
Abstract
The thrombolytic activity of tissue plasminogen activator (tPA) has undisputed benefits. However, the documented neurotoxicity of tPA raises important issues. Currently, common treatments for stroke might not be optimum if exogenous tPA can pass through the blood–brain barrier and enter the brain, thus adding to the deleterious effects of tPA within the cerebral parenchyma. Here, we determined whether tPA could damage brain microvascular endothelial cells (BMECs) during cerebral ischemia. We showed that treatment of BMECs with tPA decreased trans-endothelial electrical resistance and cell proliferation, and blocked the cell cycle at the G0–G1 phase. In addition, the Sonic hedgehog (Shh) signaling pathway was involved in tPA-induced BMECs dysfunction. However, tPA-enhanced oxygen glucose deprivation-induced BMECs dysfunction was eliminated by Shh administration and the effects could be reversed by Shh inhibitors. Taken together, these results demonstrate that tPA administration might result in damage to the endothelial barrier owing to blocked Shh signaling pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke leads to the disruption of the blood–brain barrier (BBB), which increases the permeability of the brain microvasculature and eventually results in brain edema [1]. The BBB is formed by brain endothelial cells lining cerebral microvessels and performs a combination of physical, transport, and enzymatic barrier functions [2]. Endothelial cells have the capacity to modulate barrier permeability by regulating the expression of tight and adherent junction proteins. Therefore, endothelial cell injury can influence the permeability of the BBB [3, 4]. Brain microvascular endothelial cells (BMECs) play an important role in maintaining brain vascular homeostasis, and dysfunction of the endothelial layer is recognized as an early event in the pathogenesis of cerebrovascular diseases such as subarachnoid hemorrhage and ischemic stroke [5, 6].
Tissue plasminogen activator (tPA) is a serine protease that induces the conversion of plasminogen to plasmin to dissolve fibrin-based blood clots and is routinely used in ischemic stroke pharmacotherapy [7, 8]. However, treatment via tPA thrombolysis is limited to less than 5% of stroke patients due to the narrow therapeutic time window and the potentially devastating complication of intracerebral hemorrhage [9, 10]. tPA-related intracerebral hemorrhage occurs as a consequence of severe BBB disruption during thrombolytic reperfusion [11]. Previously, in vitro studies using BMECs and astrocytes have shown that endothelial tPA expression is negatively regulated by astrocytes [12]. A growing body of evidence suggests that exacerbation of BBB injury following tPA administration, such as the induction of proteolytic degradation of the structural components of the BBB, increases the production of free radicals and induces toxicity to neurovascular cells [13,14,15]. However, the molecular effects associated with tPA administration after ischemic stroke remain unknown.
Sonic hedgehog (Shh) is a glycoprotein that has both morphogenic and mitogenic properties and is an indirect angiogenic factor in individual development and tissue repairment [16]. Shh binds to the specific receptor Patched-1 (Ptch1), thereby causing the release of the transmembrane protein Smoothened (Smo). This leads to the activation of the transcription factor Gli-1, which induces the expression of genes in downstream signaling pathways and regulates endothelial cell apoptosis [17, 18]. It has also been suggested that Shh induces cerebral angiogenesis and mediates neural-tube angiogenesis during embryonic development [19, 20]. In addition, Shh promotes the integrity and immune quiescence of the BBB by decreasing the expression of inflammatory mediators as well as the adhesion and migration of leucocytes [21].
In the present study, we investigated the effect of tPA administration on the Shh signal pathway in BMECs. This study was primarily designed to elucidate the role of Shh in the effects of tPA administration after ischemic stroke.
Materials and Methods
Ethic Statements
This study was approved by the committees on experimental animal ethics of Renmin Hospital, Wuhan University. All animal experiments were performed in accordance with the Guidelines for Experimental Animals of the Ministry of Science and Technology (Beijing, China). All dissections were performed according to recommendations of the European Commission, and all efforts were made to minimize suffering in our animals.
