
Abstract
Small scale laboratory experiments on the oxidation of liquid silicon have reproduced important features of the industrial refining of liquid silicon: active oxidation led to the formation of amorphous silica spheres as a reaction product. The boundary condition for active oxidation in terms of maximum oxygen partial pressure in the bulk gas was found to lie between 2·10−3 and 5·10−3 atm at T = 1,500 °C. The active oxidation of liquid silicon had linear kinetics, and the rate was proportional to bulk oxygen partial pressure and the square root of the linear gas flow rate, consistent with viscous flow mass transfer theory. Classical theory for unconstrained flow over a flat plate led to mass transfer rates for SiO(g) which were 2–3 times slower than observed. However, computational fluid dynamic modeling to take into account the effects of reactor tube walls on flow patterns yielded satisfactory agreement with measured volatilization rates.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
One of the main environmental and economic challenges facing the metallurgical industry is fugitive emissions of both materials and energy. In the production of metallurgical grade silicon (MG-Si), the tapping and refining processes are generally the biggest sources of fugitive emissions in the plant. Silicon refining is performed in the ladle by purging with an air–O2 mixture in order to reduce the levels of the main impurities calcium and aluminum. Oxygen in the refining bubbles is consumed through the overall reaction with the melt
The refining exchange reactions may be written respectively
where underlining denotes solute species. These exchange reactions recycle some of the silicon back to the melt and deplete the silicon of calcium and aluminum [1]. An equilibrium partial pressure of SiO(g) is simultaneously established in the bubbles [2].
In the refining process, exposure of liquid silicon to air results in the formation of large amounts of condensed silica fume (SiO2)—one of the main sources of fugitive emissions in a silicon production plant. In an earlier study [3], this fume was characterized, and its generation quantified. The fuming mechanism was concluded to be active oxidation of the liquid silicon surface, as described below. The reaction product consisted of spherical, amorphous silica particles with an average diameter of ~60 nm. The flux of silicon in this particular process was found to be 0.031–0.076 mol m−2 s−1.
In 1958, Wagner [4] published a theory regarding the active and passive oxidation of liquid silicon, where the active oxidation involves the partial oxidation of silicon to SiO-gas:
In his paper, Wagner stated that the maximum bulk oxygen partial pressure for active oxidation to occur was 6.1·10−3 atm at 1,410 °C. If the bulk oxygen pressure was higher than this, passivation would occur: an oxide layer would form on the surface, making the production of SiO-gas very slow [4]. However, Wagner did not account for the fate of the SiO(g) formed during active oxidation, and the maximum oxygen pressure he stated for active oxidation was much lower than that in ambient air. Thus this theory does not match the observation of active silicon surface oxidation in the industrial ladle refining process. Accordingly, Ratto et al. [5]. presented a modified theory regarding the occurrence of active oxidation at higher oxygen pressures: If the SiO(g) formed at the silicon surface reacts with incoming oxygen inside the hydrodynamic boundary layer, the oxygen partial pressure at the surface can be low enough for active oxidation to occur. According to their theory of a heterogeneous boundary layer (that is, with condensed matter inside the boundary layer), the surface oxygen pressure is independent of the bulk oxygen pressure. Hence active oxidation of silicon in ambient air is possible.
Hinze and Graham [6] investigated the oxidation mechanism of solid silicon at high temperatures. They used a set of equilibrium equations in the Si–O system to calculate the possible bulk oxygen partial pressures to obtain active oxidation, and calculated an experimental mass transfer coefficient from their experimental results. The results were in accordance with the theoretical calculations, but the active oxidation of solid silicon had two different stages. The first stage was the “normal” active oxidation with a bare silicon surface, and the second stage showed growth of silica whiskers at the silicon surface, but still at a high rate. Growth of these silica whiskers at the silicon surface was also found by Carter et al. [7] who studied silicon bearing alloys at 1,130 °C. The growth of these structures at the silicon surface is probably not applicable for liquid silicon, as the surface structure is so different. However, Hinze and Graham’s [6] technique for evaluating the system and calculating the experimental mass transfer coefficient may be applied to liquid silicon oxidation.
The oxidation of liquid silicon, as opposed to solid silicon, has not been extensively experimentally studied. A thorough investigation of the oxidation mechanisms and factors affecting the oxidation reaction rate is essential in order to obtain a more fundamental understanding of the reactions, and thus arrive at a solution for the fugitive emissions in the industry.
In the current work, the rate of liquid silicon oxidation has been studied through small scale experiments, where the kinetics and boundary conditions have been examined. The aims of this paper are thus to (1) find the rate of active oxidation as a function of oxygen pressure and linear gas velocity and (2) determine the boundary conditions for active oxidation of liquid silicon.
Thermodynamic and Kinetic Calculations
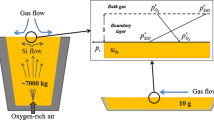
A simple schematic of the system is shown in Fig. 1. Diffusion of gaseous species both inward towards the surface and outward to the bulk gas are expected. Oxygen from the bulk, with partial pressure \( p_{{{\text{O}}_{2} }}^{ \circ } \), diffuses through a boundary layer of thickness δ, and meets the liquid silicon surface with a partial pressure \( p_{{{\text{O}}_{2} }}^{*} \). This value is very low as a result of the rapid consumption of oxygen via the reaction of Eq. (3). The resulting SiO(g), with surface partial pressure \( p_{\text{SiO}}^{*} \), diffuses out of the boundary layer. Its bulk partial pressure \( p_{\text{SiO}}^{ \circ } \) ≈ 0, due to the instability of SiO(g) and its prompt oxidation:

