
Abstract
Bisperoxovanadium (pyridine-2-carboxyl) [bpV(pic)] is a commercially available PTEN inhibitor. Previous studies from us and others have shown that bpV(pic) confers neuroprotection in cerebral ischemia injury. We set up to determine whether ERK 1/2 activation plays a role in bpV(pic)-induced neuroprotective effect in cerebral ischemia injury. We found that the phosphorylation levels of Akt (p-AKT) and ERK1/2 (p-ERK 1/2) were down-regulated after cerebral ischemia–reperfusion injury. The injection of bpV(pic) after injury not only increased the level of p-AKT but also the level of p-ERK 1/2. While the inhibition of PTEN mediated the up-regulatation of p-AKT and p-ERK 1/2 by bpV(pic). Interestingly, the ERK 1/2 activation induced by bpV(pic) was also independent of the inhibition of PTEN. Our results indicate that bpV(pic) protects against OGD-induced neuronal death and promotes the functional recovery of stroke animals through PTEN inhibition and ERK 1/2 activation, respectively. This study suggests that the effect of bpV(pic) on ERK 1/2 signaling should be considered while using bpV(pic) as a PTEN inhibitor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is induced by arterial embolism, microangiopathy or macroaniopathy, results in oxygen and glucose deprivation in brain, leading to brain damage/injury and neurologic deficit [1]. Oxidative stress, inflammation, and excitotoxicity have been considered as major contributors to ischemic neuronal injury [2]. Neuronal injury after cerebral ischemia also produces a complex train of signaling cascades that also lead to cell injury/death [3]. The complex environment and high damage/death ratio after ischemic stroke, neuroprotection becoming a related question. Thrombolytic therapy and neuroprotective drug therapy become two major potential terapeutic strategies after ischemic stroke.
PTEN (phosphatase and tensin homolog deleted on chromosome 10) a tumor suppressor, associate with several neoplastic diseases [4]. PTEN plays an important role in mediating intracellular signaling of cell proliferation and survival [5, 6]. PTEN is a phosphatase has both lipid and protein phosphatase function [7]. But unlike sevaral cellular proteins activated by phosphorylation, PTEN inactivated its phosphorylation through its specific kinases phosphorylate serine and threonine residues in C-terminal region [8]. PTEN depends on its lipid phosphatase activite to inhibit the PI3K/AKT pathway, the PI3K/AKT pathway playing a key role in promoting the cell survival and growth [6, 9]. Substantial evidenced indicate that inhibition of PTEN induces neuroprotection.
Bisperoxovanadium (pyridine-2-carboxyl) [bpV(pic)] is a commercially available PTEN inhibitor [10]. Previous studies from us and others have shown that bpV(pic) confers neuroprotection in cerebral ischemia injury [11]. Bisperoxovanadium (pyridine-2-squaramide) [bpV(pis)], also an inhibitor of PTEN, which was designed by our laboratory. In our study, we found that the bpV(pis) not only can activate AKT activity through PTEN inhibition, but also can enhance ERK 1/2 activation independent of PTEN. The bpV(pis)-induced neuroprotect through both PTEN inhibition and ERK 1/2 activation [12]. we set up to determine whether bpV(pic) exerted its neuroprotective effect in cerebral ischemia injury through both PTEN inhibition and ERK 1/2 activation.
Mitogen-activated protein kinase (MAPK) is a family of serine/threonine protein kinases that are widely expressed in all eukaryotic cells [13]. Extracellular signal-regulated kinase (ERK), one of the extensively studied MAPK family members, is classically associated with receptor protein tyrosine kinase-induced signaling cascades [14]. It consists of two isoenzymes, so namely ERK 1/2, also called p44/42 MAPK. ERK 1/2 is known to mediate neuronal plasticity, migration and cell survival [15, 16]. Up-regulation of ERK 1/2 signalling activity reveals a positive neuroprotective effect in ischaemia stroke models both in vivo and in vitro [17,18,19].
In this study, we aimed to investigate the relationship of bpV(pic) and ERK 1/2, and confirm the bpV(pic)-induced neuroprotect whether connect with ERK 1/2. Our results show that bpV(pic) confers neuroprotection after cerebral ischemia–reperfusion injury through inhibition of PTEN lipid phosphatase activity as well as ERK 1/2 activation.
Materials and Methods
Animals
Adult male Sprague-Dawley (SD) rats were housed with three rats per cage on a 12 h light/dark cycle in a temperature-controlled room (23–25 °C) with free access to water and food. Animals were allowed at least 3 days to acclimatize before experimentation. We used total 248 male rats in our in vivo expriments, 56 male rats were discarded during the operational procedures. 12 adult pregnant female rats and 45 embryos were used in our expriments for cortical neuronal cultures. All animal use and experimental protocols were approved and carried out in compliance with the IACUC guidelines and the Animal Care and Ethics Committee of Wuhan University School of Medicine. Samples to the experimental groups and to collect and process data was assigned by randomization. The experiments were performed by investigators blinded to the groups for which each animal was assigned.
Focal Cerebral Ischemia and Infarct Measurement
Transient focal cerebral ischaemia was induced using the suture occlusion technique [20, 21]. The whole process do as our lab described [22]. Male Sprague-Dawley rats weighing 250–300 g were anaesthetized with 4% isoflurane in 70% N2O and 30% O2 by using a mask. The rectal temperature was maintained at 37.0 ± 0.5 °C using a homoeothermic blanket. A midline incision was made in the neck, the right external carotid artery (ECA) was carefully exposed and dissected and a 3–0 monofilament nylon suture was inserted from the ECA into the right internal carotid artery to occlude the origin of the rightmiddle cerebral artery (approximately 22 mm). After 90 min of occlusion, the suture was removed to allow reperfusion, the ECA was ligated and the wound was closed. Sham-operated rats underwent identical surgery and/or i.c.v. injections except that the suture was inserted and withdrawn immediately. At 24 h after middle cerebral artery occlusion (MCAO), rats (n = 24) were reperfused with ice-cold 0.9% saline after anaesthetized with 4% isoflurane in 70% N2O and 30% O2, and the brains were rapidly removed for western blot analysis and 2,3,5-triphenyltetrazolium chloride (TTC) staining.
The brain was placed in a cooled matrix and 2 mm coronal sections were cut. Individual sections were placed in 10 cm petri dishes and incubated for 30 min in a solution of 2% TTC in phosphate buffered saline at 37 °C. The slices were fixed in 4% paraformaldehyde at 4 °C for 24 h. All image collection, processing and analysis were performed in a blind manner and under controlled environmental lighting. The scanned images were analyzed using image analysis software (Image-Pro Plus Version 6.0, USA). The infarct volume was calculated to correct for edema. The normal volume of contralateral hemisphere and the normal volume of ipsilateral hemisphere were measured, and the infarct percentage was calculated as % contralateral structure to avoid mismeasurement secondary to edema [23, 24].