Isolation of Rat BMECs
BMECs were isolated from Sprague–Dawley rats, according to reported protocols with some modifications [22]. Briefly, rats were sacrificed and their brains were collected. Subsequently, white matter, brain stems, surface vessels, and leptomeninges were removed carefully. The isolated cerebral cortices were placed in ice-cold phosphate-buffered saline (PBS), minced into small pieces, and homogenized. The homogenates were centrifuged at 500×g for 5 min at 4 °C. The pellet was resuspended in 20% bovine serum albumin (BSA) and centrifuged at 1000×g for 20 min at 4 °C. The microvessels in the lower layer were transferred to a new tube. After one PBS wash, the microvessel pellets were digested using 0.1% collagenase II/dispase and 1000 U/ml DNase I at 37 °C for 1 h. Following this, the microvessel pellets were centrifuged at 500×g for 5 min at 4 °C and resuspended in 10 ml of M131 medium (Invitrogen, USA) supplemented with microvascular growth supplement (Invitrogen, USA), 100 U/ml penicillin, and 100 U/ml streptomycin. The cell suspension was seeded into a 75-cm2 flask and incubated at 37 °C in humidified 5% CO2/95% air. BMECs at passages 3 to 6 were used.
Oxygen Glucose Deprivation and Drug Treatment
In order to simulate ischemic conditions, oxygen glucose deprivation (OGD) was induced in the in vitro cell study. Cells were exposed to OGD medium with low glucose (0.2 g/l) equilibrated with nitrogen (Gibco, USA). Hypoxic conditions were achieved using a chamber with 95% N2 and 5% CO2 at 37 °C for 4 h. PBS (control group, CON), tPA (300 µg/ml), Shh (3 µg/ml), or cyclopamine (20 µM) were added after the cells were subjected to OGD for 4 h, following which they were cultured under normoxic conditions for another 24 h and harvested for different experiments [23]. Each experiment was performed in triplicate.
Immunofluorescence
For immunofluorescence, cells were seeded onto 1.5-mm glass coverslips coated with 0.2% gelatin. The cells were washed with PBS and fixed in 4% paraformaldehyde for 15 min, followed by permeabilization in 0.2% Triton X-100 for 10 min. After being blocked in 5% BSA for 1 h at room temperature, the cells were incubated with primary antibodies (VIII factor or CD34) for 1 h at room temperature. The cells were washed and incubated for 1 h with Alexa Fluor-conjugated secondary antibodies and 4′-6-diamidino-2-phenylindole as a nuclear counterstain. The coverslips were washed and imaged using a ZeissAxioplan II microscope (Carl Zeiss, USA).
Measurement of Trans-endothelial Electrical Resistance (TEER)
The integrity of the BMECs was evaluated by measuring the TEER using a Millicell-ERS device (Millipore, Bedford, USA). Cells were allowed to grow until a TEER value of greater than 1000 Ω cm2 was reached, indicating the formation of a tight epithelial monolayer [24]. TEER measurements were made using an epithelial volt-ohm-meter (EVOM, World Precision Instruments Inc., Sarasota, USA). Changes in TEER were expressed as the percentage of the initial value adjusted for control cell layers according to the equation:
Proliferation Assay
The cell-counting kit 8 assay (CCK-8) was used to evaluate cell proliferation. Cells were seeded in 96-well plates with 1 × 104 cells in each well. Subsequently, 10 µl of CCK-8 reagent (5 mg/ml) was added to each well, and the plates were incubated at 37 °C for 2 h. The optical density was measured at 450 nm.
Cell Cycle Assay
The cell cycle of rat BMECs was examined using Lipofectamine 2000. Cells were washed thrice with ice-cold PBS and fixed with 70% (v/v) ethanol at − 70 °C for 1 h. Following the PBS wash, a staining solution containing 10 mM Tris (pH 7.0), 0.1% NP-40, 1 mM NaCl, 0.7 µg/ml ribonuclease A, and 5 µg/ml propidium iodide (PI) was added to the cells. After incubation for 30 min in the dark, the cellular DNA content was examined by PI-staining flow cytometry.
