
Abstract
The dynamic behaviors of prebiotic reaction networks may be critically important to understanding how larger biopolymers could emerge, despite being unfavorable to form in water. We focus on understanding the dynamics of simple systems, prior to the emergence of replication mechanisms, and what role they may have played in biopolymer formation. We specifically consider the dynamics in cyclic environments using both model and experimental data. Cyclic environmental conditions prevent a system from reaching thermodynamic equilibrium, improving the chance of observing interesting kinetic behaviors. We used an approximate kinetic model to simulate the dynamics of trimetaphosphate (TP)-activated peptide formation from glycine in cyclic wet-dry conditions. The model predicts that environmental cycling allows trimer and tetramer peptides to sustain concentrations above the predicted fixed points of the model due to overshoot, a dynamic phenomenon. Our experiments demonstrate that oscillatory environments can shift product distributions in favor of longer peptides. However, experimental validation of certain behaviors in the kinetic model is challenging, considering that open systems with cyclic environmental conditions break many of the common assumptions in classical chemical kinetics. Overall, our results suggest that the dynamics of simple peptide reaction networks in cyclic environments may have been important for the formation of longer polymers on the early Earth. Similar phenomena may have also contributed to the emergence of reaction networks with product distributions determined not by thermodynamics, but rather by kinetics.
Similar content being viewed by others

Introduction
How organic monomers developed into polymers capable of replication, metabolism, and other key behaviors of life remains unknown. Repeated reactions, such as those produced from cycling a sample between wet and dry conditions, can promote the formation of more complex molecules (Lahav and Chang 1976; Ross and Deamer 2016), but also tend to produce intractable tars that would be unlikely to support continuous replication (Shapiro 2000). Life-like behavior probably emerged in at least partially open systems that were able to exchange materials and energy with their environment (Wagner et al. 2019; Baum 2018). Open systems allow fresh reactants to be supplied while removing side products from previous reactions. Removing products from a system can also decrease the overall system complexity by favoring products that form quickly, potentially avoiding the formation of tars and supporting the proliferation of catalytic or autocatalytic reactions (Martin and Horvath 2013; Colón‐Santos et al. 2019). Experiments with open systems have been performed using various mixtures of organic molecules (Lahav et al. 1978; Maio et al. 2021; Bartolucci et al. 2022), and some have suggested the possible emergence of function, though the details of those functions remain ambiguous (Doran et al. 2019; Vincent et al. 2019). Some dynamic phenomena, such as sustained oscillations, are only thermodynamically possible in open systems (Wagner et al. 2019).
Another significant feature of open systems in the origins of life is that they can remain away from thermodynamic equilibrium indefinitely. One of the hallmarks of a life-like system is that it should remain out of equilibrium with its environment, meaning that life almost certainly originated in far-from-equilibrium conditions (Eigen and Schuster 1977; Pross 2003; Pascal et al. 2013; Mamajanov et al. 2014). The kinetic behavior of systems which are far from equilibrium may have provided the driving force necessary for the emergence of organization from an unordered system (Prigogine 1978; Astumian 2019). Chemical reaction networks in far-from-equilibrium conditions may exhibit overshoot, a dynamic phenomenon in which a species passes through its equilibrium point one or more times before actually reaching it (Jia et al. 2014). Overshoot is a kinetically driven phenomena and has been associated with the ability of biochemical systems to recover their original state after a perturbation (Jia and Qian 2016; Ma et al. 2009).
Nonlinear dynamics can emerge in relatively simple chemical reaction networks and lead to complex behaviors in open or partially open systems (Epstein and Showalter 1996), but these behaviors have not been extensively explored to evaluate their significance to the chemical origins of life. Computational models of replenished systems have been investigated, but many of the existing models either assume the presence of an autocatalytic network, are deliberately vague about the identities of the molecules and mechanisms involved, or both (Kindermann 2005; Walker et al. 2012; Wynveen et al. 2014; Peng et al. 2020). The inclusion of chemical replicators increases the diversity of potential dynamics in a model system, but these approaches gloss over the question of how chemical replicators arose in the first place.
Here we will discuss the possible significance of open-system dynamics that can arise in chemical networks prior to the emergence of catalysis or autocatalysis. We examined the kinetic behavior in fed batch systems of glycine polymerizing into oligoglycine through a combination of wet-dry cycles and activation by trimetaphosphate (TP), an inorganic phosphate that significantly enhances peptide bond formation across a wide range of environmental conditions (Sibilska et al. 2018). Using parameters fitted from experimental data, a simplified mass-action based ordinary differential equation (ODE) model predicts the emergence of overshoot in this open system. We explored the effect that cycling between two different reaction mechanisms had on the system dynamics and compared model results to experiments. The experimental results support the idea that oscillations can alter the selectivity to favor certain products. However, our work highlights the difficulty of evaluating kinetics in open systems, specifically where reactions are driven by drying into the solid state.
