
Abstract
The influence of KCl, NaCl and CaCl2 on the oxidation of 304-type (Fe18Cr10Ni) stainless steel at 600 °C in 5 %O2 + 40 %H2O was investigated. Prior to exposure, a small amount of the preferred salt (cation equivalent: 1.35 μmol/cm2) was deposited on the samples. Exposure time was 1–168 h. The oxidized samples were analyzed by SEM/EDX, XRD, FIB and IC. The presence of KCl and NaCl strongly accelerates high temperature corrosion of 304L. Corrosion attack is initiated by the formation of alkali chromate through the reaction of alkali with the protective oxide. Chromate formation is a sink for chromium in the oxide and leads to a loss of its protective properties. Subsequently a rapidly growing scale forms, consisting of an outer hematite layer with chromate particles on top and an inner spinel oxide layer. In contrast to NaCl and KCl, CaCl2 is not very corrosive. At temperature, CaCl2 is rapidly converted to CaO. Small amounts of CaCrO4 form where CaO is in direct contact with the scale. CaO also reacts with the scale to form Ca2Fe2O5.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The production of electricity from biomass and waste-fired power plants is increasing all over the world. Unfortunately, the combustion of biomass and waste results in the formation of corrosive deposits and gases. Besides water vapor and carbon dioxide, the flue gas contains high concentrations of alkali chlorides and hydrogen chloride while the sulphur dioxide content is typically low [1, 2]. Hence, the deposits formed on the superheater tubes are often rich in alkali chlorides which are known to cause accelerated corrosion of the superheater tubes [1–7]. The corrosivity of alkali chlorides in the presence of O2 is often explained by applying “the chlorine cycle” mechanism [3, 5–10] where Cl2(g) plays a seminal role, acting as a catalyst for corrosion. Thus, the mechanism supposes that Cl2 forms in a reaction between O2 and alkali chloride at the scale/metal interface. Cl2 then penetrates the scale and generates volatile transition metal chlorides at the scale/metal interface. The transition metal chlorides then diffuse back to the scale/gas interface where they are oxidized by O2, precipitating metal oxide and releasing Cl2(g) which enters the process again.
Recently, a new, electrochemical, mechanism has been proposed to explain the alkali chloride-induced corrosion of low-alloyed steel. In the new mechanism, chloride ion is the diffusing chlorine species rather than Cl2 [11]. Also, it has been reported recently that the potassium ion plays an important role in the initiation of KCl-induced corrosion of stainless steels [12–16]. Thus, it was shown that K2CO3 is equally corrosive as KCl towards stainless steel 304L at 600 °C. The corrosiveness of the potassium ion was attributed to the formation of potassium chromate(VI) by reaction with the oxide scale. The reaction depletes the oxide in chromium and destroys its protective properties [17].
The present study investigates the corrosion effects of three chloride salts (KCl, NaCl and CaCl2) on the austenitic stainless steel 304L (Fe18Cr10Ni) in 5 %O2 and 40 % H2O at 600 °C. The emphasis is on the early stages of corrosion and on elucidating the sequence of events that cause the corrosion attack.
Experimental Procedures
Sample Preparation
The material investigated is the austenitic stainless steel, 304L, for chemical composition see Table 1. The geometrical area of the samples was 5.56 cm2, (15 × 15 × 2 mm3). For handling, a hole (φ = 1.5 mm) was drilled, 2 and 7.5 mm from the edges. Before exposure the samples were grinded on 320 grit SiC with deionized water. The samples were then polished with 9, 3 and 1 μm diamond solution and lubricating liquid on a polish cloth until a mirror-like surface appeared. After polishing, the samples were degreased and cleaned in acetone and ethanol using an ultrasonic bath. The samples were coated with the preferred salt (NaCl, KCl or CaCl2) to a cation equivalent of 1.35 μmol/cm2. The salts were applied by spraying the samples with a saturated solution of the prefer salt in water/ethanol. NaCl and KCl sprayed samples were weight in order to calibrate the amount. Because of the hygroscopic nature of CaCl2, the amount was calibrated with ion chromatography. The samples were dried with air and stored in a desiccator prior to exposure. The mass change of the samples was measured prior to and after exposure using a six decimal Sartorius™ balance.