RNA Extraction and Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from samples with Trizol reagent (TaKaRa, Dalian, China) and detected by an ultraviolet spectrophotometer and agarose electrophoresis. For each sample, 1 µg of total RNA was reverse-transcribed to obtain first-strand cDNA using the PrimeScript®RT reagent Kit with gDNA Eraser (TaKaRa, Dalian, China) according to the manufacturer’s instructions. Primer Premier 5.0 was used to design the fluorescent primers before gene synthesis. The synopsis of primers for each gene is listed in Table 1. The reaction mixture (total volume of 20 µl) contained 10 µl of 2 × SYBR Premix Ex TaqTM (TaKaRa, Dalian, China), 0.4 µl of each primer, and 0.2 ± 0.02 µl of cDNA template. The following qRT-PCR reaction was performed: pre-denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 3 min, annealing at 56–60 °C for 20 s, and elongation at 72 °C for 20 s. The transcriptional levels of genes were calculated using the ΔΔCt method. The threshold cycle (Ct) was determined for each reaction using the ΔΔCt method, and each gene of interest was normalized to the endogenous control gene (GAPDH). For each group, three samples were measured and three technical replicates of each measurement were obtained.
Western Blot
Protein expression levels were analyzed by western blot. For total protein extraction, cells were washed twice with PBS and lysed at 4 °C with RIPA buffer (Beyotime, China) containing protease inhibitor. The cell lysate was centrifuged at 12,000×g for 15 min and the supernatants were collected. The protein concentration was determined by a bicinchoninic acid kit (Bioswamp, China). Equal amounts of protein (30 µg) were separated by 10% sodium dodecyl sulfate–polyacrylamide electrophoresis, after which the proteins were transferred onto polyvinylidene fluoride membranes (Millipore, USA) at 200 mA for 2 h. The membranes were blocked for 2 h at room temperature with 5% skim milk in Tris-buffered saline (20 mM Tris, 500 mM NaCl, and 0.05% Tween 20). Subsequently, the membranes were incubated with primary antibodies against Shh (ab19897, 1:1000, abcam), Ptch1 (ab53715, 1:1000, abcam), Smo (ab72130, 1:1000, abcam), Gli-1 (ab49314, 1:500, abcam), Gli-2 (ab167398, 1:1000, abcam), Gil-3 (ab69838, 1:1000, abcam), ZO-1 (Sc-8146, 1:500, Santa-Cruz Biotechnology), and claudin-3 (ab116165, 1:1000, abcam) overnight at 4 °C. GAPDH (2118, 1:10000, CST) was selected as the internal reference. After primary antibody incubation, the membranes were washed with Tris-buffered saline and incubated in biotinylated goat IgG secondary antibody (PAB160011, 1:10000, Bioswamp) for 2 h at room temperature. Immunoreactivity was visualized by colorimetric reaction using an enhanced chemiluminescence substrate buffer (Millipore, Massachusetts, USA). The membranes were scanned with the Gel Doz EZ imager (Bio-Rad, USA).
Statistical Analysis
Statistical significance of differences between the experimental data were evaluated via one-way analysis of variance (ANOVA), using the SPSS 19.0 software package. Differences were considered statistically significant at P < 0.05. All results are expressed as mean ± standard deviation (SD).
Results
Observation and Identification of Rat BMECs
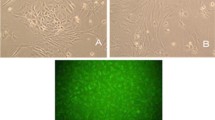
Rat BMECs were successfully isolated from brain tissue. Cells formed a confluent monolayer and exhibited a cobblestone pattern (Fig. 1a). Immunofluorescence showed that the cells were positively stained for VIII factor (Fig. 1b) and CD34 (Fig. 1c), suggesting that BMECs were present.

BMECs were isolated and identified by optical and fluorescence microscopy (100 ×). Cells formed a confluent monolayer and exhibited a cobblestone pattern. Immunofluorescence showed that the cells were positively stained for VIII factor and CD34, suggesting the presence of BMECs
tPA Decreased TEER of BMECs
The TEER of BMECs was detected by a Millicell-ERS device (Fig. 2). OGD and tPA administration significantly decreased the TEER of rat BMECs. However, Shh significantly inhibited the OGD- or tPA-induced decrease in TEER.