Methods
Experimental Methods
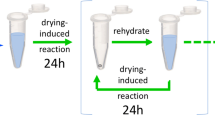
We performed several cyclic, multi-day experiments to compare with model predictions. Samples were prepared as 1 mL samples containing 0.1 M glycine, 0.1 M TP, and 0.15 M NaOH, and heated with the caps open at 90 °C for 24-h intervals. Since reaction rate parameters depend on the environment, we kept the initial conditions of each cycle as close to those conditions as possible; this required keeping the trimetaphosphate and base conditions close to their initial conditions, since they are not explicitly included in the model. To maintain these conditions without causing continuous accumulation of TP, base, or their byproducts, we used an iterative strategy, recreating samples using peptide standards to match the concentrations measured after each cycle (Fig. 1a). This ensured complete replacement of TP and base. To keep the amino acid to TP ratio constant, we also adjusted the total glycine species balance with each cycle by adding glycine monomers to compensate for any glycine lost to side products, such as 2,5-diketopiperazine or glycine oligomers with a length of seven or above, which we did not quantify. Although this may underestimate the significance of side-products, this experimental setup adheres most rigorously to the assumptions of the model.

Methods of experimental replenishment
Compartmentalization or adherence to solid surfaces can allow some molecules to be diluted less rapidly than others in an open system, but complete removal of waste and replacement of activating molecules, as occurs in iterative replenishment, is probably too idealized to be physically realistic in an origins of life context. Therefore, we also performed a set of experiments using batch replenishment, in which the reaction products from one cycle were directly transferred into the subsequent cycle (Fig. 1b). This process is a discontinuous analog of what would occur in a continuously stirred tank reactor (CSTR), where side-products from reactions can build up over time. This approach to replenishment is a more realistic representation of flow in open systems insofar as we might expect all the molecules to be equally affected by the flow rate.
For the batch replenishment experiments, all samples were initially 1 mL samples and contained 0.1 M glycine, 0.1 M TP, and 0.15 M NaOH. Samples were heated at 90 °C with the caps open for 24 h to allow them to dry, then rehydrated with 1 mL water and vortexed until the solid was fully dissolved. The term ‘replenishment rate’ refers to the percentage of fresh material that is replaced in each subsequent cycle; for example, a 75% replenishment rate indicates that 250 μL of the dissolved reaction product is transferred to the next generation, and 750 μL of a solution of 0.1 M glycine, 0.1 M TP, and 0.15 M NaOH solution is added. In practice, the glycine, TP, and NaOH solutions were stored separately and mixed only during the preparation of the subsequent cycles to prevent them from reacting in storage. We compared the results of multiple replenishment rates – 50%, 75%, and 90%.
All samples were analyzed using fluorenylmethyloxycarbonyl chloride (FMOC) derivatization and high-performance liquid chromatography (UV-HPLC) for improved retention and quantitation, as in our previous work (Boigenzahn and Yin 2022; Boigenzahn et al. 2023). FMOC derivatization was used to improve the retention time and signal strength of the peptide analytes. For the FMOC derivatization procedure, 25 μL of sample was diluted with 75 μL milliQ water and mixed with 100 μL 0.1 M sodium tetraborate, which acted as a buffer. Finally, 800 μL 0.0391 M FMOC dissolved in acetone was added to each sample, equating to 25% excess FMOC to possible amino acid.
Between cyclic transfers and prior to derivatization, samples were vortexed at maximum speed until there were no visible solids remaining (Pulsing Vortex Mixer, Fischer Scientific), usually approximately two minutes. pH was measured using an Apera Instruments PH8500-MS Portable pH microelectrode. For the batch replenishment samples, the pH at the start of each heating cycle was measured using duplicate samples prepared from the remaining material after the volume needed for HPLC analysis and transfer to subsequent generations was removed. Since a small amount of sample tends to stay on the pH probe due to surface tension, the use of duplicate samples prevented effects due to volume changes.
Derivatized samples were analyzed using a Shimadzu Nexera HPLC with a C-18 column (Phenomenex Aeris XB-C18, 250 mm × 4.6 mm, 3.6μL) and quantified using calibration curves generated from laboratory standards (Supplemental Section 1). All analysis was carried out using Solvent A: milliQ water with 0.01 v/v trifluoroacetic acid (TFA) and Solvent B: acetonitrile with 0.01% v/v TFA. Replenished samples with serial transfer were analyzed using the following gradient: 0–3 min, 30% B; 3–16 min, 30–100% B, 16–19 min, 100% B; 19–21 min, 100–30% B; 21–24 min, 30% B. Iteratively recreated samples were analyzed using the following gradient for improved resolution of G6: 0–3 min, 30% B; 3–16 min, 30–70% B, 16–19 min, 70% B; 19–21 min, 70–30% B; 21–24 min, 30% B. The solvent flow rate was 1 mL/min. Peak integration was performed in LabSolutions with the ‘Drift’ parameter set to 1000.
Materials
All materials were of analytical grade purity and used without additional purification. Materials were obtained from the following suppliers: Glycine and triglycine (G3) from Alfa Aesar (Heysham, LA3 2XY, England), diglycine, TP and TFA from Sigma-Aldrich (St. Louis, MO, USA), tetraglycine (G4), pentaglycine (G5), and hexaglycine (G6) from Bachem (Torrance, CA, USA), acetone and sodium hydroxide from Fisher Scientific (Fair Lawn, NJ, USA), acetonitrile from VWR International (Radnor, PA, USA), and FMOC from Creosalus (Louisville, KY, USA).