Intraventricular Injection (i.c.v) Administration
Rats were anaesthetized with a mixture of 4% isoflurane in 30% O2 and 70% N2O in a sealed perspective box. When the rats were deeply anaesthetized, we would use the ear bars and upper incisor bar to secure the rat’s head in a stereotaxic frame, and the rats were continuously under anaesthesia with 4% isoflurane using a mask. Next, making a small sagittal incision, and bregma was located as the anatomical reference point. The cerebral ventricle (from the bregma: lateral, 1.5 mm; anteroposterior, − 0.8 mm; depth, 3.5 mm) was performed using a 23 gauge needle attached via polyethylene tubing to a Hamilton microsyringe, and drug infusion at a rate of 1.0 µL/min. Proper needle placement was verified via withdrawing a few microlitres of clear cerebrospinal fluid into the Hamilton microsyringe.
Cortical Neuron Culture and OGD Insult
The cortical neuronal cultures were prepared from gestation 17 day’s female SD rats [25]. The pregnant rats were killed by cervical dislocation after anaesthetized with 4% isoflurane in 70% N2O and 30% O2. The embryos were sprayed with 70% ethanol that were removed after the rats. The embryos were rapidly excised, the embryo skull was removed, and the brain tissue was compeletely removed. The cerebral cortex tissue was separated under a microscope and placed in a fresh frozen liquid and placed in ice-cold plating medium (Neurobasal medium, 0.5% FBS, 2% B-27 supplement, 25 mM glutamic acid and 0.5 mM l-glutamax, specifically for culturing primary neurons). The cortical neurons were plated on Petri dishes coated with poly-d-lysine (PDL) and suspended in plating medium. Half of the plating medium was removed and replaced with maintenance medium (Neurobasal medium, 0.5 mM l-glutamine and 2% B-27 supplement) in the same manner every 3 days. The cultured neurons were used for the experiments after 12 days [25].
Oxygen–glucose deprivation (OGD) challenge, cells were transferred to glucose-free extracellular solution (116 mM NaCl, 0.8 mM MgSO4, 5.4 mM KCl, 1.0 mM NaH2PO4, 26 mM NaHCO3 and 1.8 mM CaCl2) and deoxygenated environment, placed in a humidified chamber (Plas-Labs, Lansing, USA), and maintained at 37 °C in H2/5% CO2 85% N2/10% for 60 min. Next, replacing with fresh maintenance medium containing an appropriate concentration of reagents for 24 h during the recovery period in a 5% CO2/95% O2 incubator. The control cultures were firstly transferred to another extracellular solution (5.4 mM KCl, 116 mM NaCl, 0.8 mM MgSO4, 1.8 mM CaCl2, 1.0 mM NaH2PO4, 26 mM NaHCO3 and 33 mM glucose), and placed in the humidified chamber, which was maintained at 37 °C in 95% O2/5% CO2 for 60 min [26]. Last, replacing with fresh maintenance medium for the whole period at 37 °C in a 95% O2/5% CO2 incubator.
Western Blotting Analysis
Western blotting was performed as previously described [22]. Briefly, the polyvinylidene difluoride (PVDF) membrane by Millipore (USA) was used incubate with a first antibody against ERK 1/2 (Rabbit, 1:2000), phospho-ERK 1/2 (Thr202/Tyr204) (Rabbit, 1:2000), PTEN (Rabbit, 1:1000), AKT (Mouse, 1:1000), phospho-AKT (Ser473) (Rabbit, 1:2000), Actin (Rabbit, 1:2000) from Cell Signaling Technology (MA, USA). First antibodies were labelled with secondary antibody, protein bands were imaged using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA). The EC3 Imaging System (UVP, LLC, Uplant, USA) was used to obtained blot images directly from the PVDF membrane. The data of western blot were quantified using Image J software.
Fluoro-Jade C (FJC) Staining
Rats were treated with an over dose of isoflurane, then intracardiac perfusion with 0.9% saline, next put in 4% paraformaldehyde (PFA) at 4 °C for 24 h, and followed by transfered into 30% sucrose solution in 100 mol/mL phosphate buffer at 4 °C for 72 h. Then the brains tissue were kept in 4% paraformaldehyde solution at 4 °C overnight. Brains tissues were cut into 16 µm coronal sections by a Leica VT1000S vibratome (Leica Micro-systems AG, Nussloch, Germany). FJC labelling was performed using by the standard protocol [27]. Brain sections were first immersed in 1% sodium hydroxide in 80% ethanol for 5 min, then rinsing in 70% ethanol for 2 min, then 2 min in distilled water, and followed by incubated in 0.06% potassium permanganate solution for 10 min. Following a 2 min distilled water rinse, the brain sections were transferred into 0.0001% solution of FJC (Sigma-Aldrich, USA) 10 min which was dissolved in 0.1% acetic acid. Brain sections were rinsed with water for 1 min three times, then air dried on a warmer at 50 °C at least 5 min, last immersed in xylene at least 1 min. The brain sections were mounted with DPX media (Sigma Aldrich, USA). The brain sections were photographed by a blinded investigator using an Olympus fluorescent microscope (IX51, Olympus, Japan). Series of microphotographs were taken from three region of the ipsilateral cerebral cortex, with a ×20 objective and FJC-positive cells were counted by Image J software (ImageJ, USA). The data were expressed as cells/mm2.
Immunofluorescence Analysis
The immunostaining brain sections was prepared as FJC staining, the immunofluorescence staining steps based on the description of the prior execution [28]. The brain sections were treated with primary antibody rabbit anti- phospho-AKT (Ser473) (1:250), phospho-ERK 1/2 (Thr202/Tyr204) (1:250) from Cell Signaling Technology, mouse anti- NeuN (neuronal-specific nuclear protein) from Chemicon. The secondary antibody goat anti- Rabbit 594, goat anti- Mouse 488 from Molecular Probes (Eugene, USA). The hochest probe from Life Technologies. The sections were photographed by a blinded investigator using an Olympus fluorescent microscope (IX51, Olympus, Japan). Analysed by Image J software (Image J, USA).
Cell Culture, Transfection and Treatment
SH-SY5Y cells and U251 cells were purchased from the Chinese Academy of Sciences Cell Bank. SH-SY5Y cells and U251 cells were sowed in a 6-well plate (8 × 105 cells per well) in DMEM supplemented with 10% heat-inactivated FBS, penicillin G (100 U/mL), streptomycin (100 mg/mL) and l-glutamine (2.0 mM) and incubated at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air.