The TEER of BMECs was detected by a Millicell-ERS device. Data are shown as mean ± SD (n = 3); *P < 0.05 versus CON group, #P < 0.05 versus OGD group, △P < 0.05 versus tPA group, ▲P < 0.05 versus Shh group
tPA Inhibited the Proliferation of BMECs
To explore the viability of BMECs following tPA administration, a CCK-8 assay was performed to evaluate the cytotoxicity of tPA toward cells. We found that the proliferation of BMECs was significantly inhibited by OGD or tPA. However, tPA administration combined with Shh treatment significantly inhibited the tPA-induced decrease in BMEC proliferation. Conversely, after treatment with cyclopamine, cell proliferation decreased significantly (Fig. 3).

The proliferation of BMECs was detected by CCK-8 assay. Data are shown as mean ± SD (n = 3); *P < 0.05 versus CON group, #P < 0.05 versus OGD group, △P < 0.05 versus tPA group, ▲P < 0.05 versus Shh group
tPA Affected the Cell Cycle of BMECs
Because we observed that tPA inhibited cell proliferation in BMECs, we investigated the effect of tPA on the cell cycle of BMECs. As shown in Fig. 4, significant G0–G1 cell cycle arrest was detected in BMECs treated with OGD or tPA, accompanied by a reduction in cell number in the S-phase. Shh repaired the arrest and sped up the cell process. Unfortunately, the recovery function of Shh could be offset by tPA administration. These results suggest that Shh is involved in OGD- or tPA-induced BMEC cycle arrest in the G0-G1 phase.

tPA induced cell cycle arrest in G0–G1 phase. The cell cycle distribution of BMECs was measured by flow cytometry. Representative histograms and the cell number in each cell cycle are plotted as mean ± SD (n = 3). *P < 0.05 versus CON group, #P < 0.05 versus OGD group, △P < 0.05 versus tPA group, ▲P < 0.05 versus Shh group
tPA Blocked the Shh Signaling Pathway in BMECs and Upregulated the Expression of ZO-1 and Claudin-3
Western blot and qRT-PCR were performed to evaluate the effect of tPA on the expression levels of Shh signaling components in BMECs. As shown in Fig. 5, the protein and mRNA levels of Shh, Smo, Ptch1, Gli-1, Gli-2, and Gli-3 were significantly increased in BMECs after OGD or Shh administration, but significantly decreased with tPA treatment. The protein and mRNA levels of ZO-1 and claudin-3 in BMECs subjected to OGD were lower than those in normal BMECs, but increased significantly after Shh administration.

a Western blot and qRT-PCR analysis of Shh, Smo, Ptch1, Gli-1, Gli-2, Gli-3, ZO-1, and claudin-3 expression in BMECs. Bands were quantified using Quantity One 5.0. Claudin-3 (b) and ZO-1 (c) expressions were detected by immunofluorescence assay in BMECs (200 × amplification). Data are shown as mean ± SD (n = 3); *P < 0.05 versus CON group, #P < 0.05 versus OGD group, △P < 0.05 versus tPA group, ▲P < 0.05 versus Shh group
Discussion
The BBB is composed of endothelial cells, pericytes, and the end-feet of astrocytes. Among these, the intact tight junctions of brain capillary cells are critical for normal blood–brain barrier function, so that the endothelial cell barrier line is critical for preventing toxic substances from entering the brain [25]. BMECs can be detected and used as a cellular index of BBB damage [26]. This study described BBB disruption in BMECs subjected to OGD, as evidenced by the decreased TEER of cells, and tPA administration promoted OGD-induced BBB damage. Furthermore, we demonstrated that administration of high-concentration tPA acted on the Shh signaling pathway in BMECs, inhibiting it to enhance OGD-induced cell damage.