Schematic of active oxidation in the Si–O system, where oxygen diffuses into the surface and reacts with liquid silicon to form SiO(g), which diffuses out through the boundary layer of thickness δ
These processes generate the concentration gradients of gaseous species within the boundary layer shown schematically in Fig. 1. The diffusion of SiO(g) out to the bulk will be a counter flux to the oxygen coming in, and the production of SiO(g) is controlled by the rate of the oxygen flux. In this discussion, it is assumed that SiO(g) oxidizes to SiO2 outside the boundary layer.
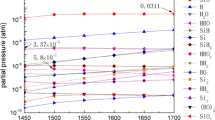
Thermodynamic equilibrium calculations using HSC Chemistry [8] were used to establish the partial pressure of Si-bearing species at 1,500 °C. The results are shown in Fig. 2, which illustrates the principal features of the Si–O system. The partial pressure of SiO is controlled by the equilibrium of Eq. (3) at oxygen activities too low for solid silica formation.

Thermodynamically calculated partial pressures of Si(g), SiO(g) and SiO2(g) as a function of \( p_{{{\text{O}}_{2} }} \) at 1,500 °C
The variation of \( p_{\text{SiO}}^{ * } \) with \( p_{{{\text{O}}_{2} }}^{ * } \) is represented by the solid line in Fig. 2, and reaches a maximum at a surface oxygen pressure of \( p_{{{\text{O}}_{2} }}^{ * } \) (max) = 2.86·10−18 atm, where SiO2 becomes stable. At higher \( p_{{{\text{O}}_{2} }}^{ * } \) values, the equilibrium in Eq. (1) leads to formation of a stable SiO2 phase, and passivation of the silicon surface. The maximum oxygen partial pressure in the bulk gas permitting active oxidation, \( p_{{{\text{O}}_{2} }}^{ \circ } \) (max), is now calculated.
Boundary value gas partial pressures are related to the rate of mass transport across the hydrodynamic boundary layer. Assuming that the melt surface is clean, and Si(l) freely available, then if chemical reaction at the liquid–gas interface is fast enough to establish local equilibrium, the molar flux of SiO(g), J SiO, from the silicon surface can be calculated:
Here k SiO is the mass transfer coefficient for SiO(g) (m s−1), R is the gas constant (8.21·10−5 m3 atm K−1 mol−1), T is the absolute temperature and the stoichiometric balance between SiO and O2 fluxes is recognized.
The mass transfer coefficient, k, is calculated from the relationship [6]:
where D i is the diffusion coefficient of species i (m2 s−1), ν i is the kinematic viscosity (viscosity, μ divided by density, ρ) (m2 s−1), v g is the velocity of the gas in the vicinity of the surface (m s−1), and l is the length of the sample (m). A condition [9] for the applicability for Eq. (6), can be expressed in terms of the Schmidt number, Sc,
The diffusion coefficients and viscosities are calculated according to the Chapman–Enskog method, as described by Bird et al. [10]. The Schmidt number for the present conditions is 0.71 for oxygen in argon, thus Eq. (6) is valid.
Experimental conditions were selected to yield the maximum rate of SiO volatilization in order to improve measurement accuracy. For T = 1,500 °C and \( p_{{{\text{O}}_{2} }}^{*} \) = 1·10−18 atm (just below the maximum \( p_{{{\text{O}}_{2} }}^{*} \)), \( p_{\text{SiO}}^{ * } \) is calculated to be 2.1·10−2 atm. This value allows calculation of the outward SiO flux from Eq. (5), as 1.47·10−3 mol m−2 s−1 at a linear gas velocity of 0.047 m s−1. The stoichiometric equivalent inward flux of oxygen of 7.34·10−4 mol m−2 s−1 then leads via the mass transfer equation:
to an estimated maximum \( p_{{{\text{O}}_{2} }}^{ \circ } \;({ \max }) \) of 8.6·10−3 atm. Higher values of \( p_{{{\text{O}}_{2} }}^{ \circ } \) are expected to produce faster rates of inward mass transfer and silicon passivation. Lower values of \( p_{{{\text{O}}_{2} }}^{ \circ } \) would reduce the rate of SiO(g) volatilization. This estimate of the maximum \( p_{{{\text{O}}_{2} }}^{ \circ } \) is independent of v g, because both k SiO and \( k_{{{\text{O}}_{2} }} \) depend in the same way on gas flow rate, and the effects cancel out.
It is of interest that this detailed mass transfer calculation leads to a result similar to that of 6.1·10−3 atm for T = 1,410 °C, calculated by Wagner [4] using an approximate estimate of mass transfer rates.
Experimental
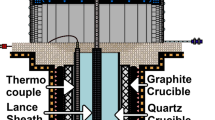
The material used in the experiments was photovoltaic grade silicon (9 N). The experiments were performed in a resistance heated furnace, fitted with an alumina tube with an inner diameter of 5.2 cm. Silicon charges of about 10 g were placed in pre-fired alumina boats of 9 cm length and 1 cm width and height, inserted parallel to the tube axis. The gas flow was parallel to the silicon surface. During heating and cooling, a gas-mixture of 5 vol.% H2 in Ar was used to avoid oxidation. The reaction gas used was premixed 1 vol.% O2 in Ar which was further diluted with high purity Ar in order to obtain the desired bulk oxygen partial pressure. Gas flow rates were controlled by mass flow controllers, and were in the range 0.023–0.14 m s−1, corresponding to Reynolds numbers of 8.5–51, well below the turbulent flow limit of 2,000. All experiments were performed at 1,500 °C. Temperatures in the sample location were uniform within ±3 °C.
Experimental conditions used are summarized in Table 1. The kinetics of SiO(g) volatilization were investigated at \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 0.5·10−3 atm and v g = 0.047 m s−1. For the other exposure conditions a single reaction time of 16 h was used in each case. Post-melting sample surface areas were measured manually.
Weight changes were measured after each exposure, and the samples were metallographically sectioned and examined by X-ray diffraction (XRD) and scanning electron microscopy (SEM). Silica found deposited on the furnace wall was removed from the alumina tube and examined by XRD and SEM.
Glancing angle XRD on the surface of a passive and an active sample was performed at angles of 2.35° and 5.5°, respectively, using a PANalytical Xpert Materials Research Diffractometer. Conventional XRD on the silica collected from the tube was conducted with a D8 Focus Advance from Bruker.
The SEM used was a Zeiss Ultra 55 Limited Edition field emission microscope, operated at 5 kV.
Results
Initial experiments at \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 1·10−2 atm resulted in passivation. Oxidation of the Si-surface is clearly shown in the cross section as a thick oxide scale in Fig. 3a. The structure of the surface layer was identified by low angle XRD as a mixture of amorphous silica and the crystalline phase of SiO2 cristobalite (syn), shown in Fig. 4a. Passivation also resulted at \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 5·10−3 atm but was avoided at lower oxygen pressures. The samples where active oxidation occurred had a visually clean surface.