Exposures
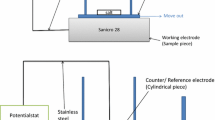
All exposures were isothermal and performed in silica tube furnaces with ex-situ recording of the weight (Fig. 1). The samples, three at time, were mounted on a sample holder and positioned parallel to the gas flow direction. The temperature was kept at 600 °C (±1 °C) and was calibrated before each exposure. The experiments were carried out in 5 %O2 + 40 %H2O and the flow rate was 2.5 cm/s. Nitrogen was used as carrier gas. The flow rate was controlled with a Bios DC2 Flow Calibrator. The dry gas was led through a humidifier and a temperature controlled condenser to obtain the correct water concentration. The experiments lasted 1, 24, 72 or 168 h.

Experimental setup for exposures in a horizontal silica tube furnace
Corrosion Product Characterization
X-ray Diffraction, XRD
The crystalline corrosion products were analysed with Grazing-Incidence X-Ray Diffraction (GI-XRD) using a Siemens D5000 powder diffractometer, equipped with grazing—incidence beam attachment and a Göbel mirror. Cu K. ψ radiation was used and the angle of incidence was 2°–5°. The measurement range was 20° < 2θ < 70°.
Scanning Electron Microscopy, SEM/EDX
SEM imaging was performed using an FEI Quanta 200 ESEM FEG. The EDX detector is linked with the SEM and an Oxford Inca EDX system was used. The microscope was operated at 15–20 kV.
Ion Chromatography, IC
To determine the amount of water-soluble anions (Cl− and CrO4 2−) a Dionex ICS-90 system was used. The anions were analysed with an IonPac AS4A-SC analytic column and 1.8 mM Na2CO3/1.7 mM NaHCO3 was used as eluent. To determine the amount of water-soluble cations (K+, Na+ and Ca2+) a Dionex ICS-900 system was used. The anions were analysed with an IonPac CS12A analytic column and 20 mM sulfonic acid was used as eluent. The flow rate was 2 mL/min for both anions and cations analyses.
Results
Gravimetry
Figure 2 shows mass gain as a function of exposure time for 304L steel in 5 %O2 + 40 %H2O environment at 600 °C in the presence of NaCl, KCl and CaCl2. The graph includes a reference exposure performed in the absence of salt. The results show that both NaCl and KCl strongly accelerate corrosion. While the two mass gain curves are similar during later stages of the exposures, NaCl initially causes higher mass gain than KCl. In contrast, exposure in the presence of CaCl2 does not result in a significantly increased mass gain.

Mass gain versus exposure time for samples exposed in the presence of NaCl, KCl, CaCl2 in 5 %O2 + 40 %H2O at 600 °C. Reference exposures in 40 %H2O at 600 °C are also included
Surface Morphology
The surface morphology was investigated by Scanning Electron Microscope (SEM). Figure 3 shows plan view images after 1 and 24 h of exposure in the presence and in the absence of salt. Figure 3a shows a SEM image of a reference sample exposed for 1 h in O2 + H2O at 600 °C. The environment results in the formation of large iron-rich oxide island on the surface. Similar oxide islands were observed in previous investigations of the oxidation of 304L in O2 + H2O environment at 600 °C. The earlier studies show that the oxide islands have formed in the middle of the underlying steel grains while the areas close to the steel boundaries remain protective. Figure 3b and c show samples exposed for 1 h in the presence of NaCl and KCl, respectively. In both cases, the salt particles are partly reacted and have started to become overgrown by oxide. At this stage the NaCl treated sample exhibit considerably more corrosion between the salt particles than in the case of KCl. Figure 3d shows a sample exposed for 1 h in the presence of CaCl2. The salt agglomerates have a roughly circular form and at this stage CaCl2 is partially decomposed (see XRD and IC analysis below). The surface between the salt agglomerations is relatively even. Oxide islands have started to form, similar to the ones observed in the reference run (compare Fig. 3a). Furthermore, there is no tendency for the salt agglomerations to become overgrown by oxide.

SEM images of samples exposed for 1 and 24 h at 600 °C in a and e: 5 %O2 + 40 %H2O, b and f: 5 %O2 + 40 %H2O + NaCl, c and g: 5 %O2 + 40 %H2O + KCl, d and h: 5 %O2 + 40 %H2O + CaCl2
Figure 3e shows the surface after 24 h exposure in the absence of salt (reference sample). The oxide islands have increased both in number and size while the areas near the steel borders remain protective. Figure 3f and g show samples after 24 h exposure in the presence of NaCl and KCl, respectively. At this stage the surface is covered by thick scale, the former salt crystals being replaced by irregularly shaped oxide agglomerations. The surface between the oxide agglomerations is very uneven. The KCl treated samples exhibits an island-like scale morphology that reflects the substrate grain structure. In the case of NaCl, the corrosion affects the entire surface and no preferential corrosion of the grain centers is observed.