On the day of treatment, when the confluence of SH-SY5Y and U251 cells were reached 80–90%, cells were washed with standard ECS for 60 min and then treated with bpV (pic) for 30 min. The cells were then collected for western blot analysis.
When the confluence of SH-SY5Y cells reached 60–70% on the treatment day, cells were transfected with human PTEN siRNA (siRNApten) and non-targeting control siRNA (NsiRNA) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 8 h. The sequence of human PTEN siRNA (siRNApten) was 5′-CTGCTAGCCTCTGGATTTGA-3′ and non-targeting control siRNA (NsiRNA) was 5′-CTTCTGGCATCCGGTTTAGA-3′, as previously described [29]. The medium was then replaced with normal growth medium for 24 h. On the following day, the cells were treated with standard ECS for 60 min and then treated with bpV(pic) for 30 min. The cells were then collected for western blot analysis.
We established the PTEN WT and phosphatase domain mutant: PTEN G129E, in which Glu replaces Gly129, which lacks lipid phosphatase activity (TaiHe Biotechnology Co, LTD) [30].When the confluence of U251 cells reached 60–70% on the treatment day, cells were transfected with human PTEN plasmid pCDNA3.1(+)-PTEN-WT (WT PTEN), pCDNA3.1(+)-PTEN-G129E (PTEN G129E) and pCDNA3.1(+) [Empty vector (EV)] for 8 h. The medium was then replaced with normal growth medium for 24 h. On the following day, the cells were treated with standard ECS for 60 min and then treated with bpV(pic) for 30 min. The cells were then collected for western blot analysis.
Analysis of LDH Release and Cell Viability
Lactate dehydrogenase (LDH) release was analysed by using a colorimetric CytoTox 96 Cytotoxicity kit (Promega, USA). Cell viability in the neuronal cultures was evaluated by the ability of take up thiazolyl blue tetrazolium bromide (MTT) (PowerWave X, Bio-Tek, Winooski, State, USA). Two methods are following by the manufacturer’s instructions.
Neurological Severity Scores
The rats were subjected to a modified neurological severity score test as reported previously. These tests are a battery of reflex, sensory, motor and balance tests, which are similar to the contralateral neglect tests in humans. Neurological function was graded on a scale of 0–18 (normal score, 0; maximal deficit score, 18) [31].
Beam Walk Test
The beam walk test measures the animals’ complex neuromotor function. The animal was timed as it walked a (100 × 2 cm) beam. A box for the animal to feel safe was placed at one end of the beam. A loud noise was created to stimulate the animal to walk towards and into the box. Scoring was based upon the time it took the rat to go into the box. The higher the score, the more severe the neurological deficit [32].
Adhesive-Removal Test
A modified sticky-tape test was performed to evaluate forelimb function. A sleeve was created using a 3.0 × 1.0 cm piece of yellow paper tape and was subsequently wrapped around the forepaw so that the tape attached to itself and allowed the digits to protrude slightly from the sleeve. The typical response is for the rat to vigorously attempt to remove the sleeve by either pulling at the tape with its mouth or brushing the tape with its contralateral paw. The rat was placed in its cage and observed for 30 s. Two timers were started: the first ran without interruption and the second was turned on only while the animal attempted to remove the tape sleeve. The ratio of the left (affected)/right (unaffected) forelimb performance was recorded. The contralateral and ipsilateral limbs were tested separately. The test was repeated three times per test day, and the best two scores of the day were averaged. The lower the ratio, the more severe the neurological deficit [33].
Statistics
In this study, on experimental design and analysis, the data and statistical analysis comply with the recommendations. All datas are expressed as mean ± SE. Student’s t-test and variance analysis was used in differences among groups. P < 0.05 was considered statistically significant.
Materials
Bisperoxovanadium (pyridine-2-carboxyl) [bpV(pic)] and AKT Inhibitor IV (ChemBridge 5233705) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ERK 1/2 inhibitor U0126 was purchased from Delleckchem (Houston, TX).
Results
The Level of Phospho-AKT and Phospho-ERK 1/2 are Down-Regulated After Cerebral Ischemia–Reperfusion Injury
Ischemic stroke results from thrombotic blockage, which in brain causes the oxygen and glucose deprivation, leading to brain damage and neurological deficit. We generated an experimental stroke model induced by MCAO [34, 35] for 1.5 h followed by various periods of reperfusion to simulate ischemia stroke. AKT activation was quantified by measuring AKT phosphorylation (p-AKT) on Ser473 [25] and the activation of ERK 1/2 was measured by quantifying ERK 1/2 phosphorylation (p-ERK 1/2) on Thr202/Tyr204 [36]. Immunofluorescence staining of p-AKT and p-ERK 1/2 in the peri-infarct area of cortex after ischemia–reperfusion (I/R), results show that a decrease p-AKT and p-ERK 1/2 signals were obtained after I/R compared with sham (Fig. 1a, b). Neurons are more vulnerable to stroke insult than other cells in the brain [3]. Double-immunofluorescence labelling detected a down-regulate of p-AKT and p-ERK 1/2 signal in neurons, and did not show any significant difference at I/R 24 h or 72 h (Fig. 1a, b).

After ischemic stroke p-AKT and P-ERK 1/2 levels are decreased. a and b Double-immunofluorescence staining of p-AKT or p-ERK 1/2 with NeuN in the peri-infarct area of cortex 24 h or 72 h after I/R compared with the ipsilateral sham, NeuN performes green, P-AKT and p-ERK 1/2 is shown in red and hochest is shown in blue. Scale bar, 20 µm. c and d Western blots showing a decreasing expression in p-AKT (c) and p-ERK 1/2 (d) at the indicated time points after I/R at rats (left). Right: quantification analysis of normalized p-AKT and p-ERK 1/2 levels (n = 6 per time points, *P < 0.05 vs. the sham). e and f Western blots of p-AKT (e) and p-ERK 1/2 (f) in OGD-treated cultured primary neurons (left) and quantification analysis of p-AKT and p-ERK 1/2 levels (right), showing a similar down-regulation with rats sample (n = 6, *P < 0.05 vs. the control). The data are expressed as mean ± SE. Statistical analysis was implemented by student’s t-test and variance analysis. (Color figure online)
Western boltting analysis of ipsilateral brain homogenates at the indicated time point after I/R revealed a down-regulate p-AKT and p-ERK 1/2 levels as time goes on (Fig. 1c, d). Furthermore, we cultured primary neurons from the rat cortex, and used a OGD insult model. Western boltting analysis of p-AKT and p-ERK 1/2 in cultured cortical neurons after OGD insult, results showed a similar p-AKT and p-ERK 1/2 levels pattern with the animals (Fig. 1g, h). These data shows that the neuronal p-AKT and p-ERK 1/2 are down-regulated after ischemic stroke.