Model Formulation
Peptide concentrations were modeled using mass-action ordinary differential equations (ODEs) for four reversible reactions that describe how a single amino acid (glycine) forms peptides of up to four amino acids in length (Scheme 1). Although we observed and quantified G5 and G6 in our experiments, their empirical concentrations are low during the time frame that used to estimate parameters for the network, so we chose to exclude them and instead focus on species that could be clearly quantified (Boigenzahn and Yin 2022). Parameters were estimated from experimental data according to the procedure outlined in (Boigenzahn et al. 2023). As in Boigenzahn et al. (2023), the effects of volume change due to drying and the presence of any reaction intermediates were excluded for simplicity.

Reaction network for peptide formation and hydrolysis. ODEs and parameters are detailed in Supplemental Section 2
In Boigenzahn and Yin (2022), we described two mechanisms of trimetaphosphate (TP) activated peptide formation, which are differentially dependent on pH and water activity. We fit two sets of parameters to the network in Scheme 1, representing the two different reaction mechanisms. Briefly, Mechanism 1 occurs in alkaline conditions and proceeds readily in water (Chung et al. 1971), and Mechanism 2 occurs in neutral conditions and proceeds as the sample approaches the solid state due to drying (Yamanaka et al. 1988). Mechanism 1 lowers the pH of the samples when it occurs, so samples containing amino acids and TP dried in alkaline conditions naturally transition from Mechanism 1 to Mechanism 2. This transition can be used to explore how environmental oscillations can alter kinetic behavior within a reaction network. The code and data for this manuscript can be found at https://github.com/haboigenzahn/Cyclic-Environments.
Initial Parameter Estimation
To estimate parameters for each mechanism, we selected experimental data in which one mechanism strongly dominated over the other. The parameters for Mechanism 1 were generated based on 8-h time courses obtained from 0.1 M glycine or 0.05 M diglycine (GG) heated at 90 °C with 0.1 M trimetaphosphate and 0.15 M base without drying (closed caps). Additional data came from the first four hours of equivalent experiments that were allowed to dry (open caps), since during the early stages of drying there is still bulk water present such that Mechanism 1 dominates (Boigenzahn and Yin 2022). Finally, to predict behavior in second and subsequent drying cycles while minimizing the number of experiments required, we included peptide concentrations from the first four hours of only the first day of iterative experiments. We deliberately kept the time span of the training data relatively short so that later experimental cycles could be used to assess the predictive accuracy of the model.
The parameters for Mechanism 2 were estimated from 0.1 M glycine and 0.1 M TP with and without 0.15 M base, starting after the samples had been drying for 4 h and including the next 20 h. Additional results from samples of 0.1 M glycine dried with 0.1 M TP and 0.15 M base were prepared and measured after 4 h of heating in 24-h intervals for up to 72 h to improve the long-term estimates of Mechanism 2. Finally, several measurements from the first day of the iterative experiments were also taken after the first four hours of heating and included in the training data for Mechanism 2. The specific fitted parameter values for Mechanism 1 and Mechanism 2 are detailed in Supplemental Section 2.
Results and Discussion
To evaluate the accuracy of the model predictions using separate parameters for Mechanism 1 and Mechanism 2, we compared the predictions to peptide concentration profiles measured in Boigenzahn and Yin (2022) (Fig. 2). We used the assumption that Mechanism 1 occurred during the first 4 h and Mechanism 2 accounted for the following 20 h after verifying the timing of this transition by testing parameters fit to alternative timings (Supplemental Section 3). Although the model tended to slightly underestimate peptide formation, we determined that this could be interpreted as a conservative estimate of the rates of polymer formation, and was acceptable for the behaviors we were aiming to explore.

Comparison of two-step model to analogous experimental data. Experimental data is from Boigenzahn and Yin (2022)
We used these parameters to model peptide formation in a cyclic environment and observed that for certain combinations of parameters and initial conditions, the average yield of the longer glycine polymers exceeded the ‘thermodynamic equilibrium’ predicted by the fitted parameters of either mechanism (Fig. 3). It should be noted that the model system was formulated with the same structure as mass action kinetics models and each parameterized model inherently has a fixed-point attractor, or a state which the system approaches as time approaches infinity. In an ideal mass action kinetics system, the attractor is equivalent to the concentrations of the species at thermodynamic equilibrium. However, since the reference model is not a true mass-action representation of the system, we will henceforth use the term ‘attractor’ when referring to the steady state solutions to the model system; this allows us to distinguish the mathematical predictions from the simplified model from concepts of true thermodynamic equilibrium.

Cycling conditions where the average yield of G3 and G4 at steady state exceeds the value of the attractor due to overshoot. The model is initialized with 0.1 M G. The approach of each mechanism to its attractor in a non-cyclic system is shown for comparison. The cyclic trajectory alternates between 4 h of Mechanism 1 and 20 h of Mechanism 2. Parameters are estimated from experimental data
Many simple chemical reactions exhibit monotonic kinetics in all species. In such systems, the maximum yield of each species is limited by the level of the attractor. In the system predicted by our model, G3 and G4 surpass the attractor of both Mechanism 1 and Mechanism 2 due to a dynamic phenomenon called overshoot. Overshoot is a non-monotonic behavior which occurs in a wide variety of systems, including biological and man-made control networks (Ogata 1995; Jia et al. 2014; Chen et al. 2016). Systems with overshooting kinetics will eventually return to the attractor, however, in this example the environmental cycling between the two mechanisms cause overshoot to occur repeatedly.