Figure 3h shows a sample after 24 h of exposure in the presence CaCl2, showing roughly circular areas corresponding to the deposited salt. Between the circular formations, the oxidation morphology resembles the reference sample (see Fig. 3e), showing oxide islands. In the position of the former CaCl2 agglomerations the scale tends to spall, see Fig. 4 for 45° tilted SEM image.

45° tilted SEM image showing spallation of a former CaCl2 salt crystal on the sample exposed for 24 h in the presence of CaCl2 at 600 °C in 5 %O2 + 40 %H2O
Increasing the exposure time from 24 to 168 h only produced minor changes in corrosion morphology.
Corrosion Product Characterization
The surface of the exposed samples was investigated with respect to elemental distribution and crystalline corrosion products using SEM/EDX together with XRD. EDX analyses (not shown) reveal that much of the NaCl and KCl added remains unreacted after 1 h at 600 °C. Accordingly, XRD (Figs. 5, 6) features strong diffraction peak from the salts. The crystalline corrosion products identified after 1 h of exposure were hematite (Fe2O3), spinel type oxide (Me3O4) and alkali chromate (Na2CrO4 or K2CrO4) indicating that the protective oxide has started to be converted to a poorly protective iron-rich oxide. However, strong diffraction from the steel substrate indicates that much of the surface is still covered by very thin oxide.

Combined diffractogram for 304L samples exposed in 5 %O2 + 40 %H2O + NaCl at 600 °C after 1 and 24 h. The upper diffractogram shows the sample exposed for 24 h and the under diffractogram shows the sample exposed for 1 h. The matching peaks in both diffractogram are marked in the upper diffractogram. The peaks that are only presence after 1 h are marked in the under one

Combined diffractogram for 304L samples exposed in 5 %O2 + 40 %H2O + KCl at 600 °C after 1 and 24 h. The upper diffractogram shows the sample exposed for 24 h and the under diffractogram shows the sample exposed for 1 h. The matching peaks in both diffractogram are marked in the upper diffractogram. The peaks that are only presence after 1 h are marked in the under one
In contrast to the behavior of NaCl and KCl, the EDX analyses shows that CaCl2 has largely decomposed after 1 h, the former CaCl2 agglomerates exhibiting just a few percent of chlorine (not shown). XRD (Fig. 7) shows no evidence for CaCl2 or its hydrates. There was very weak diffraction from CaO and the corrosion product detected were Fe2O3, spinel type oxide (Me3O4) and small amounts of dicalcium ferrate (III), Ca2Fe2O5. Also in this case there was strong diffraction from the steel indicating the presence of a very thin oxide scale.

Combined diffractogram for 304L samples exposed in 5 %O2 + 40 %H2O + CaCl2 at 600 °C after 1 and 24 h. The upper diffractogram shows the sample exposed for 24 h and the under diffractogram shows the sample exposed for 1 h. The matching peaks in both diffractogram are marked in the upper diffractogram. The peaks that are only presence after 1 h are marked in the under one
The fact that only small amounts of crystalline calcium-containing substances (Ca2Fe2O5 and CaO) were identified implies that other, non-crystalline or poorly crystalline, calcium containing compounds is present at this stage. It is proposed that these compounds are products of the thermal decomposition of CaCl2, consisting of hydroxy-chlorides (e.g., Ca(OH)Cl) or oxy-chlorides (e.g., Ca2OCl2) [18].
Figures 8 and 9 show the surface after 24 h exposure in the presence of NaCl and KCl, respectively. In both cases, the surface is covered by iron-rich oxide. While the XRD results (Figs. 5, 6) show the same corrosion products as after 1 h, there is no diffraction from KCl or NaCl at this stage, indicating that the salts have reacted completely and/or evaporated. There is no or very weak diffraction from the steel substrate which indicates that most of the surface is covered by a thick oxide scale. Morphological features reminiscent of the original salt particles and consisting of corrosion product agglomerations are present on the surface. Areas rich in sodium and potassium are found in and between these features. Both elements are correlated to chromium and oxygen in the EDX maps. Sodium chromate (Na2CrO4) and potassium chromate (K2CrO4) were identified by XRD on the NaCl- and KCl-treated samples, respectively. Hence, the Na- and K-rich areas are attributed to sodium chromate and potassium chromate. In addition, EDX point analyses of NaCl treated samples (not shown) reveal that the amount of Na detected exceeds the amount of chromate in some areas. Furthermore, EDX and XRD analysis gave no evidence for transition metal chlorides.