BpV(pic) Induces Not Only the Level of P-AKT Up-Regulation But Also P-ERK 1/2 and Protects Against Ischemia-Induced Brain Damage and Neuronal Death
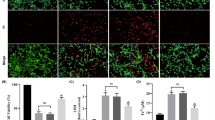
BpV(pic) can activate AKT through inhibition of PTEN. Previous studies have shown that bpV(pic) confers neuroprotection in cerebral ischemia injury [11]. In the Fig. 1, we have shown that the level of p-AKT decreased after I/R, therefore, we used the PTEN inhibitor bpV(pic) to treat the animals. Rats were given bpV(pic) at a dose of 100 µM (5 µL) 1.0 h after MCAO by intracerebroventricular (i.c.v) injection (Fig. 2a). Rats in control group received i.c.v injection of vehicle (0.9% saline). Western blotting analysis of p-AKT after I/R 24 h showed that the level of p-AKT up-regulated after bpV(pic) treatment (Fig. 2b). Interestingly, the level of p-ERK 1/2 also up-regulated (Fig. 2c). To further confilm the neuroprotective of bpV(pic), TTC straining analysis was performed in the mouse brain tissues, results show that the bpV(pic) was against the ischemia-induced brain damage (Fig. 2d). Further, we used the Fluoro-Jade C (FJC) straining to determine the neurons status. Results show that the number of FJC-positive degenerating neurons in the cortex of peri-infarct area were significantly decreased after bpV(pic) treatment (Fig. 2e, f). These results further comfirm the neuroprotective effect of bpV(pic) in the neurons. Together, we confilm the neuroprotective effect of bpV(pic) in cerebral ischemia injury, and we further reveal that the bpV(pic) induces not only the level of p-AKT up-regulation but also p-ERK 1/2.

BpV(pic) up-regulated the p-AKT and p-ERK 1/2 level in rats and protects against ischemia–reperfusion injury. a A time points diagram shows rat ischemia–reperfusion injury and IV (AKT inhibitor), U0126 (ERK 1/2 inhibitor), bpV(pic) treatment procedure. b and c Western blots showing an increased expression in p-AKT (b) and p-ERK 1/2 (c) after i.c.v inject bpV(pic) (100 µM, 5 µL) 24 h after ischemia–reperfusion injury comparing with I/R + vehicle group (left). Right: quantification analysis of p-AKT and p-ERK 1/2 levels (n = 6, *P < 0.05 vs. the sham, #P < 0.05 vs. the I/R + vehicle). d Sample images of TTC staining brain sections show that bpV(pic) decreases the infarct volume in brain 24 h after ischemia onset comparing with I/R + vehicle group (left), and quantification analysis of the infarct volume on right (n = 6, *P < 0.05 vs. the I/R + vehicle). e Fluoro-Jade C (FJC) staining brain sections collected at 24 h after ischemia onset. BpV(pic) was injected at 1 h after MCAO. Scale bar, 100 µm. f Quantitative analysis of Fluoro-Jade C positive degeneration neurons shows an up-regulation numbers after I/R compare with sham, and presents a positive down-regulation while is injected bpV(pic) (n = 6, *P < 0.05 vs. the sham, #P < 0.05 vs. the I/R + vehicle). The datas are expressed as mean ± SE. Statistical analysis was implemented by student’s t-test and variance analysis
BpV(pic)-Mediated AKT Activation Get Through by Inhibition of PTEN Lipid Phosphatase Activity
To further investigate the effect of bpV(pic). We used the human neuroblastoma SH-SY5Y cells, which were treated with different concentrations of bpV(pic) (10, 50, 100, 200 and 500 nM). Results show that bpV(pic) increased the level of p-AKT in SH-SY5Y cells (Fig. 3a). BpV(pic) mostly performs as a PTEN inhibitor to activate AKT activity, therefore, we used a PTEN knockdown approach in SH-SY5Y cells (Fig. 3b) to further confilm the relationship of PTEN with AKT activation. Results show that the level of p-AKT increased after PTEN consumption (Fig. 3c). Moreover, the bpV(pic) (200 nM) was traeated in SH-SY5Y cells after transfection of siRNApten, bpV(pic) did not cause a significant increase of p-AKT level in SH-SY5Y cells after transfection of siRNApten (Fig. 3c). To further investigate whether the bpV(pic)-induced p-AKT up-regulation depends only on PTEN. We used a PTEN-deficient cell line, human glioblasoma U251 cells (Fig. 3d). The level of p-AKT did not up-regulation after bpV(pic) treatment (Fig. 3e). Together, these results indicate that the bpV(pic) enhances AKT activation by inhibition of PTEN.

BpV(pic) up regulates p-AKT level through inhibiting PTEN lipid phosphatase activity. a Western blots analysis of p-AKT levels in SH-SY5Y cells treated with bpV(pic) (10–500 nM) in the right. Left: quantification analysis of p-AKT levels treated with bpV(pic) shown an increased expression of normalized p-AKT compare with vehicle group (n = 6 independent cultures, *P < 0.05 vs. the vehicle). b In SH-SY5Y cells PTEN expression decreased after transfected with siRNApten (n = 6 independent cultures, *P < 0.05 vs. the NsiRNA). c The p-AKT levels in SH-SY5Y cells transfect with NsiRNA or siRNApten then treated with bpV(pic) (200 nM) (n = 6 independent cultures, *P < 0.05 vs. the NsiRNA + vehicle). d Western blots analysis of PTEN expression in SH-SY5Y and U251 cells. e The level of p-AKT in U251 cells treatment with bpV(pic) (50–1000 nM) did not change (n = 6 independent cultures). f Western blots of PTEN expression in U251 cells transfected with PTEN-cDNA WT and G129E. g The levels of p-AKT in U251 cells transfected with PTEN-cDNA WT and G129E, then treatment with bpV(pic) (200 nM) (n = 6 independent cultures, *P < 0.05 vs. the EV, #P < 0.05 vs. the WT). The data are expressed as mean ± SE. The datas are expressed as mean ± SE. Statistical analysis was implemented by student’s t-test and variance analysis
We know from a previous study that PTEN can negatively regulate AKT activation through PI3K signal pathway because of it’s lipid phosphatase activity. To further reveal the mechanism of bpV(pic) inhibition function in PTEN, we established the PTEN WT and phosphatase domain mutant: PTEN G129E, in which Glu replaces Gly129, which lacks lipid phosphatase activity [30]. Because U251 cell line has no PTEN expression, we first confirmed that PTEN-cDNA was expressed in U251 cells (Fig. 3f). Next, we tested the level of p-AKT after transfected with EV, WT or G129E. The resultsshow that the level of p-AKT was down-regulated in WT group compared with EV, and did not show any significant changes after transfection of PTEN G129E (Fig. 3g). Furthermore, we treated bpV(pic) (200 nM) in U251 cells after transfection of PTEN plasmid. Results show that the level of p-AKT did not increase in the EV + bpV(pic) and G129E + bpV(pic) group compared with EV, however, increased in the WT + bpV(pic) group compared with WT group (Fig. 3g). These results show that bpV(pic) increases AKT activation through inhibiting PTEN lipid phosphatase activity.