Mathematical Definition of Overshoot
Consider a high-dimensional system with many chemical species. Overshoot occurs when a system passes through the coordinates of its stable point in any dimension before the stable point is reached. For a mathematical definition of overshoot, consider a dynamical system whose state at time t is given by a vector of real numbers \(x(t)\) such that
where \(F(x)\) is the rate vector for the system. Assume the system has at least one stable fixed-point \(\underline x\), such that
Starting from an initial condition in the vicinity of the fixed point \(x(0)\), the system overshoots in dimension \(j\) if there exists a time \(t \in (0,\infty )\) for which the state at \(t\) is further away from but lies in the same direction of the fixed point as the initial condition,
This characterization provides a straightforward algorithm for determining if a dynamical system is capable of overshooting in any direction around a stable fixed point. First, discretely sample a small sphere centered around the stable fixed point. Then from each point on the sphere, integrate the dynamical system backwards in time and track the distance of the state from the fixed point. If the maximum distance is not monotonic in any dimension, then the system overshoots.
Significance of Overshoot
It should also be noted that whether overshoot occurs in an experimental system can be restricted by thermodynamics. All reactions still must obey the Second Law of Thermodynamics, so systems which are close to thermodynamic equilibrium will rarely overshoot. Overshoot requires a system to be under kinetic control, which is more likely in systems which are further from equilibrium (Epstein and Showalter 1996).
Overshoot depends on the kinetic reaction network and initial conditions of a system. In the example with alternating mechanisms shown in Fig. 3, Mechanism 2 approaches its attractor monotonically for a system initialized with pure glycine monomer, but the formation of diglycine by Mechanism 1 creates initial conditions where Mechanism 2 tends to overshoot G3 and G4. One factor is that Mechanism 1 hydrolyzes some of the longer polymers produced, but also generates more GG. Repeated cycles of Mechanism 1 and Mechanism 2 in a 24-h pattern created a dynamic steady state for which the average yields of G3 and G4 exceed the predicted attractor values for either mechanism. We also found that for some initial conditions, this behavior was possible using a single reaction mechanism with batch replenishment. This demonstrates that these dynamics can occur within a single reaction mechanism, though there still needs to be oscillations (Supplemental Section 4).
To understand why it is significant that the longer species exceed the system attractors, it is useful to revisit the perspective of thermodynamic equilibrium. The formation of longer peptides in water is thermodynamically limited (Ross and Deamer 2016). Allowing samples to dry favors polymerization, but the lack of molecular mobility in the dry state limits the potential for long-term reactions. Overshoot provides an explanation for how systems subjected to wet-dry cycling drying could not only form and maintain populations of longer polymers, but could also exhibit selectivity, since the distribution of polymers formed depends on the reaction kinetics and the timing of the cycles. Species that form quickly can more easily recover from dilution or hydrolysis. In many cases there may be an ‘optimal’ cycle timing, or resonance time, for each species, which maximizes the yield of a species of interest (Haugerud et al. 2023).
Repeated overshoot can allow a system to remain away from its predicted attractor for long periods of time, and may help provide the dynamic, non-equilibrium conditions that have been proposed as being critical for the origin of life (Pross 2011; Pascal et al. 2013). Therefore, overshoot and far-from-equilibrium conditions may be mutually reinforcing: overshoot pushes systems away from their attractor and alters system compositions, creating the potential for the system to experience overshoot again. These paired behaviors could begin to move the system towards a regime of kinetic control, in which product compositions are influenced more heavily by reaction rates than by product energy levels. Autocatalysis, one of the essential features of a life-like system, is an example of a behavior which is strongly under kinetic control (Pross 2003).
Overshoot has been observed in multiple experimental systems containing designed peptide replicators (Dadon et al. 2015; Miao et al. 2021). Similar dynamics have also been linked to the emergence of biochemical adaptation, which refers to the ability of a system to return to its original state after an environmental perturbation (Jia et al. 2014; Ma et al. 2009; François and Siggia 2008). It is interesting that our simple system of just four reversible reactions shows qualitatively similar behavior to these much more complex cases.
Experimental Comparison
Since the kinetic parameters estimated for Mechanism 1 and Mechanism 2 were primarily based on short term data, we wanted to determine how accurately the model captured the long-term behavior of the system. Therefore, we performed experiments to evaluate the capacity of the kinetic model to make predictions about the yields in multi-day experiments. We sought to identify potential signs of nonlinear dynamics, which may include features like non-monotonic concentration profiles, sigmoidal growth, or evidence of multiple steady states.
Due to the various simplifications included in the model formulation, we used the iterative replenishment approach to replicate the model assumptions as closely as possible. While the method is somewhat contrived, it has the advantage of eliminating as many known confounding variables as possible to facilitate the comparison of the model and the experimental data. Variables not included in the model that might impact the reactivity of the system included the concentrations of TP, base, and orthophosphate side products.