tPA, which affects neurite outgrowth, is expressed in many types of neural cells in the developing brain, including astrocytes and BMECs [27, 28]. To date, tPA is the only approved therapy for stroke, whereby intravenous injection of the thrombolytic agent promotes early reperfusion [29]. However, tPA potentiates neuronal death both in vitro and in vivo, even when administered intravenously. Cerebral hemorrhage after tPA infusion is relatively common, and in rare cases, tPA infusion may result in fragmentation of a cardiac thrombus, leading to an ischemic cerebral stroke [30]. Accordingly, despite clear overall benefits from its fibrinolytic action, there exists a legitimate need to explain how exogenous tPA can contribute to brain damage. In the present study, the mechanism of tPA-induced ischemia reperfusion damage was explored using an OGD model established in BMECs. Results revealed that administration of 300 µg/mL tPA significantly enhanced OGD and induced BMEC injury. The TEER and proliferation of BMECs were decreased and the cell cycle was blocked at the G0–G1 phase. Furthermore, the Shh signaling pathway was inhibited in rat BMECs by tPA treatment, which decreased the protein levels of Shh, Smo, Ptch1, Gli-1, Gli-2, and Gli-3.
Shh, as a mitogen and morphogen, is involved in vascular proliferation, differentiation, and maturation [31]. Recent researches have shown that the elements of the Shh signaling pathway are increased after ischemia in some tissues, including brain and myocardium [32, 33]. Moreover, blocked Shh signaling aggregates the level of brain edema in ischemic stroke [34]. However, whether Shh is involved in tPA-related intracerebral hemorrhage and BBB disruption during thrombolytic reperfusion remains unclear. In this study, we found that tPA-enhanced OGD-induced BMEC damage was eliminated by treatment with Shh, and the effects were reversed by cyclopamine. Cyclopamine is an antagonist of Smo that specifically inhibits the Shh pathway [35]. The Shh signaling pathway regulates apoptosis through the Smo protein in endothelial cells, and Shh administration accelerates wound healing by enhancing endothelial progenitor cell-mediated microvascular remodeling [18, 36]. The results of the present study indicated that tPA decreased cell proliferation and permeability transition by decreasing the TEER in BMECs. The underlying mechanism was mediated, at least partially, through the regulation of the expression of Shh signaling pathway components.
Previous studies have suggested that tPA could diffuse in the brain parenchyma and its deleterious effects such as BBB disruption would counteract its beneficial thrombolytic action [37, 38]. Moreover, the Shh signaling pathway is considered to regulate BBB permeability by downregulating tight junction proteins in astrocytes [39]. The present study further suggests that Shh signaling provides a barrier-promoting effect and endogenous anti-inflammatory balance with respect to central nervous system-directed immune attacks against endothelial cells and perivascular astrocytes, which compose the BBB. Here, we first demonstrated that Shh was involved in tPA-induced reduction of TEER in BMECs. Inhibition of the Shh signaling pathway in BMECs might contribute to the tPA-induced disruption of the endothelial barrier and promote cell injury.
In conclusion, this study demonstrates the direct involvement of tPA in the process of OGD-induced injury to BMECs. Blockage of the Shh signaling pathway might be one of the mechanisms by which tPA induces ischemia reperfusion injury. The precise mechanism by which tPA enhances ischemic injury requires further investigation.