BpV(pic) Increases ERK 1/2 Activition Through Both Inhibition of PTEN Lipid Phosphatase and Independent of PTEN
From Fig. 2c, we found that the level of p-ERK 1/2 increased after bpV(pic) treatment in rats. To further inverstigate the mechanism of bpV(pic) regulates ERK 1/2 activation, we firstly increased the level of p-ERK 1/2 in SH-SY5Y cells after treatment different concentrations of bpV(pic) (Fig. 4a). Furthermore, we also measured the effect of PTEN knockdown on ERK 1/2 phosphorylation in SH-SY5Y cells. After PTEN suppression, we found the level of p-ERK 1/2 was increased (Fig. 4b). These results show that the ERK 1/2 activation can be mediate by PTEN. Moreover, we tested the level of p-ERK 1/2 in U251 cells after transfection of PTEN plasmid, results show that the level of p-ERK 1/2 decreased in WT group and did not show any significant difference in G129E group compared with EV (Fig. 4c). These results indicate that the ERK 1/2 can be activared by inhibition PTEN lipid phosphase activity. We further tested the effect of bpV(pic) (50, 100, 200, 500 and 1000 nM) in U251 cells, interestingly, the level of p-ERK 1/2 also up-regulated (Fig. 4d). These data indicate that bpV(pic) activates ERK 1/2 can independently of PTEN. Together, BpV(pic) increases ERK1/2 activition through both inhibition of PTEN lipid phosphatase and independent of PTEN.

BpV(pic) not only through inhibit PTEN lipid phosphatase activity but also independently of PTEN to up-regulation p-ERK 1/2 level. a Western blots analysis of p-ERK 1/2 levels in SH-SY5Y cells treated with bpV(pic) (10–500 nM) on right. Left: quantification analysis of p-ERK 1/2 levels treated with bpV(pic) shows an increased expression of normalized p-ERK 1/2 compare with vehicle group (n = 6 independent cultures, *P < 0.05 vs. the vehicle). b The p-ERK 1/2 levels in SH-SY5Y cells transfected with NsiRNA or siRNApten then treated with bpV(pic) (200 nM) (n = 6 independent cultures, *P < 0.05 vs. the NsiRNA + vehicle). c The levels of p-ERK 1/2 in U251 cells transfected with PTEN-cDNA WT and G129E (n = 6 independent cultures, *P < 0.05 vs. the EV). d The levels of p-ERK 1/2 increased in PTEN-deficient cell U251 cells when treated with bpV(pic) (50–1000 nM). Quantification analysis of p-ERK 1/2 levels on the right (n = 6 independent cultures, *P < 0.05 vs. the vehicle). e the ERK 1/2 levels have no change by bpV(pic) in vitro (n = 6 independent cultures). The data is expressed as mean ± SE. Statistical analysis was implemented by student’s t-test and variance analysis
We set up to determine whether bpV(pic) directly activates of ERK 1/2, so we used the recombinant ERK 1/2 to test the activation effect of bpV(pic) in it [12]. Our results did not show alterations in the ERK 1/2 activity (Fig. 4e), from which we know that bpV(pic) can not directly increase ERK 1/2 activity.
BpV(pic) Protects Against OGD-Induced Neuronal Death Through Both AKT and ERK 1/2 Activation
From Fig. 1e, f, we found the level of p-AKT and p-ERK 1/2 decreased in the cultured cortical neurons after OGD insult. We up-regulated the level of p-AKT and p-ERK 1/2 in cultured cortical neurons after 24 h of OGD insult (Fig. 5a, b). Furthermore, we used LDH and MTT assay measure neuronal death/viability in cultured cortical neurons after 24 h of OGD insult. Results show that the bpV(pic) decreased LDH release and increased cell viability of neurons (Fig. 5c, d). We have confirmed that bpV(pic) can activation both of AKT and ERK 1/2, an AKT inhibitor IV (1.0 µM) and/ or an ERK 1/2 inhibitor U0126 (10 µM) were treated in the cultured cortical neurons 30 min after re-oxygenation, then the bpV(pic) (200 nM) was treated 1 h after re-oxygenation, we obtained a down-regulation neuroprotective effect of bpV(pic) after inhibition of AKT and/or ERK 1/2 (Fig. 5e, f). Therefore, BpV(pic) protects against OGD-induced neuronal death through both AKT and ERK 1/2 activation.

BpV(pic) protect against OGD induced neuronal death through ERK 1/2 activation and PTEN lipid phosphatase activity inhibition. a and b Western blots analysis of p-AKT (a) and p-ERK 1/2 (b) levels in cultured primary neurons, bpV(pic) (200 nM) againsts the OGD-induced p-AKT and p-ERK 1/2 down-regulation. Quantification analysis of the levels are on the right (n = 6 independent cultures, *P < 0.05 vs. the control, #P < 0.05 vs. the OGD + vehicle). c and d During OGD induced neuronal death the LDH release (c) decreased and the cell viability (d) increased after treated with bpV(pic) (10–500 nM). This tests were performed in quintuplicate (n = 6 independent cultures, *P < 0.05 vs. the OGD + Vehicle). e and f IV and/or U0126 were treated 30 min after re-oxygenation, bpV(pic) was treated for 1.0 h followed by re-oxygenation in the cultured primary neurons. The LDH release (e) and the cell viability (f) results show that the neuroprotective effect of bpV(pic) was blocked by IV and U0126 [n = 6 independent cultures, *P < 0.05 vs. the OGD + Vehicle, #P < 0.05 vs. the OGD + bpV(pic)]. The data are expressed as mean ± SE. Statistical analysis was implemented by student’s t-test and variance analysis
BpV(pic) Reduces the Infarct Volume and Promotes Functional Recovery in Ischemic Stroke Animals Through AKT and ERK 1/2 Activation
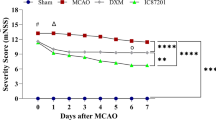
On the basis of Fig. 5e, f, we further confirmed the neuronprotective effect role of bpV(pic) in vivo. Followed the Fig. 2a, we i.c.v injected IV (100 µM, 2 µL) and/or U0126 (500 µM, 2 µL) 30 min after MCAO, and bpV(pic) (100 µM, 5 µL) was i.c.v injected 1.0 h after MCAO [37, 38]. Results show that inhibition of AKT and/or ERK 1/2 activation prevented the bpV(pic)-induced infarct volume down-regulation (Fig. 6a, b). To further provide evidence for the bpV(pic)-induced functional recovery after MCAO model rats, we performed a train of neurobehavioral tests. As show in Fig. 7a–c, results show that bpV(pic)-induced stroke animals functional recovery were mediated through both AKT and ERK 1/2 signalling pathways.