There was reasonably good agreement between the model predictions and the experimental results, especially the final yields of the peptides (Fig. 4). Significantly more G4 forms during the first few cycles than the model predicts (Fig. 4d), but the model underestimating the yield of G4 is consistent with the results of the training data (Fig. 2). However, there are some discrepancies between the first few days of the model trajectories and the experimental results. These discrepancies could be the result of inaccuracies in parameter estimation, experimental variance, or a consequence of confounding variables such as those caused by drying or the formation of longer polymers in the system.

Comparison of experimental and model results for 7 days. Plots show concentrations for a glycine, b diglycine, c triglycine, and d tetraglycine. Experimental data was generated using the iterative approach. Models use an initial condition of 0.1 M Gly. Error bars represent the sample standard deviation of experimental triplicates
One noteworthy discrepancy is that the experimentally measured maximum yield of GG and G3 occurs after two days, then the yield drops in subsequent days, which is not predicted by the model (Fig. 4b, c). These experimental trajectories are notable since a non-monotonic result would not be expected from systems with monotonic underlying kinetics. Non-monotonic trajectories like those observed in GG and G3 can occur in species that are initially overshooting, but the overshoot is damped and decreases as the reaction cycles progress. However, damped overshoot behavior was not predicted by the model. Although GG and G3 appear to slightly exceed their eventual final yields then gradually drop in concentration, this is not necessarily experimental evidence of overshoot.
Although we can evaluate the accuracy of the model and look for evidence of nonlinear dynamics, we cannot confirm the existence or absence of overshoot in the experimental system. Maintaining repeated overshoot relative to a mathematically predicted attractor requires an open system, but in practice overshoot is not well defined for open systems, since there is no experimentally measurable point which is equivalent to the attractor. In closed systems, the attractor represents the thermodynamic equilibrium of the system, but open systems violate a key principle of equilibrium, namely that there cannot be any exchange of energy or mass with the environment.
Instead, the real systems reach a dynamic steady state, in which the overall species concentrations remain constant from cycle to cycle, but there are bonds which form and break during each cycle. This state may have been a precursor to the emergence of dynamic kinetic stability, a concept introduced by Pross to describe the behavior of systems of replicators which maintain their population while experiencing a continuous flux of energy (Pross 2009).
Ideally, evidence of overshoot might be found experimentally by allowing Mechanism 2 to continue and looking for evidence of non-monotonic behavior. However, simply allowing the system to continue heating indefinitely would not result in the continuation of Mechanism 2 with its estimated rate constants, since those parameters were estimated primarily using data from samples likely still contained some residual water. It is therefore unsurprising that experiments which were continuously heated did not show non-monotonic behavior (Fig. 5), since once the samples were completely dry, they were unlikely to undergo any significant hydrolysis.

Replenishment affects species selectivity. Plots show concentrations for a glycine, b diglycine, c triglycine, and d tetraglycine. Samples carried out using iterative replenishment have higher yields of G4, while samples that were continuously dried have higher yields of G3. Samples were analyzed at the end of each 24-h cycle
When comparing the steady states reached by the iteratively cycled experiments to those of non-cycled experiments, we found the iterative experiments had higher yields of G4, but lower yields of G3 (Fig. 5c, d). This supports the idea that cyclic environmental conditions can select for certain species over others. This type of selectivity is interesting because it is driven primarily by kinetics, with the actual thermodynamic stability of each species playing a secondary role. This type of behavior may contribute to the ability of some chemical reaction networks to move away from thermodynamic equilibrium within their local environment, even in the absence of chemical replicators. Further experiments using different replenishment procedures, such as varying the cycle timing to study how it affects selectivity, may produce valuable insights. It may also be possible to observe additional kinetic effects by diluting a species relative to the others with each cycle, creating artificial pressure against it.
Complete Replenishment
Since the iteratively recreated experiments are not entirely physically realistic owing to the complete removal of waste and replacement of food without any dilution of the desired products, we also investigated the behavior of the system using replenishment conditions in which a portion of the dissolved products from the previous cycle are transferred and replaced with fresh reactants, as shown in Fig. 1b. We examined three different replenishment rates, or three different fractions of products to be transferred from sample to sample and compared them to one another. This method creates a trade-off between the costs and benefits of dilution and replenishment. High replenishment rates have high dilution rates, so less of the product formed in previous cycles is transferred into subsequent generations. Low replenishment rates transfer more previously formed products, but have limited access to fresh reactants, which is detrimental when the reactants include activating agents, like TP, that are needed for reactions to occur. Since the model does not include the effects of reduced TP or base, it predicts higher peptide yields for lower replenishment rates (Supplemental Section 5).