EDX images of a sample exposed for 24 h in presence of NaCl at 600 °C in 5 %O2 + 40 %H2O

EDX images of a sample exposed for 24 h in presence of KCl at 600 °C in 5 %O2 + 40 %H2O
After 24 h exposure in the presence of CaCl2, the corrosion morphology is very different from that of the NaCl and KCl treated samples, see Fig. 10. In this case there are no corrosion product agglomerations. The area between the original salt particles is partly covered by thin, iron-rich oxide islands. The image shows a network pattern consisting of thin oxide scale, corresponding to the grain boundaries in the steel substrate. The presence of thin oxide is in accordance with the detection of strong X-ray diffraction from the steel substrate (see Fig. 7). EDX indicated that there is no Ca in this area. XRD shows very strong diffraction from CaO and relatively strong diffraction from Ca2Fe2O5 at this stage. This implies that the decomposition of calcium chloride is now complete. EDX mapping shows that CaO forms patches that correspond to the original agglomerations of CaCl2 particles (compare also Fig. 3h). Ca2Fe2O5 is located in the periphery of the CaO patches. CaO is absent in some of the smaller circular areas, implying that it has been consumed by reaction with the scale, forming Ca2Fe2O5. The overlap between Ca and Cr at the edges of the circular patches indicates the presence of CaCrO4.

EDX images of a sample exposed for 24 h in presence of CaCl2 at 600 °C in 5 %O2 + 40 %H2O
In order to further investigate the oxide scale, FIB cross sections were prepared from samples exposed for 24 h in the presence of salt, see Figs. 11, 12, and 13. In the case of NaCl and KCl, the FIB cross sections were prepared through a former salt particle. In the case of CaCl2, the FIB cross section was performed at the edge of the former CaCl2 agglomerates including both the area originally covered by salt and the surrounding area. A platinum layer was first deposited on the selected area in order to protect the oxide.

EDX images of a FIB cross section through a former salt crystal on a sample exposed for 24 h in presence of NaCl at 600 °C in 5 %O2 + 40 %H2O

EDX images of a FIB cross section through a former salt crystal on a sample exposed for 24 h in presence of KCl at 600 °C in 5 %O2 + 40 %H2O

45° tilted EDX images of a FIB cross section partly through a former salt crystal on a sample exposed for 24 h in presence of CaCl2 at 600 °C in 5 %O2 + 40 %H2O
Figure 11 shows a FIB cross section prepared through a former NaCl crystal after 24 h exposure at 600 °C. The shape of the former NaCl crystal can still be discerned but neither Na nor Cl is detected within its shape. A thick layered oxide scale has formed beneath the former NaCl crystal. The upper part of the scale consists of almost pure iron oxide while the bottom part shows high concentrations of chromium, iron and nickel. While iron and chromium are evenly distributed in the bottom part of the scale the nickel distribution is more uneven. Local enrichments of metallic nickel are also present in the alloy substrate immediately below the scale. No chlorine was detected. Based on this information and on the evidence from XRD (see Fig. 5) it is concluded that the upper iron-rich oxide corresponds to hematite while the bottom part of the scale is made up of spinel type oxide.
Figure 12 shows a corresponding sample exposed in presence of KCl. The corrosion morphology is similar to the NaCl case. Thus, a two layered oxide scale has formed beneath the former salt particle, consisting of an outer iron-rich layer and an inner layer containing iron, chromium and nickel. Again, the distribution of Ni in the bottom part of the scale is very uneven. In this case the oxygen map implies that at least some of the Ni in the lower part of the scale is metallic. An area rich in potassium, chromium and oxygen is detected within the former salt particle. Based on XRD (see Fig. 6) and EDX point analysis it is concluded that this is potassium chromate (K2CrO4). Again, no chlorine was detected.