SEM image of sample cross section after a passive and b active oxidation. Experimental conditions: a v g = 9.3 cm s−1, t = 16 h, \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 1·10−2 atm, b v g = 4.7 cm s−1, t = 16 h, \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 0.5·10−3 atm

Glancing XRD diffraction patterns of the surface of a passive sample (a) and an active sample (b). Experimental conditions: a v g = 9.3 cm s−1, t = 16 h, \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 1·10−2 atm, b v g = 4.7 cm s−1, t = 16 h, \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 1·10−3 atm
Cross section analysis by SEM showed no deposits formed on the silicon surface (Fig. 3b), and the low-angle XRD did not detect any silica, only silicon (Fig. 4b). Closer examination of the Si diffraction seen in Fig. 4a and b reveals that the silicon had developed different textures during solidification with and without a silica coating.
Weight loss kinetics are shown in Fig. 5 for \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 0.5·10−3 atm and v g = 0.047 m s−1. The regression line has an R 2 value of 0.74.

Weight loss kinetics at T = 1,500 °C, \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 0.5·10−3 atm and v g = 0.047 m s−1. Regression line has an R 2 value of 0.74
The rate of weight loss (r) as a function of bulk oxygen partial pressure, \( p_{{{\text{O}}_{2} }}^{ \circ } \) at a flow rate of v g = 0.047 m s−1 is displayed in Fig. 6, with an R 2 value of the regression line of 0.90.

Rate of weight loss, r, as a function of bulk oxygen pressure at 1,500 °C and v g = 0.047 m s−1
The linear dependence of rate on \( p_{{{\text{O}}_{2} }}^{ \circ } \) confirms the applicability of Eq. (8). The slope of the curve is related through Eq. (9) to the mass transfer coefficient for oxygen, \( k_{{{\text{O}}_{2} }} \):
The experimental mass transfer coefficient estimated this way is \( k_{{{\text{O}}_{2} }}^{\text{ex}} \) = 3.06 cm s−1 for a linear velocity of 0.047 m s−1 and T = 1,500 °C. The variations in the rate of weight loss with (v g /l)0.5 [see Eq. (6)] is shown in Fig. 7, where the linear dependency is apparent. The R 2 value for the linear regression in Fig. 7 is 0.73.

Rate of weight loss as a function of the square root of linear velocity divided by sample length, measured at T = 1,500 °C and \( p_{{{\text{O}}_{2} }}^{ \circ } \) = 0.5·10−3 atm
The experiments which gave active oxidation produced a silica layer deposited on the tube wall. The XRD spectrum of this deposit is presented in Fig. 8, and an SEM image of the fume particles is shown in Fig. 9. It is shown that the deposit is amorphous silica particles in the shape of spheres with a rather uniform size of 50–200 nm.