BpV(pic) through PTEN inhibition and ERK 1/2 activation reduces the infarct volume in ischemic stroke animals. a Sample images of TTC staining brain sections show that bpV(pic) decreases the infarct volume in brain 24 h after ischemia onset was prevented by IV and U0126. b Quantification analysis of the infarct volume [n = 6, *P < 0.05 vs. the I/R + bpV(pic)]. The data are expressed as mean ± SE. Statistical analysis was implemented by student’s t-test and variance analysis

BpV(pic) induces the functional recovery in ischemic stroke animals through PTEN inhibition and ERK 1/2 activation. a Animals treated with bpV(pic) have lower scores in mNSS test at day 7 and 14 after ischemia–reperfusion injury compared with I/R + Vehicle group. Animals injected with IV and/or U0126 before injected with bpV(pic) show a higher scores in mNSS test at day 7 and 14 after ischemia–reperfusion than I/R + bpV(pic) group [n = 6 for each group, *P < 0.05 vs. the I/R + Vehicle,#P < 0.05 vs. the I/R + bpV(pic)]. b Animals treated with bpV(pic) have lower scores in beam-walking test at day 3, 7 and 14 after ischemia–reperfusion injury compared with I/R + Vehicle group. Animals injected with IV and/or U0126 before injected bpV(pic) show a higher scores in beam-walking test at day 3, 7 and 14 after ischemia–reperfusion than I/R + bpV(pic) group [n = 6 for each group, *P < 0.05 vs. the I/R + Vehicle,#P < 0.05 vs. the I/R + bpV(pic)]. c Animals treated with bpV(pic) have a highter ratio in MST test at day 14 after ischemia–reperfusion injury compared with I/R + Vehicle group. Animals injected with IV and/or U0126 before injected bpV(pic) show lower ratio in MST test at day 14 after ischemia–reperfusion than I/R + bpV(pic) group [n = 6 for each group, *P < 0.05 vs. the I/R + Vehicle,#P < 0.05 vs. the I/R + bpV(pic)]. The datas are expressed as mean ± SE. Statistical analysis was implemented by student’s t-test and variance analysis
Discussion
PTEN is widely known as a tumor suppressor that is mostly associated with several neoplastic diseases. PTEN also plays a pivotal role in neuropathic diseases [39, 40]. Previous study shows that PTEN have both protein and lipid phosphatase activity [7]. When the PTEN protein phosphatase activity is suppressed, the extrasynapic GluN2B containing NMDA receptors (NMDARs) would be inhibited [41]. While inhibiting the lipid phosphatase activity of PTEN, the AKT pathway is activated. No matter which of the phosphatase activity is inhibited, it would induce neuroprotection in ischemic stroke. We further showed that inhibiting PTEN induces the nuclear TDP-43 (TAR DNA-binding protein-43) increase [42], and also produce the GABAA receptor expression and function enhancement to protect against the ischemic stroke induced neuronal death in vivo and in vitro [43, 44]. PTEN inhibition-induced neuroprotection is also confirmed by other studies [45,46,47].
BpV(pic) is a commercially available PTEN inhibitor, and previous studies from us and others have shown that bpV(pic) confers neuroprotection in cerebral ischemia injury [47, 48]. We have recently reported a new compound, the bisperoxovandium (pyridin-2-squaramide) [bpV(pis)], an inhibitor of PTEN, the structure is similar to bpV(pic), that confers neuroprotection in the ischemic injury model in vitro and in vivo through suppressing PTEN and activating ERK 1/2 [12]. In this study, we set up to determine whether bpV(pic) exerted its neuroprotective effect in cerebral ischemia injury through both PTEN inhibition and ERK 1/2 activation. We indicate that the neuropective effect of bpV(pic) get through by activation of AKT and ERK 1/2, and the bpV(pic) activation of AKT dependent on inhibition of PTEN lipid phosphatase activity only, activation of ERK 1/2 get through by both inhibition of PTEN and independent of PTEN (Fig. 8).

The mechanism of bpV(pic)-mediateded neuroprotect in ischaemia–reperfusion cerebral injury. After ischaemia–reperfusion, the phospho-AKT (Ser473) and phospho-ERK 1/2 (Thr202/Tyr204) were down-regulated, inducing the increase of neuronal death and cerebral injury (left). When treated with bpV(pic), we found that bpV(pic) can not only enhance the level of p-AKT and p-ERK 1/2 through inhibiting PTEN lipid phosphatase activity, but also in a PTEN independent pathway to up regulation of ERK 1/2 activity, leading to neuronal survival and animal functional recovery
Previous studies have demonstrated that the AKT activity can be increased by bpV(pic) [49]. Our finding confirms that the bpV(pic) enhences the AKT activity through the PTEN lipid activity inhibition. After cerebral ischaemia–reperfusion, the p-AKT and p-ERK 1/2 were down-regulated, inducing the increase of neuronal death and cerebral injury. After treated with bpV(pic), results show a positive improvement in the above phenomenon. But we found that the p-ERK 1/2 activity also increased after injected bpV(pic). As previous reports show that ERK 1/2 activation also have a neuroprotective effect [50, 51]. A number of pathways, growth factors and hormones are shown to confer protection through up-regulation of ERK 1/2 activity [52, 53]. Our data shows that bpV(pic) induced neuroprotection after ischemic stroke also depends on enhancement of ERK 1/2 activity, supports the ERK 1/2 neuroprotective role in ischaemic stroke. BpV(pic) always perform as a inhibitor of PTEN to inhibit PTEN lipid phosphatase activity. Thus, we further inverstigated the mechanism of bpV(pic) increasing ERK 1/2 activity. We found that bpV(pic) enhances ERK 1/2 activity not only by inhibiting of PTEN lipid phosphatase activity, but also by increasing of ERK 1/2 activity independently of PTEN. Our data also shows that the bpV(pic) is not directly affect the activity of ERK 1/2. As reported previously, ERK 1/2 has many up-stream signaling, like MEK, Raf, PKA and etc [54,55,56]. Thus, it is possible that ERK 1/2 activation by bpV(pic) might depend on PKA or others. The underlying mechanisms remain to be inverstigated. Furthermore, it is known that BpV(pic) inhibits the dephosphorylation of autophosphorylated insulin receptors [57, 58]. As phosphorylation of ERK1/2 is intimately connected to insulin signalling [59, 60]. Therefore, when we use bpV(pic) to act on brain insulin receptors, we should consider the effects of ERK 1/2 signaling.