References
Nag S (2002) The blood-brain barrier and cerebral angiogenesis: lessons from the cold-injury model. Trends Mol Med 8:38–44
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25
Liu N, Li AL, Zhou XP, Chen Q, Cao W (2015) P120 catenin attenuates lipopolysaccharide-induced blood-brain barrier dysfunction and inflammatory responses in human brain microvascular endothelial cells. Int J Clin Exp Pathol 8:4204–4212
Wosik K, Cayrol R, Dodelet-Devillers A, Berthelet F, Bernard M, Moumdjian R, Bouthillier A, Reudelhuber TL, Prat A (2007) Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J Neurosci 27:9032–9042
Ma J, Zhao S, Gao G, Chang H, Ma P, Jin B (2015) Probucol protects against asymmetric dimethylarginine-induced apoptosis in the cultured human brain microvascular endothelial cells. J Mol Neurosci 57:546–553
Xiao X, Zhang C, Ma X, Miao H, Wang J, Liu L, Chen S, Zeng R, Chen Y, Bihl JC (2015) Angiotensin-(1–7) counteracts angiotensin II-induced dysfunction in cerebral endothelial cells via modulating Nox2/ROS and PI3K/NO pathways. Exp Cell Res 336:58–65
Wang X, Tsuji K, Lee SR, Ning M, Furie KL, Buchan AM, Lo EH (2004) Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke 35:2726–2730
Yang F, Liu S, Yu C, Wang SJ, Paganini-Hill A, Fisher MJ (2012) PDE4 regulates tissue plasminogen activator expression of human brain microvascular endothelial cells. Thromb Res 129:750–753
Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA (2003) Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest 112:1533–1540
Alberts MJ (1998) tPA in acute ischemic stroke: United States experience and issues for the future. Neurology 51:S53–S55
Leigh R, Jen SS, Hillis AE, Krakauer JW, Barker PB (2014) Pretreatment blood-brain barrier damage and post-treatment intracranial hemorrhage in patients receiving intravenous tissue-type plasminogen activator. Stroke 45:2030–2035
Kim JA, Tran ND, Wang SJ, Fisher MJ (2003) Astrocyte regulation of human brain capillary endothelial fibrinolysis. Thromb Res 112:159–165
Yamashita T, Kamiya T, Deguchi K, Inaba T, Zhang H, Shang J, Miyazaki K, Ohtsuka A, Katayama Y, Abe K (2009) Dissociation and protection of the neurovascular unit after thrombolysis and reperfusion in ischemic rat brain. J Cereb Blood Flow Metab 29:715–725
Abu Fanne R, Nassar T, Yarovoi S, Rayan A, Lamensdorf I, Karakoveski M, Vadim P, Jammal M, Cines DB, Higazi AA (2010) Blood-brain barrier permeability and tPA-mediated neurotoxicity. Neuropharmacology 58:972–980
Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA (2011) Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by Increasing superoxide production. Ann Neurol 70:583–590
He QW, Xia YP, Chen SC, Wang Y, Huang M, Huang Y, Li JY, Li YN, Gao Y, Mao L et al (2013) Astrocyte-derived sonic hedgehog contributes to angiogenesis in brain microvascular endothelial cells via RhoA/ROCK pathway after oxygen-glucose deprivation. Mol Neurobiol 47:976–987
Osterlund T, Kogerman P (2006) Hedgehog signalling: how to get from Smo to Ci and Gli. Trends Cell Biol 16:176–180
Zhu SL, Luo MQ, Peng WX, Li QX, Feng ZY, Li ZX, Wang MX, Feng XX, Liu F, Huang JL (2015) Sonic hedgehog signalling pathway regulates apoptosis through Smo protein in human umbilical vein endothelial cells. Rheumatology 54:1093–1102
Kanda S, Mochizuki Y, Suematsu T, Miyata Y, Nomata K, Kanetake H (2003) Sonic hedgehog induces capillary morphogenesis by endothelial cells through phosphoinositide 3-kinase. J Biol Chem 278:8244–8249
Nagase T, Nagase M, Yoshimura K, Fujita T, Koshima I (2005) Angiogenesis within the developing mouse neural tube is dependent on sonic hedgehog signaling: possible roles of motor neurons. Genes Cells 10:595–604
Han BW, Layman H, Rode NA, Conway A, Schaffer DV, Boudreau NJ, Jackson WM, Healy KE (2015) Multivalent conjugates of Sonic hedgehog accelerate diabetic wound healing. Tissue Eng Part A 21:2366–2378
Diglio CA, Grammas P, Giacomelli F, Wiener J (1982) Primary culture of rat cerebral microvascular endothelial cells. Isolation, growth, and characterization. Lab Invest 46:554–563