XRD-pattern from fume deposits collected from inside the furnace tube

SEM image of the fume deposits collected from inside the tube
Discussion
Experiments performed at 5·10−3 and 10·10−3 atm bulk oxygen pressures led to passive oxidation of the liquid silicon surface (Fig. 3a). This was observed as an apparently glassy layer, identified as a mixture of amorphous silica and cristobalite by XRD (Fig. 4a).
At oxygen partial pressures of 2·10−3 atm and lower, the active oxidation regime was obtained, as evidenced by the clean silicon surface (Fig. 3b), absence of crystalline oxide detectable by glancing angle XRD (Fig. 4b) and linear oxidation kinetics. Thus the critical value for the active–passive transition, \( p_{{{\text{O}}_{2} }}^{ \circ } \) (max), was between 2·10−3 and 5·10−3 atm. Active oxidation rates were proportional to \( p_{{{\text{O}}_{2} }}^{ \circ } \) and (v g/l)0.5, in agreement with the viscous flow regime mass transfer Eqs. (6) and (8). However, the critical value \( p_{{{\text{O}}_{2} }}^{ \circ } \) (max) calculated from this theory was 8.6·10−3 atm, approximately twice the experimental value. Reasons for this quantitative failure are investigated first.
In an evaluation of mass transfer, the boundary layer thickness, δ, is an important factor. The elementary mass transfer theory described earlier applies to boundary layer flow over a flat plate in an otherwise unconstrained space. However, the experiments were performed in a tubular furnace where the velocity profile is not accurately predicted by the boundary layer theory, because δ is significant compared to the tube cross-section. For a proper assessment of the experiments, it is therefore necessary to employ computational fluid mechanics (CFD).
Mathematical modeling of the experiments was performed using the CFD-software ANSYS/Fluent 13.0. The experimental geometry and conditions were simulated as isothermal laminar gas flow with species tracking of Ar, SiO(g) and O2. Gas properties were specified as described above at the experimental temperature of 1,500 °C. At the tube inlet, velocity and gas composition are set by the experimental conditions. At the metal surface, all O2 is assumed to be consumed and a SiO(g) concentration is specified. Calculations of flow field and mass transfer coefficient are not sensitive to the chosen SiO(g) surface concentration. All simulations were carried out with higher order numerical schemes and showed good convergence.
Results of interest are the velocity field and calculations of the overall mass transfer coefficient. The mass transfer coefficient is given by the flux of O2 and the concentration difference of O2 between the surface and the bulk gas (inlet value). These values can be extracted from the CFD simulations. The mass transfer coefficient results are discussed below. A typical flow field is seen in Fig. 10, where the average linear gas velocity is 4.7 cm s−1. The mathematical assessment shows that the flow profile deviates from that of simple boundary layer theory, but that the flow is laminar for all experimental settings.

Flow pattern in a tube modeled with CFD. Linear velocity color codes shown in cm s−1. The average linear velocity of the gas is 4.7 cm s−1
Mass transfer coefficients extracted from the CFD modeling, \( k_{{{\text{O}}_{2} }}^{\text{CFD}} \), are compared to values calculated from Eqs. (6) and (8), \( k_{{{\text{O}}_{2} }}^{\text{th}} \), in Fig. 11. The CFD results are seen to be consistently higher than those calculated for unconstrained flow over a flat plate. The difference increases with gas velocity, but is not large in the region examined. At v g = 4.7 cm s−1, \( k_{{{\text{O}}_{2} }}^{\text{CFD}} \) ≈ 3.9 cm s−1. Use of this higher value in Eq. (8) leads to a prediction of \( p_{{{\text{O}}_{2} }}^{ \circ } ({ \max }) \) ≈ 3·10−3 atm, in satisfactory agreement with the experimental finding of 2–5·10−3 atm.