Figure 13 shows a sample exposed for 24 h in presence of CaCl2. The FIB cross section includes part of the former CaCl2 agglomerate and the surrounding area. The metal not covered by CaCl2 has formed a two-layered oxide scale consisting of an iron oxide top layer and a bottom layer containing oxygen, iron, chromium and nickel. Again, it is suggested that the top and bottom parts are made up of hematite and spinel oxide respectively, both phases being detected by XRD. The area formerly covered by CaCl2 exhibits a scale consisting of three distinctive layers. The outer part is rich in iron and calcium, the middle part is iron-rich together with small amounts of chromium while the bottom part contains iron, chromium and nickel. The combination of SEM/EDX and the XRD analysis (see Fig. 7) allows us to attribute the upper iron-calcium-rich oxide to dicalcium ferrate(III) (Ca2Fe2O5). Similarly, it is considered that the middle, iron-rich oxide most consists of hematite while the bottom part of the scale is made up of spinel type oxide. As in the case of NaCl and KCl treated samples, nickel is unevenly distributed in the scale while there is an even distribution of chromium and iron. According to the EDX analysis of the FIB cross section, some areas at the metal/oxide interface contain chlorine and no calcium. These areas are suggested to contain transition metal chlorides although no such compounds were detected by XRD.
Analysis of Water Soluble Compounds
Tables 2, 3, 4 show the results from the ion chromatography (IC) analysis of the salt treated samples after 1, 24 and 168 h exposure. Tables 2 and 3 show the results for the samples exposed in the presence NaCl and KCl. After 1 h of exposure, the results are similar for the two salts. About 80 % of the applied alkali and about 60 % of the added chlorine is detected. The discrepancy between the amounts of alkali and chlorine found is attributed to the formation of alkali chromate with the simultaneous release of HCl(g) (see “Discussion” section).
After 24 h of exposure 44 % of the applied Na+ was retrieved while the corresponding value for K+ was only 19 %. Very little chlorine remained on the two samples. In the case of KCl the amount of potassium ions found after 24 h corresponds to the amount of chromate detected. In contrast, after 24 h the amount of sodium ions found on the NaCl treated samples exceeds the amount of chromate. After 168 h the NaCl treated samples showed essentially the same amounts of soluble ions as after 24 h. In contrast, the KCl treated samples show a decrease in the amount of chromate and of potassium from 24 to 168 h of exposure.
Table 4 shows the IC analysis of the samples exposed in the presence CaCl2. After 1 h of exposure 52 % of the applied Ca2+ was detected. There is a strong decrease in the amount of chloride ions (12 % of the added amount being detected) which is attributed to the thermal decomposition and reaction of CaCl2, forming CaO, Ca2Fe2O5 and HCl(g). A small amount of CrO4 2− has formed, corresponding to 2 % of the theoretical yield of CaCrO4. After 24 h the amount of chromate has increased slightly but is still just a few percent of the theoretical yield. It is suggested that CaCl2 has decomposed completely at this stage. Thus, the small amount of chlorine found is attributed to the transition metal chlorides detected by SEM/EDX (see Fig. 13). Extending the exposure time to 168 h does not change the results appreciably.
Discussion
Previous studies show that 304L initially forms a smooth, thin (<200 nm) Cr-rich (Fe, Cr)2O3 protective scale in dry O2 at 600 °C [19–21]. In the presence of water vapor chromium is volatilized by formation of CrO2(OH)2 on the oxide surface [19, 20, 22]:
Chromium volatilization depletes the protective oxide and the steel substrate in chromium. If the chromium content drops below a critical level the oxide loses its protective properties and an iron-rich, rapidly growing oxide forms. Therefore, the supply of chromium to the oxide by diffusion in the metal must match the evaporation rate to maintain the protective properties of the oxide. At the alloy grain boundaries the diffusion is higher compared to the center of the steel grains. Hence, the oxidation behavior of the steel surface depends on the distance to a grain boundary. This explains why “islands” consisting of thick oxide form on central parts of the underlying steel grains while the areas close to a grain boundary retain a thin slow-growing oxide (see Fig. 3a, e). The oxide islands consist of an outer hematite (Fe2O3) and inner spinel-type ((Fe, Cr, Ni)3O4) layer [23].