Xia YP, He QW, Li YN, Chen SC, Huang M, Wang Y, Gao Y, Huang Y, Wang MD, Mao L et al (2013) Recombinant human sonic hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoietin-1 in stroke damage. PLoS One 8:e68891
Farcal L, Torres Andon F, Di Cristo L, Rotoli BM, Bussolati O, Bergamaschi E, Mech A, Hartmann NB, Rasmussen K, Riego-Sintes J et al (2015) Comprehensive in vitro toxicity testing of a panel of representative oxide nanomaterials: first steps towards an intelligent testing strategy. PLoS ONE 10:e0127174
Wolburg H, Lippoldt A (2002) Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol 38:323–337
Huang SH, Wang L, Chi F, Wu CH, Cao H, Zhang A, Jong A (2013) Circulating brain microvascular endothelial cells (cBMECs) as potential biomarkers of the blood-brain barrier disorders caused by microbial and non-microbial factors. PLoS ONE 8:e62164
Haile WB, Wu J, Echeverry R, Wu F, An J, Yepes M (2012) Tissue-type plasminogen activator has a neuroprotective effect in the ischemic brain mediated by neuronal TNF-alpha. J Cereb Blood Flow Metab 32:57–69
Schreiber SS, Tan Z, Sun N, Wang L, Zlokovic BV (1998) Immunohistochemical localization of tissue plasminogen activator in vascular endothelium of stroke-prone regions of the rat brain. Neurosurgery 43:909–913
Qureshi N (1996) Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 334:1406
Chang GY (2001) An ischemic stroke during intravenous recombinant tissue plasminogen activator infusion for evolving myocardial infarction. Eur J Neurol 8:267–268
Weidemann A, Krohne TU, Aguilar E, Kurihara T, Takeda N, Dorrell MI, Simon MC, Haase VH, Friedlander M, Johnson RS (2010) Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia 58:1177–1185
Sims JR, Lee SW, Topalkara K, Qiu J, Xu J, Zhou Z, Moskowitz MA (2009) Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke 40:3618–3626
Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, Shapiro R, Taylor FR, Baker DP, Asahara T et al (2001) The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med 7:706–711
Ji H, Miao J, Zhang X, Du Y, Liu H, Li S, Li L (2012) Inhibition of sonic hedgehog signaling aggravates brain damage associated with the down-regulation of Gli1, Ptch1 and SOD1 expression in acute ischemic stroke. Neurosci Lett 506:1–6
Incardona JP, Gaffield W, Kapur RP, Roelink H (1998) The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 125:3553–3562
Asai J, Takenaka H, Kusano KF, Ii M, Luedemann C, Curry C, Eaton E, Iwakura A, Tsutsumi Y, Hamada H et al (2006) Topical sonic hedgehog gene therapy accelerates wound healing in diabetes by enhancing endothelial progenitor cell-mediated microvascular remodeling. Circulation 113:2413–2424
Benchenane K, Berezowski V, Ali C, Fernandez-Monreal M, Lopez-Atalaya JP, Brillault J, Chuquet J, Nouvelot A, MacKenzie ET, Bu G et al (2005) Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein receptor-related protein-mediated transcytosis. Circulation 111:2241–2249
Benchenane K, Berezowski V, Fernandez-Monreal M, Brillault J, Valable S, Dehouck MP, Cecchelli R, Vivien D, Touzani O, Ali C (2005) Oxygen glucose deprivation switches the transport of tPA across the blood-brain barrier from an LRP-dependent to an increased LRP-independent process. Stroke 36:1065–1070
Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, Kawanokuchi J, Mizuno T, Takeuchi H, Suzumura A (2014) Interleukin-1beta induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 9:e110024
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81371272).
Author information
Authors and Affiliations
Contributions
Mingchang Li designed the experiment and approved the final version of the manuscript. Pian Gong wrote the manuscript. Pian Gong, Changlin Zhou, and Qi Tian performed the experiments. Zhou Xu and Changlin Zou analyzed the data.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gong, P., Li, M., Zou, C. et al. Tissue Plasminogen Activator Causes Brain Microvascular Endothelial Cell Injury After Oxygen Glucose Deprivation by Inhibiting Sonic Hedgehog Signaling. Neurochem Res 44, 441–449 (2019). https://doi.org/10.1007/s11064-018-2697-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2697-2