (2013).
P. Viklund, A. Hjörnhede, P. Henderson, A. Stålenheim, and R. Pettersson, Fuel Processing Technology 105, 106 (2013).
M. Montgomery and A. Karlsson, Materials and Corrosion 50, 579 (1999).
C. Pettersson, L.-G. Johansson, J.-E. Svensson, and Oxidation of metals 70, 241 (2008).
S. Karlsson, J. Pettersson, L.-G. Johansson, and J.-E. Svensson, Oxidation of Metals 78, 83 (2012).
M. Halvarsson, J. E. Tang, H. Asteman, J.-E. Svensson, and L.-G. Johansson, Corrosion Science 48, 2014 (2006).
Y. S. Li, Y. Niu, and M. Spiegel, Corrosion Science 49, 1799 (2007).
Y. S. Li, M. Sanchez-Pasten, and M. Spiegel, Materials Science Forum 461–464, 1047 (2004).
N. Israelsson, K. A. Unocic, K. Hellström, T. Jonsson, M. Norell, J. E. Svensson, and L. G. Johansson, Oxidation of Metals 2015. doi:10.1007/s11085-015-9546-3).
N. Israelsson, K. Hellström, J. E. Svensson, and L. G. Johansson, Oxidation of Metals 83, 1 (2015).
N. Israelsson, J. Engkvist, K. Hellström, M. Halvarsson, J. E. Svensson, and L. G. Johansson, Oxidation of Metals 83, 29 (2015).
K. Hellström, N. Israelsson, M. Halvarsson, S. Canovic, J. E. Svensson, and L. G. Johansson, Oxidation of Metals 2015. doi:10.1007/s11085-015-9538-3.
K. Hellström, N. Israelsson, N. Mortazavi, S. Canovic, M. Halvarsson, J. E. Svensson, and L. G. Johansson, Oxidation of Metals 2015. doi:10.1007/s11085-015-9534-7).
T. Jonsson, N. Folkeson, J.-E. Svensson, L.-G. Johansson, and M. Halvarsson, Corrosion Science 53, 2233 (2011).
R. Prescott and M. J. Graham, Oxidation of Metals 38, 233 (1992).
J. Engkvist, S. Canovic, K. Hellström, A. Järdnäs, J.-E. Svensson, L.-G. Johansson, M. Olsson, and M. Halvarsson, Oxidation of Metals 73, 233 (2010).
C. Badini and F. Laurella, Surface & Coatings Technology 135, 291 (2001).
G. Berthomé, E. N’ Dah, Y. Wouters, and A. Galerie, Mater. Corrosion 56, 389 (2005).
T. Jonsson, J. Froitzheim, J. Pettersson, J.-E. Svensson, L.-G. Johansson, and M. Halvarsson, Oxidation of Metals 72, 213 (2009).
J. Lehmusto, P. Yrjas, B. J. Skrifvars, and M. Hupa, Fuel Processing Technology 104, 253 (2012).
S. Enestam, D. Bankiewicz, J. Tuiremo, K. Mäkelä, and M. Hupa, Fuel 104, 294 (2013).
C. Pettersson, J. Pettersson, H. Asteman, J.-E. Svensson, and L.-G. Johansson, Corrosion Science 48, 1368 (2006).
M. A. Uusitalo, P. M. J. Vuoristo, and T. A. Mäntylä, Corrosion Science 46, 1311 (2004).
H. J. Grabke, E. Reese, and M. Spiegel, Corrosion Science 37, 1023 (1995).
N. Folkeson, L.-G. Johansson, and J.-E. Svensson, journal of the electrochemical society 154, C515 (2007).
C. W. Bale, E. Bélisle, P. Chartrand, S. A. Decterov, G. Eriksson, K. Hack, I. H. Jung, Y. B. Kang, J. Melançon, D. Pelton, C. Robelin and S. Petersen, Calphad 33, 295 (2009).
F. Beniere, 2 Diffusion in alkali and alkaline earth halides. (Springer Materials - Landolt-Börnstein Database. doi:10.1007/10542761_6).
R. G. Fuller, Physical Review 142, 524 (1966).
J. F. Shackelford and R. H. Doremus, Ceramic and Glass Materials (Springer, New York, 2008), pp. 14–17.
J. Öijerholm, J. Pan, Q. Lu, and C. Leygraf, Oxidation of Metals 68, 253 (2007).
Acknowledgments
TEM research was supported by the Center for Nanophase Materials Sciences (CNMS), which is sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. The authors would like to thank D.W. Coffey for assistance with sample preparations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Israelsson, N., Unocic, K.A., Hellström, K. et al. Cyclic Corrosion and Chlorination of an FeCrAl Alloy in the Presence of KCl. Oxid Met 84, 269–290 (2015). https://doi.org/10.1007/s11085-015-9554-3
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-015-9554-3