However, when we tried these tests experimentally, we found that the opposite was true; higher replenishment rates had higher peptide yields (Fig. 6). This implies that the yield per drying is not limited by the availability of oligopeptides, but by the pH and concentration of TP. The difference in initial pH is particularly significant because it determines whether both reaction mechanisms occur during drying. There are two factors which change the pH of the samples between the initial setup and the subsequent replenishment steps. First, less base is added during the replenishment cycles than was added during the initial setup, since the samples are only replenished with a fraction of their original reactants. Second, some phosphate byproducts from the TP reactions are transferred between cycles, which changes the buffering capacity of the samples. The initial pH of the samples was 10.78 ± 0.06, and the pH of the samples at the start of the first cycle was 9.72 ± 0.06 for 90% replenishment, 9.24 ± 0.15 for 75% replenishment and 7.01 ± 0.01 for 50% replenishment (Supplemental Section 6). Mechanism 1 requires the deprotonated amine groups, and the pH at which most of the amine groups are deprotonated for glycine is 9.60. Therefore, we expect to see reduced GG formation when the initial pH of the solution is significantly below that point. This likely explains why samples with 75% replenishment have disproportionately lower GG formation than samples with 90% replenishment relative to the difference in their initial pH.

Higher replenishment rates have higher peptide yields in replenished systems with serial transfer. Replenishment percentages indicate how much of the sample was replaced with fresh reactant mixture each day. Each data set is normalized relative to the results from the first day
Although it has a less profound impact on the underlying reaction mechanism, peptide yields tend to decrease when the amino acid-to-TP ratio is less than equimolar (Supplemental Section 7), so the availability of TP likely also contributes lowering peptide yields at lower replenishment rates. This is likely to pose a challenge in any systems which include activating agents that are consumed. It is still theoretically possible for these systems to accumulate higher peptide concentrations even when the activating agent is only partially replaced; for example, a system with slow reverse kinetics whose forward rate constants were minimally impacted by the presence of side products could potentially accumulate polymers despite having less available activating agent after the first cycle. However, in many cases the combination of decreased reactivity and sample dilution is difficult to overcome, which causes the yields of longer products at steady state to be lower than their yields after one reaction cycle.
Activating materials that act as catalysts, like solid surfaces or metal salts, may help maintain a more constant reactivity in replenished systems since they are not depleted after the first cycle (Bujdák et al. 1995; Erastova et al. 2017). However, even with reusable activating agents, recursive reactions can be inhibited by waste products that are never fully removed. Thus, there is significant interest in methods that selectively retain certain products while removing most other materials. Realistic scenarios that have been suggested to produce this behavior include absorption onto mineral surfaces or containment in coacervates (Bedoin et al. 2020; Fares et al. 2020). These scenarios allow cycling of energy sources and waste products without equal dilution of biopolymers.
Given the number of possible scenarios, thorough experimental design of replenishments can be a difficult task. Moreover, the fact that kinetic reaction networks may be highly nonlinear means that relatively minor differences in experimental design may have significant consequences for the product distribution. Fully understanding the details of how system members interact with one another and with the environment is often challenging and may not currently be possible for some complex systems. Approximations of system behavior can be useful but need to be applied carefully. Despite these difficulties, it is worthwhile to develop an understanding of the dynamics of short monomers and their biopolymers undergo hydrolysis and condensation reactions, as these interactions must have been involved in the origins of long, functional biopolymers such as enzymes.
Conclusion
Although the dynamics of chemical reaction networks prior to the emergence of replicating chemical systems have not been extensively explored, they may provide valuable insights into how biopolymer systems begin to develop complex behaviors. The behaviors of open systems, which are consistently held away from equilibrium and therefore heavily influenced by kinetics, are of particular interest. However, studying chemical reaction networks in an open system can be challenging because environmental changes can make the results more difficult to interpret and experimental variables such as the method and rate of replenishment, as well as the cycle timing, which can greatly expand the experimental parameter space. As we have shown, despite these challenges, interesting behaviors can be captured using simple experimental systems paired with kinetic models.
We found that a simple ODE model of glycine polymerization using parameters fit from experimental data displayed overshoot, a dynamic phenomenon in which a species passes through its attractor in some dimension at least once before reaching it. Simulated environmental cycles that alternated between two reaction mechanisms resulted in yields of trimers and tetramers that were well above the calculated attractors of the two reaction mechanisms involved. Since equilibrium as represented by the attractors is not defined for an open system, we were unable to look for overshoot directly, but iterative experiments found relatively good agreement between the model and experimental results. It is noteworthy that different experimental cycling conditions indicate the possibility of species selectivity. These findings illustrate that even simple reaction networks, when pushed by repeated environmental changes, can start showing kinetic control, which is an important feature of reactions in life-like systems. Future modeling and experimental studies of non-linear reaction systems in open environments, thus, seem critical to help us understand the origins of complex, life-like dynamics in chemical reaction networks.
Data Availability
The data shown in this study is available from John Yin upon reasonable request.