Mass transfer coefficients estimated from classical theory (square) and from CFD modeling (diamond), compared to the experimentally measured mass transfer coefficient (triangle). l = 6 cm was used
The experimental mass transfer coefficient, \( k_{{{\text{O}}_{2} }}^{\text{ex}} \), deduced from Fig. 6 is shown as a single point on Fig. 11. The three different mass transfer coefficients are in the same order of magnitude at 0.047 m s−1 gas flow rate. However it is seen that \( k_{{{\text{O}}_{2} }}^{\text{ex}} \) (=3.06 cm s−1) is better predicted from \( k_{{{\text{O}}_{2} }}^{\text{CFD}} \) (=3.93 cm s−1) than from the classical flat plate flow model, \( k_{{{\text{O}}_{2} }}^{\text{th}} \) (=1.24 cm s−1).
The similarity between \( k_{{{\text{O}}_{2} }}^{\text{ex}} \) and \( k_{{{\text{O}}_{2} }}^{\text{CFD}} \) is significant, as it indicates that the flow in the tube was indeed laminar and that the simple mathematical model of Eqs. (6) and (8) describes reasonably well the common laboratory experiments conducted at low flow rates in tube furnaces. Nonetheless, maximum gas velocities are accelerated by the formation of a hydrodynamic layer attached to the tube wall, and in the present case the rate of silicon oxidation is thereby increased.
With this information it is possible to predict changes in the system as the temperature varies. In Table 2, the calculated SiO(g) pressure and flux plus theoretical mass transfer coefficients for oxygen temperatures are shown for three different temperatures. The minor differences in the values demonstrate that temperature is not an important factor in this system, provided that the conditions for active oxidation are achieved.
The formation of an oxidation product made up of fine, amorphous, spherical silica particles can be understood in light of the strongly exothermic nature of SiO(g) oxidation:
An adiabatic temperature rise calculation is performed using heat capacities and phase change enthalpies from HSC Chemistry [8] to show that the silica particles have been formed via the liquid state of SiO2, hence the spherical shape:
A final gas temperature of 4,935 °C is calculated from the heat balance
and it is recognized that the product is silica vapor. Rapid cooling and condensation leads to formation of liquid droplets, which grow until they ultimately solidify. This accounts for the spherical shape of the particles. Their near uniformity of size can be explained by particle growth theory [11].
Summary and Conclusions
Active oxidation of liquid silicon surfaces to produce very fine spherical particles of amorphous silica was observed in small scale laboratory experiments, successfully reproducing the essential features of fuming in the industrial oxidative ladle refining of metallurgical grade silicon.
The rate of active oxidation was controlled by gas phase mass transfer, as evidenced by the linear kinetics and the dependence of the rate on both oxygen pressure and the square root of linear gas velocity. Classical theory for mass transfer in the viscous flow regime above a flat plate yielded order of magnitude agreement with observed rates and the critical value for oxygen pressure separating active and passive oxidation regimes.
An improved description of mass transfer was provided by CFD-modeling which took into account the constraints of the reactor tube wall on gas flow. Formation of a boundary layer attached to the tube wall accelerates linear gas flow in the tube interior. This has the effect of increasing the mass transfer coefficient relative to that calculated from classical theory for unconstrained flow over a flat plate.
Calculations showed that temperature is not an important factor in determining the thermodynamics and kinetics of SiO(g) volatilization at temperatures of 1,450–1,650 °C, and laboratory flow conditions.
References
A. Schei, J. K. Tuset, and H. Tveit, Production of High Silicon Alloys (Tapir Forlag, Trondheim, 1998).
A. Ashrafian, S. T. Johansen, S. Gaal, B. Andresen, and O. S. Klevan, in 6th international Conference on CFD in Oil & Gas, Metallurgical and Process Industries (SINTEF/NTNU, Trondheim, Norway, 10–12 June, 2008).
M. K. Næss, G. Tranell, J. E. Olsen, N. E. Kamfjord and K. Tang, Oxidation of Metals, doi:10.1007/s11085-012-9303-9 (2012).
C. Wagner, Journal of Applied Physics 29, 1295 (1958).
M. Ratto, E. Ricci, E. Arato, and P. Costa, Metallurgical and Materials Transactions B 32, 903 (2001).
J. W. Hinze, and H. C. Graham, Journal of the Electrochemical Society 123, 1066 (1976).
P. Carter, B. Gleeson, and D. J. Young, Oxidation of Metals 56, 375 (2001).
HSC Chemistry 7.0, Outotec. Research Oy, Helsinki (2009).
D. J. Young, and B. A. Pint, Oxidation of Metals 66, 137 (2006).
R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena, Chaps. 1 & 17 (Wiley, New York, ISBN 0-471-41077-2 2002).
N. E. Kamfjord, H. Tveit, M. K. Næss and E. H. Myrhaug, in TMS (The Minerals, Metals & Materials Society, March 11–15, Walt Disney World, Orlando, FL, USA, 2012).
Acknowledgments
The experimental work presented in this paper was performed at the School of Materials Science and Engineering at The University of New South Wales in Sydney, Australia. The funding was provided by Norwegian Research Council and FFF (Ferro Alloys Industries Research Association) through the FUME project (Fugitive emissions of Materials and Energy). The funding for the research exchange was provided by UNIFOR, Elkem’s research fund and Norsk Hydro’s research fund.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Næss, M.K., Young, D.J., Zhang, J. et al. Active Oxidation of Liquid Silicon: Experimental Investigation of Kinetics. Oxid Met 78, 363–376 (2012). https://doi.org/10.1007/s11085-012-9312-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-012-9312-8