In general, any process that depletes the protective oxide and the steel substrate in chromium is expected to make the stainless steel more vulnerable to high temperature corrosion. The present study explores chromate formation as a mechanism for chromium depletion, showing that both NaCl or KCl can trigger a breakdown of the protective oxide on 304L by formation of Na2CrO4 and K2CrO4, respectively (see Figs. 5, 6, 8, 9 and Tables 2, 3) [24]:
The results show that chromate formation starts during the first exposure hour. After 24 h, chromates can be detected all over the surface which indicates that both KCl and NaCl diffuses out from the salt particle and react with the protective oxide that initially forms. It is proposed that chromium depletion of the protective oxide by reactions (2) and (3) result in a breakdown of the protective properties of the oxide, triggering the formation of a rapidly growing iron-rich scale. This is in accordance with earlier reports on the influence of KCl on the high temperature corrosion of 304L [12–14]. The resulting thick scale consists of an outer hematite layer and an underlying (Fe, Cr, Ni) spinel type oxide (see Figs. 11, 12) [16]. It is expected that this two-layered scale is poorly protective and the formation appears to be much faster compared to the breakdown triggered by chromium oxide hydroxide volatilization in the absence of salt.
As shown in Tables 2 and 3, about 60 % of the salts is still unreacted after 1 h. Furthermore, considerable amounts of alkali chromate have formed already during this first hour. However, about 20 % of the salts are not accounted for by the IC analysis and it is suggested that this is because of vaporization of the salts. The equilibrium partial pressure of NaCl and KCl at 600 °C are 1.4 × 10−6 bar and 3.3 × 10−6 bar, respectively [24]. The results show that chromate formation and volatilization of the salt continues during the first 24 h. It may be noted that the amount of chromate on the surface is similar after 24 and 168 h in the case of NaCl. In contrast, the amount of chromate decreases significantly with time in the case of KCl, suggesting that K2CrO4 tends to decompose. The decomposition of potassium chromate by reaction with water is not thermodynamically favoured: permeable for oxide and chloride ions. Thus, the scale structure is similar to the oxide islands formed on 304L in O2 + H2O environment in the absence of salt. The destruction of the protective scale by chromate [24]
Decomposition to Cr2O3 by water is also not favoured [24]:
Instead, it is argued that potassium chromate decomposes electrochemically. The oxidation of metals in oxygen gas at high temperature is usually described in terms of a mechanism where molecular oxygen is reduced on the scale surface while metal is being oxidized at the scale/metal interface, the two reactions being coupled by electronic and ionic currents through the scale. In this scenario oxygen is thus cathodically reduced at the scale surface. It is argued that in the present case the reduction of alkali chromate provides an alternative cathodic process:
The cathodic current is provided by metal oxidation:
The potassium hydroxide formed in this way can either react with the surface oxide or be volatilized [24]:
Because of its high equilibrium vapour pressure, KOH is not likely to be present in pure form at 600 °C.
Considering its similarity to K2CrO4 it is expected that Na2CrO4 would decompose in a similar way (i.e. forming NaOH):
However, the results show that the amount of sodium chromate does not decrease with time as in the case of potassium chromate. Another significant difference between the effects of KCl and NaCl is that while the amount of K+ corresponds to the number of moles of chromate and chloride detected, the amount of Na+ exceeds the number of moles of chromate and chloride found, suggesting alkaline condition on the surface. The disparity between Na and chromate + chlorine was confirmed by EDX point analysis. It is suggested that these differences are explained by the lower volatility of NaOH compared to KOH. Thus, the equilibrium vapour pressure over the corresponding pure condensed phase at 600 °C is 5.3 × 10−7 and 4.9 × 10−5 bar for NaOH and KOH respectively. This means that while the decomposition of K2CrO4 produces KOH, which is easily vaporized, the NaOH formed by decomposition of Na2CrO4 is expected to accumulate on the surface, in accordance with our observations. The alkaline environment created in this way is suggested to slow down the decomposition of Na2CrO4 (11).
The sodium hydroxide accumulated on the surface may react with iron oxide, forming sodium ferrate(III) [24]:
Alkali ferrate(III) may also form by reaction of the applied salt with hematite [24]:
It may be noted that the formation of sodium ferrate is much more favoured compared to potassium ferrate. It is therefore suggested that the greater stability of sodium chromate in comparison to potassium chromate can be attributed to the more alkaline conditions that prevail on the NaCl-treated samples.
In spite of these differences regarding corrosion chemistry, it is important to note that the corrosion effect of NaCl and KCl are generally speaking quite similar in these conditions.