Our finding demonstrates that bpV(pic) can activate AKT activity through the inhibition of PTEN lipid phosphatase activity. Interestingly, we show that ERK 1/2 can be also activated by bpV(pic). The bpV(pic)-induced ERK 1/2 activation is not only through the inhibition of PTEN lipid phosphatase activity, but is also independent of PTEN inhibition. Consequently, bpV(pic)-induced neuroprotection in ischemic stroke is depend on the inhibition of PTEN lipid phosphatase activity and the enhancement of ERK 1/2 activity that is independent of PTEN inhibition. Therefore, the effect of bpV(pic) on ERK 1/2 signaling should be considered while using bpV(pic) as a PTEN inhibitor in experimental conditions.
References
Han Z, Li L, Wang L, Degos V, Maze M, Su H (2014) Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J Neurochem 131:498–508
Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198
Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4:399–415
Muniyan S, Ingersoll MA, Batra SK, Lin MF (2014) Cellular prostatic acid phosphatase, a PTEN-functional homologue in prostate epithelia, functions as a prostate-specific tumor suppressor. Biochim Biophys Acta 1846:88–98
Sylvester PW, Shah SJ, Samant GV (2005) Intracellular signaling mechanisms mediating the antiproliferative and apoptotic effects of gamma-tocotrienol in neoplastic mammary epithelial cells. J Plant Physiol 162:803–810
Yamada KM, Araki M (2001) Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci 114:2375–2382
Schmid AC, Byrne RD, Vilar R, Woscholski R (2004) Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett 566:35–38
Boosani CS, Agrawal DK (2013) PTEN modulators: a patent review. Expert Opin Ther Pat 23:569–580
Milella M, Falcone I, Conciatori F, Cesta Incani C, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F et al (2015) PTEN: multiple functions in human malignant tumors. Front Oncol 5:24
Spinelli L, Lindsay YE, Leslie NR (2015) PTEN inhibitors: an evaluation of current compounds. Adv Biol Regul 57:102–111
Shi GD, OuYang YP, Shi JG, Liu Y, Yuan W, Jia LS (2011) PTEN deletion prevents ischemic brain injury by activating the mTOR signaling pathway. Biochem Biophys Res Commun 404:941–945
Zhang Z-F, Chen J, Han X, Zhang Y, Liao H-B, Lei R-X, Zhuang Y, Wang Z-F, Li Z, Chen J-C et al (2017) Bisperoxovandium (pyridin-2-squaramide) targets both PTEN and ERK1/2 to confer neuroprotection. Br J Pharmacol 174:641–656
Graves JD, Campbell JS, Krebs EG (1995) Protein serine/threonine kinases of the MAPK cascade. Ann N Y Acad Sci 766:320–343
Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF (2015) Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res 35:600–604
Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M (2010) Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 29:1091–1104
Kim MJ, Dunah AW, Wang YT, Sheng M (2005) Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46:745–760
Zhang F, Chen J (2008) Leptin protects hippocampal CA1 neurons against ischemic injury. J Neurochem 107:578–587
Atif F, Yousuf S, Sayeed I, Ishrat T, Hua F, Stein DG (2013) Combination treatment with progesterone and vitamin D hormone is more effective than monotherapy in ischemic stroke: the role of BDNF/TrkB/Erk1/2 signaling in neuroprotection. Neuropharmacology 67:78–87
Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188
Wang L, Lu Y, Guan H, Jiang D, Guan Y, Zhang X, Nakano H, Zhou Y, Zhang Y, Yang L et al (2013) Tumor necrosis factor receptor-associated factor 5 is an essential mediator of ischemic brain infarction. J Neurochem 126:400–414
Wang L, Lu Y, Deng S, Zhang Y, Yang L, Guan Y, Matozaki T, Ohnishi H, Jiang H, Li H (2012) SHPS-1 deficiency induces robust neuroprotection against experimental stroke by attenuating oxidative stress. J Neurochem 122:834–843
Chen J, Hu R, Liao H, Zhang Y, Lei R, Zhang Z, Zhuang Y, Wan Y, Jin P, Feng H et al (2017) A non-ionotropic activity of NMDA receptors contributes to glycine-induced neuroprotection in cerebral ischemia-reperfusion injury. Sci Rep 7:3575
Liu F, Schafer DP, McCullough LD (2009) TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. J Neurosci Methods 179:1–8
Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR (1990) A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab 10:290–293
Shan Y, Liu B, Li L, Chang N, Wang H, Wang D, Feng H, Cheung C, Liao M, Cui T et al (2009) Regulation of PINK1 by NR2B-containing NMDA receptors in ischemic neuronal injury. J Neurochem 111:1149–1160
Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, El-Hayek YH, Gomez E, Zukin RS, Fehlings MG et al (2006) Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci 26:5309–5319
Schmued LC, Stowers CC, Scallet AC, Xu L (2005) Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res 1035:24–31
Mason DY, Micklem K, Jones M (2000) Double immunofluorescence labelling of routinely processed paraffin sections. J Pathol 191:452–461
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT et al (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6:117–127
Ning K (2004) Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci 24:4052–4060
Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M (2001) Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 32:2682–2688
Clifton GL, Jiang JY, Lyeth BG, Jenkins LW, Hamm RJ, Hayes RL (1991) Marked protection by moderate hypothermia after experimental traumatic brain injury. J Cereb Blood Flow Metab 11:114–121
Sughrue ME, Mocco J, Komotar RJ, Mehra A, D’Ambrosio AL, Grobelny BT, Penn DL, Connolly ES Jr (2006) An improved test of neurological dysfunction following transient focal cerebral ischemia in rats. J Neurosci Methods 151:83–89
Wexler EJ, Peters EE, Gonzales A, Gonzales ML, Slee AM, Kerr JS (2002) An objective procedure for ischemic area evaluation of the stroke intraluminal thread model in the mouse and rat. J Neurosci Methods 113:51–58