References
Astumian RD (2019) Kinetic asymmetry allows macromolecular catalysts to drive an information ratchet. Nat Commun 10(1):1–14. https://doi.org/10.1038/s41467-019-11402-7
Bartolucci G, Serrão AC, Schwintek P, Kühnlein A, Rana Y, Janto P, Hofer D, Mast CB, Braun D, Weber CA (2022) Selection of prebiotic oligonucleotides by cyclic phase separation. http://arxiv.org/abs/2209.10672
Baum DA (2018) The origin and early evolution of life in chemical composition space. J Theor Biol 456:295–304. https://doi.org/10.1016/j.jtbi.2018.08.016
Bedoin L, Alves S, Lambert J (2020) Origins of life and molecular information : selectivity in mineral surface induced prebiotic amino acids polymerization. ACS Earth Space Chem https://doi.org/10.1021/acsearthspacechem.0c00183
Boigenzahn H, González L, Thompson J, Zavala V, Yin J (2023) Kinetic modeling and parameter estimation of a prebiotic peptide reaction network. J Mol Evol Manuscript in revision. https://link.springer.com/article/10.1007/s00239-023-10132-1
Boigenzahn H, Yin J (2022) Glycine to Oligoglycine via Sequential Trimetaphosphate Activation Steps in Drying Environments. Orig Life Evol Biosph 52(4):249–261. https://doi.org/10.1007/s11084-022-09634-7
Bujdák J, Faybíková K, Eder A, Yongyai Y, Rode BM (1995) Peptide chain elongation: A possible role of montmorillonite in prebiotic synthesis of protein precursors. Orig Life Evol Biosph 25(5):431–441. https://doi.org/10.1007/BF01581994
Chen X, Wang Y, Feng T, Yi M, Zhang X, Zhou D (2016) The overshoot and phenotypic equilibrium in characterizing cancer dynamics of reversible phenotypic plasticity. J Theor Biol 390:40–49. https://doi.org/10.1016/j.jtbi.2015.11.008
Chung NM, Lohrmann R, Orgel LE, Rabinowitz J (1971) The mechanism of the trimetaphosphate-induced peptide synthesis. Tetrahedron 27(6):1205–1210. https://doi.org/10.1016/S0040-4020(01)90868-3
Colón-Santos S, Cooper GJT, Cronin L (2019) Taming the Combinatorial Explosion of the Formose Reaction via Recursion within Mineral Environments. ChemSystemsChem 1(3):1–5. https://doi.org/10.1002/syst.201900014
Dadon Z, Wagner N, Alasibi S, Samiappan M, Mukherjee R, Ashkenasy G (2015) Competition and cooperation in dynamic replication networks. Chem Eur J 21(2):648–654. https://doi.org/10.1002/chem.201405195
Doran D, Abul-Haija YM, Cronin L (2019) Emergence of Function and Selection from Recursively Programmed Polymerisation Reactions in Mineral Environments. Angewandte Chemie - International Edition 58(33):11253–11256. https://doi.org/10.1002/anie.201902287
Eigen M, Schuster P (1977) The Hypercyde. Naturwissenschaften 64:541–565
Epstein IR, Showalter K (1996) Nonlinear chemical dynamics: Oscillations, patterns, and chaos. J Phys Chem 100(31):13132–13147. https://doi.org/10.1021/jp953547m
Erastova V, Degiacomi MT, Fraser DG, Greenwell HC (2017) Mineral surface chemistry control for origin of prebiotic peptides. Nat Commun 8(1):1–9. https://doi.org/10.1038/s41467-017-02248-y
Fares HM, Marras AE, Ting JM, Tirrell MV, Keating CD (2020) Impact of wet-dry cycling on the phase behavior and compartmentalization properties of complex coacervates. Nat Commun 11(1). https://doi.org/10.1038/s41467-020-19184-z
François P, Siggia ED (2008) A case study of evolutionary computation of biochemical adaptation. Phys Biol 5(2). https://doi.org/10.1088/1478-3975/5/2/026009
Haugerud IS, Jaiswal P, Weber CA (2023) Wet-dry cycles away from equilibrium catalyse chemical reactions. arXiv e-prints, arXiv-2304. https://doi.org/10.48550/arXiv.2304.14442
Jia C, Qian M (2016) Nonequilibrium Enhances Adaptation Efficiency of Stochastic Biochemical Systems. PLoS ONE 11(5):1–19. https://doi.org/10.1371/journal.pone.0155838
Jia C, Qian M, Jiang D (2014) Overshoot in biological systems modelled by Markov chains: a non-equilibrium dynamic phenomenon. IET Syst Biol 8(4):138–145. https://doi.org/10.1049/iet-syb.2013.0050
Kindermann M, Stahl I, Reimold M, Pankau WM, von Kiedrowski G (2005) Systems chemistry: kinetic and computational analysis of a nearly exponential organic replicator. Angew Chem 117(41):6908–6913. https://doi.org/10.1002/ange.200501527
Lahav N, Chang S (1976) The possible role of solid surface area in condensation reactions during chemical evolution: Reevaluation. J Mol Evol 8(4):357–380. https://doi.org/10.1007/bf01739261
Lahav N, White D, Chang S (1978) Peptide formation in the prebiotic era: Thermal condensation of glycine in fluctuating clay environments. Science 201(4350):67–69. https://doi.org/10.1126/science.663639
Ma W, Trusina A, El-Samad H, Lim WA, Tang C (2009) Defining Network Topologies that Can Achieve Biochemical Adaptation. Cell 138(4):760–773. https://doi.org/10.1016/j.cell.2009.06.013
Mamajanov I, Macdonald PJ, Ying J, Duncanson DM, Dowdy GR, Walker CA, Engelhart AE, Fernández FM, Grover MA, Hud NV, Schork FJ (2014) Ester formation and hydrolysis during wet-dry cycles: Generation of far-from-equilibrium polymers in a model prebiotic reaction. Macromolecules 47(4):1334–1343. https://doi.org/10.1021/ma402256d
Martin O, Horvath JE (2013) Biological Evolution of Replicator Systems: Towards a Quantitative Approach. Origins of Life and Evolution of Biospheres 43(2):151–160. https://doi.org/10.1007/s11084-013-9327-4
Miao X, Paikar A, Lerner B, Diskin-Posner Y, Shmul G, Semenov SN (2021) Kinetic Selection in the Out-of-Equilibrium Autocatalytic Reaction Networks that Produce Macrocyclic Peptides. Angewandte Chemie - International Edition 60(37):20366–20375. https://doi.org/10.1002/anie.202105790
Ogata K (1995) Discrete-time control systems. Prentice-Hall Inc.