In contrast to NaCl and KCl, CaCl2 is only slightly corrosive under the exposure conditions. One explanation is that CaCl2 is readily converted to CaO [24]:
Accordingly, the quantitative analysis of the samples after oxidation (Table 4) show that only 12 % of the added chloride remains on the surface after 1 h of oxidation. This indicates that most of the CaCl2 has been converted to CaO at this stage. However, XRD analysis gave only weak diffraction from CaO while no evidence was found for CaCl2 or its hydrates. The thermal decomposition of CaCl2 to CaO is known to proceed through intermediate steps and several calcium-hydroxychlorides and -oxychlorides have been described (e.g. Ca(OH)Cl) [18]. These compounds are metastable and tend to be poorly crystalline. Thus, the chloride remaining on the surface after 1 h of exposure is mainly attributed to calcium-hydroxychlorides and –oxychlorides. After 24 h only 2 % of the added chloride remains. At this stage there is strong diffraction from CaO and Ca2Fe2O5. The latter compound corresponds to the iron- and calcium-rich band seen in Fig. 10, encircling a former CaCl2 agglomeration. Apparently, reaction (15) is now completed and parts of the CaO formed has reacted further with iron oxide at the scale/gas interface (reaction 16) [24].
Because of its rapid thermal decomposition, CaCl2 does not have the opportunity to react extensively with the chromium-rich oxide as is the case for NaCl and KCl. However, small amounts of calcium chromate are detected around the former CaCl2 agglomerates (reaction 17).
Calcium chromate can also form by reaction with CaO (reaction 18). Because of the poor mobility of CaO, Ca does not spread over the surface and the reaction between Ca and the protective oxide is limited [24].
Because of the small extent of chromate formation, the protective oxide is not chromium depleted in a major way, explaining why exposure in the presence of CaCl2 does not result in accelerated corrosion, compared to the reference exposure, in the absence of salt. Accordingly, the oxidation morphology is similar to that observed in 5 %O2 + 40 %H2O environment in the absence of salt [23]. Thus, Fig. 13 shows a two-layered oxide consisting of an outer iron-rich part and an inner layer that contains iron, chromium and nickel. In the area previously covered by CaCl2, three scale layers can be detected. The outer part is rich in Fe and Ca (corresponding to Ca2Fe2O5). The middle and bottom parts correspond to the scale formed away from the salt agglomeration. Thus, the middle part is iron-rich together with small amounts of chromium (probably corresponding to Fe2O3) while the inner part contains iron, chromium and nickel (spinel oxide, (Fe, Cr, Ni)3O4). Noticeable is that the bottom spinel oxide layer is thinner than the corresponding oxide formed adjacent to the former salt agglomerate. This may indicate that the top Ca2Fe2O5 layer acts as a barrier. The slightly lower mass gain of the CaCl2 exposed samples compared to the salt free exposures is not considered to be significant because of the occurrence of substantial scale spallation at the location of the former CaCl2 agglomerations (see Fig. 4). Spallation is also suggested to explain why the Ca2+ content decreases with time, see Table 4. In addition, the thermal decomposition of CaCl2 forming CaO and HCl(g) also contributes to a lower mass gain. All the chlorine added corresponds to 0.09 mg/cm2.
Figure 13 also shows two small areas enriched in chlorine at the metal/oxide interface. This corresponds to sub-scale transition metal chlorides formed by the alloy. It is suggested that the small amounts of chloride detected by IC analysis can be attributed to such sub-scale chlorides. It appears that the chloride accumulations do not influence corrosion in a major way in the present case.
Conclusions
The presence of KCl and NaCl strongly accelerates the high temperature corrosion of 304L stainless steel in 5 %O2 + 40 %H2O environment. The corrosion is initiated by the formation of alkali chromate(VI) through the reaction of alkali with the protective oxide. Chromate formation is a sink for chromium in the oxide and leads to a loss of its protective properties. Subsequently a rapidly growing scale forms, consisting of an outer hematite layer with chromate particles on top and an inner layer consisting of spinel oxide, (Fe, Cr, Ni)3O4. While the extent of corrosion attack is similar for samples exposed to NaCl and KCl, some differences are observed. Thus, the amount of Na2CrO4 formed was higher compared to K2CrO4, the latter compound decomposing with time. Furthermore, the samples exposed in the presence of NaCl formed a corrosion product containing Na, Fe and O, possibly Na2Fe2O4. In contrast, the only alkali-containing product forming on the KCl-treated samples was K2CrO4. These differences are attributed to the greater volatility of KOH compared to NaOH.