Uluc K, Miranpuri A, Kujoth GC, Akture E, Baskaya MK (2011). Focal cerebral ischemia model by endovascular suture occlusion of the middle cerebral artery in the rat. J Vis Exp. https://doi.org/10.3791/1978
Yang BH, Son H, Kim SH, Nam JH, Choi JH, Lee JS (2004) Phosphorylation of ERK and CREB in cultured hippocampal neurons after haloperidol and risperidone administration. Psychiatry Clin Neurosci 58:262–267
Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Gala R, Simon RP (2008) In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab 28:232–241
Liu J, Xu Q, Wang H, Wang R, Hou XY (2013) Neuroprotection of ischemic postconditioning by downregulating the postsynaptic signaling mediated by kainate receptors. Stroke 44:2031–2035
Huang SY, Sung CS, Chen WF, Chen CH, Feng CW, Yang SN, Hung HC, Chen NF, Lin PR, Chen SC et al (2015) Involvement of phosphatase and tensin homolog deleted from chromosome 10 in rodent model of neuropathic pain. J Neuroinflamm 12:59
Endersby R, Baker SJ (2008) PTEN signaling in brain: neuropathology and tumorigenesis. Oncogene 27:5416–5430
Ning K, Pei L, Liao M, Liu B, Zhang Y, Jiang W, Mielke JG, Li L, Chen Y, El-Hayek YH et al (2004) Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci 24:4052–4060
Zheng M, Liao M, Cui T, Tian H, Fan DS, Wan Q (2012) Regulation of nuclear TDP-43 by NR2A-containing NMDA receptors and PTEN. J Cell Sci 125:1556–1567
Liu B, Li L, Zhang Q, Chang N, Wang D, Shan Y, Wang H, Feng H, Zhang L, Brann DW et al (2010) Preservation of GABAA receptor function by PTEN inhibition protects against neuronal death in ischemic stroke. Stroke 41:1018–1026
Liu B, Li L, Chen J, Wang Z, Li Z, Wan Q (2013) Regulation of GABAA receptors by fragile X mental retardation protein. Int J Physiol Pathophysiol Pharmacol 5:169–176
Chen J, Zhuang Y, Zhang ZF, Wang S, Jin P, He C, Hu PC, Wang ZF, Li ZQ, Xia GM et al (2016) Glycine confers neuroprotection through microRNA-301a/PTEN signaling. Mol Brain 9:59
Kitagishi Y, Matsuda S (2013) Diets involved in PPAR and PI3K/AKT/PTEN pathway may contribute to neuroprotection in a traumatic brain injury. Alzheimers Res Ther 5:42
Mao LL, Hao DL, Mao XW, Xu YF, Huang TT, Wu BN, Wang LH (2015) Neuroprotective effects of bisperoxovanadium on cerebral ischemia by inflammation inhibition. Neurosci Lett 602:120–125
Chen Y, Luo C, Zhao M, Li Q, Hu R, Zhang JH, Liu Z, Feng H (2015) Administration of a PTEN inhibitor BPV(pic) attenuates early brain injury via modulating AMPA receptor subunits after subarachnoid hemorrhage in rats. Neurosci Lett 588:131–136
Sury MD, Vorlet-Fawer L, Agarinis C, Yousefi S, Grandgirard D, Leib SL, Christen S (2011) Restoration of Akt activity by the bisperoxovanadium compound bpV(pic) attenuates hippocampal apoptosis in experimental neonatal pneumococcal meningitis. Neurobiol Dis 41:201–208
Ulbrich F, Kaufmann KB, Coburn M, Lagreze WA, Roesslein M, Biermann J, Buerkle H, Loop T, Goebel U (2015) Neuroprotective effects of Argon are mediated via an ERK-1/2 dependent regulation of heme-oxygenase-1 in retinal ganglion cells. J Neurochem 134:717–727
Wang D, Guo TQ, Wang ZY, Lu JH, Liu DP, Meng QF, Xie J, Zhang XL, Liu Y, Teng LS (2014) ERKs and mitochondria-related pathways are essential for glycyrrhizic acid-mediated neuroprotection against glutamate-induced toxicity in differentiated PC12 cells. Braz J Med Biol Res 47:773–779
Pratsinis H, Constantinou V, Pavlakis K, Sapkas G, Kletsas D (2012) Exogenous and autocrine growth factors stimulate human intervertebral disc cell proliferation via the ERK and Akt pathways. J Orthop Res 30:958–964
Meller R, Stevens SL, Minami M, Cameron JA, King S, Rosenzweig H, Doyle K, Lessov NS, Simon RP, Stenzel-Poore MP (2005) Neuroprotection by osteopontin in stroke. J Cereb Blood Flow Metab 25:217–225
Yu Z, Ye S, Hu G, Lv M, Tu Z, Zhou K, Li Q (2015) The RAF-MEK-ERK pathway: targeting ERK to overcome obstacles to effective cancer therapy. Future Med Chem 7:269–289
Hu Y, Mintz A, Shah SR, Quinones-Hinojosa A, Hsu W (2014) The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis 35:1491–1499
Goto A, Nakahara I, Yamaguchi T, Kamioka Y, Sumiyama K, Matsuda M, Nakanishi S, Funabiki K (2015) Circuit-dependent striatal PKA and ERK signaling underlies rapid behavioral shift in mating reaction of male mice. Proc Natl Acad Sci USA 112:6718–6723
Cummings C, Zhu L, Sorisky A, Liu XJ (1996) A peroxovanadium compound induces Xenopus oocyte maturation: inhibition by a neutralizing anti-insulin receptor antibody. Dev Biol 175:338–346
Yu ZW, Posner BI, Smith U, Eriksson JW (1996) Effects of peroxovanadate and vanadate on insulin binding, degradation and sensitivity in rat adipocytes. Biochim Biophys Acta 1310:103–109
Perrault R, Wright B, Storie B, Hatherell A, Zahradka P (2011) Tyrosine kinase-independent activation of extracellular-regulated kinase (ERK) 1/2 by the insulin-like growth factor-1 receptor. Cell Signal 23:739–746
Zhang R, Liu S, Guo B, Chang L, Li Y (2014) Chemerin induces insulin resistance in rat cardiomyocytes in part through the ERK1/2 signaling pathway. Pharmacology 94:259–264
Acknowledgements
This work was supported by China Key Project of Basic Research (“973” Project; 2014CB541606), Natural Science Foundation of China (NSFC; 81470599) and The Fund of Collaborative Innovation Center for Brain Science to Q.W.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
We have recently reported a new compound, the bisperoxovandium (pyridin-2-squaramide) [bpV(pis)] that confers neuroprotection in the ischemic injury model in vitro and in vivo through suppressing PTEN and activating ERK 1/2, respectively.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Liu, R., Tang, JC., Pan, MX. et al. ERK 1/2 Activation Mediates the Neuroprotective Effect of BpV(pic) in Focal Cerebral Ischemia–Reperfusion Injury. Neurochem Res 43, 1424–1438 (2018). https://doi.org/10.1007/s11064-018-2558-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-018-2558-z