Pascal R, Pross A, Sutherland JD (2013) Towards an evolutionary theory of the origin of life based on kinetics and thermodynamics. Open Biol 3(11):130156. https://doi.org/10.1098/rsob.130156
Peng Z, Plum AM, Gagrani P, Baum DA (2020) An ecological framework for the analysis of prebiotic chemical reaction networks. J Theor Biol 507:110451. https://doi.org/10.1016/j.jtbi.2020.110451
Prigogine I (1978) Time, structure, and fluctuations. Science 201(4358):777–785
Pross A (2003) The driving force for life’s emergence: Kinetic and thermodynamic considerations. J Theor Biol 220(3):393–406. https://doi.org/10.1006/jtbi.2003.3178
Pross A (2009) Seeking the chemical roots of Darwinism: Bridging between chemistry and biology. Chem Eur J 15(34):8374–8381. https://doi.org/10.1002/chem.200900805
Pross A (2011) Toward a general theory of evolution: Extending Darwinian theory to inanimate matter. J Syst Chem 2(1):1–14. https://doi.org/10.1186/1759-2208-2-1
Ross DS, Deamer D (2016) Dry/wet cycling and the thermodynamics and kinetics of prebiotic polymer synthesis. Life 6(3):1–12. https://doi.org/10.3390/life6030028
Shapiro R (2000) A replicator was not involved in the origin of life. IUBMB Life 49(3):173–176. https://doi.org/10.1080/152165400306160
Sibilska I, Feng Y, Li L, Yin J (2018) Trimetaphosphate activates prebiotic peptide synthesis across a wide range of temperature and pH. Orig Life Evol Biosph 48:277–287. https://doi.org/10.1007/s11084-018-9564-7
Vincent L, Berg M, Krismer M, Saghafi SS, Cosby J, Sankari T, Vetsigian K, Cleaves HJ, Baum DA (2019) Chemical ecosystem selection on mineral surfaces reveals long-term dynamics consistent with the spontaneous emergence of mutual catalysis. Life 9(4). https://doi.org/10.3390/life9040080
Wagner N, Hochberg D, Peacock-Lopez E, Maity I, Ashkenasy G (2019) Open prebiotic environments drive emergent phenomena and complex behavior. Life 9(2). https://doi.org/10.3390/life9020045
Walker SI, Grover MA, Hud NV (2012) Universal sequence replication, reversible polymerization and early functional biopolymers: A model for the initiation of prebiotic sequence evolution. PLoS ONE 7(4):31–37. https://doi.org/10.1371/journal.pone.0034166
Wynveen A, Fedorov I, Halley JW (2014) Nonequilibrium steady states in a model for prebiotic evolution. Phys Rev E Stat Nonlinear Soft Matter Phys 89(2):1–10. https://doi.org/10.1103/PhysRevE.89.022725
Yamanaka J, Inomata K, Yamagata Y (1988) Condensation of Oligoglycines with Trimeta- and tetrametaphosphate in aqueous solution. Orig Life Evol Biosph 18(31):165–179. https://doi.org/10.1007/bf01804669
Acknowledgements
Thank you to David Baum for reviewing the manuscript, and members of the Yin and Baum labs for their feedback and support.
Funding
This research was funded by the Vilas Distinguished Achievement Professorship, the Office of the Vice Chancellor for Research and Graduate Education, the Wisconsin Institute for Discovery, all at the University of Wisconsin-Madison; and the Wisconsin Alumni Research Foundation (WARF); and DMS-2151959 from the US National Science Foundation. Funding for PG was provided by the National Science Foundation, Division of Environmental Biology (Grant No: 2218817).
Author information
Authors and Affiliations
Contributions
All experimental data and model figures were prepared and analyzed by HB. The mathematical formulation of overshoot was written by PG. The manuscript was drafted by HB with input from PG and JY, and all authors contributed to the subsequent editing.
Corresponding author
Ethics declarations
Ethics Approval
No approvals required.
Consent to Publish
All authors read and approved the final manuscript.
Conflict of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Boigenzahn, H., Gagrani, P. & Yin, J. Enhancement of Prebiotic Peptide Formation in Cyclic Environments. Orig Life Evol Biosph 53, 157–173 (2023). https://doi.org/10.1007/s11084-023-09641-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-023-09641-2