In contrast to NaCl and KCl, CaCl2 is not very corrosive. Under the exposure conditions CaCl2 is rapidly converted to CaO. Because of the poor mobility of CaO, only small amounts of CaCrO4 form where CaO is in direct contact with the scale. CaO also reacts with the scale to form Ca2Fe2O5.
References
K. O. Davidsson, L. E. Amand, B. Leckner, B. Kovacevik, M. Svane, M. Hagstrom, J. B. C. Pettersson, J. Pettersson, H. Asteman, J. E. Svensson, and L. G. Johansson, Energy & Fuels 21, 71 (2007).
J. Pettersson, C. Pettersson, N. Folkeson, L.-G. Johansson, E. Skog, and J.-E. Svensson, Materials Science Forum 522–523, 563 (2006).
H. P. Nielsen, F. J. Frandsen, K. Dam-Johansen, and L. L. Baxter, Progress in Energy and Combustion Science 26, 283 (2000).
N. Folkeson, J. Pettersson, C. Pettersson, L. G. Johansson, E. Skog, B. Å. Andersson, S. Enestam, J. Tuiremo, A. Jonasson, B. Heikne, and J. E. Svensson, Materials Science Forum 595–598, 289 (2008).
H. P. Michelsen, F. Frandsen, K. Dam-Johansen, and O. H. Larsen, Fuel Processing Technology 54, 95 (1998).
H. J. Grabke, E. Reese, and M. Spiegel, Corrosion Science 37, 1023 (1995).
M. Montgomery and A. Karlsson, Materials and Corrosion 50, 579 (1999).
H. P. Nielsen, F. J. Frandsen, and K. Dam-Johansen, Energy & Fuels 13, 1114 (1999).
C. J. Wang and T. T. He, Oxidation of Metals 58, 415 (2002).
M. Montgomery, A. Karlsson, and O. H. Larsen, Materials and Corrosion 53, 121 (2002).
N. Folkeson, T. Jonsson, M. Halvarsson, L.-G. Johansson, and J.-E. Svensson, Materials and Corrosion (2010). doi:10.1002/maco.201005942.
C. Pettersson, L. G. Johansson, and J. E. Svensson, Oxidation of Metals 70, 241 (2008).
C. Pettersson, J. Pettersson, H. Asteman, J. E. Svensson, and L. G. Johansson, Corrosion Science 48, 1368 (2006).
J. Pettersson, H. Asteman, J.-E. Svensson, and L.-G. Johansson, Oxidation of Metals 64, 23 (2005).
C. Proff, T. J., C. Pettersson, J.-E. Svensson, L.-G. Johansson, and M. Halvarsson, Submitted to Microscopy of Oxidation (2008).
T. Jonsson, et al., Oxidation of Metals 72, 213 (2009).
J. Pettersson, N. Folkeson, J.-E. Svensson, and L.-G. Johansson, Oxidation of Metals 76(1–2), 93 (2011).
L. Gmelin, Gmelins Handbuch der anorganischen Chemie (1924–1997).
H. Asteman, J. E. Svensson, L. G. Johansson, and M. Norell, Oxidation of Metals 52, 95 (1999).
H. Asteman, J. E. Svensson, M. Norell, and L. G. Johansson, Oxidation of Metals 54, 11 (2000).
J. E. Tang, et al., Micron 32, 799 (2001).
B. Pujilaksono, T. Jonsson, M. Halvarsson, I. Panas, J.-E. Svensson, and L.-G. Johansson, Oxidation of Metals 70, 163 (2008).
M. Halvarsson, J. E. Tang, H. Asteman, J.-E. Svensson, and L.-G. Johansson, Corrosion Science 48, 2014 (2006).
I. Barin, Thermodynamic Data of Pure Substances, 3rd edn. (1995).
Acknowledgment
This work was carried out within the High Temperature Corrosion Centre (HTC) at Chalmers University of Technology.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Karlsson, S., Pettersson, J., Johansson, L.G. et al. Alkali Induced High Temperature Corrosion of Stainless Steel: The Influence of NaCl, KCl and CaCl2 . Oxid Met 78, 83–102 (2012). https://doi.org/10.1007/s11085-012-9293-7
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-012-9293-7