
Abstract
Purpose
Review of the clinicopathologic and genetic features of early ependymal tumor with MN1-BEND2 fusion (EET MN1-BEND2), classical astroblastomas, and recently described related pediatric CNS tumors. I also briefly review general mechanisms of gene expression silencing by DNA methylation and chromatin remodeling, and genomic DNA methylation profiling as a powerful new tool for CNS tumor classification.
Methods
Literature review and illustration of tumor histopathologic features and prenatal gene expression timelines.
Results
Astroblastoma, originally descried by Bailey and Cushing in 1926, has been an enigmatic tumor. Whether they are of ependymal or astrocytic derivation was argued for decades. Recent genetic evidence supports existence of both ependymal and astrocytic astroblastoma-like tumors. Studies have shown that tumors exhibiting astroblastoma-like histology can be classified into discrete entities based on their genomic DNA methylation profiles, gene expression, and in some cases, the presence of unique gene fusions. One such tumor, EET MN1-BEND2 occurs mostly in female children, and has an overall very good prognosis with surgical management. It contains a gene fusion comprised of portions of the MN1 gene at chromosomal location 22q12.1 and the BEND2 gene at Xp22.13. Other emerging pediatric CNS tumor entities demonstrating ependymal or astroblastoma-like histological features also harbor gene fusions involving chromosome X, 11q22 and 22q12 breakpoint regions.
Conclusions
Genomic DNA profiling has facilitated discovery of several new CNS tumor entities, however, traditional methods, such as immunohistochemistry, DNA or RNA sequencing, and cytogenetic studies, including fluorescence in situ hybridization, remain necessary for their accurate biological classification and diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Epigenetic regulation of gene expression and genomic DNA methylation analysis
Nuclear chromatin is comprised of histones and other proteins wrapped within coils of genomic DNA forming nucleosome structures that are either transcriptionally active (euchromatin) or inactive (heterochromatin). Differential gene expression occurs through multiple mechanisms but is largely due to epigenetic silencing of genes by DNA methylation [1]. Methylation of cytosine residues at multiple CpG dinucleotide sites within gene promoters, and adjacent first exons, effectively turns off gene expression by directly hindering binding of some transcription factors, and by recruiting proteins that alter chromatin structure and further interfere with transcription. Such proteins include methyl-CpG binding proteins that both repress transcription directly and recruit histone deacetylases (HDACs) and other transcriptional corepressors. Deacetylation of specific histone amino-terminal lysine residues restricts transcription factor access to DNA, while acetylation is permissive to transcription [2]. Histone lysine methylationFootnote 1 by histone methyltransferases (HMTs), e.g., histone 3 (3.1, 3.2 and 3.3) lysine 28Footnote 2 trimethylation (H3K28me3), represses gene transcription by promoting heterochromatin assembly [4], or can activate transcription when less histone methylation, e.g., histone 3 lysine 28 monomethylation (H3K28me1), and/or concomitant acetylation occurs [5,6,7]. Missense mutation of H3K28 to methionine (H3K28M, aka H3K27M) in diffuse midline glioma leads to decreased histone methylation at this site [8].
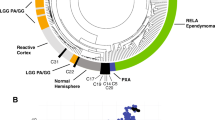
Several new central nervous system (CNS) tumor entities have been defined based on genomic DNA methylation profiling using Illumina’s Methylation450K and MethylationEPIC BeadChips [9,10,11]. This technology detects methylated cytosine residues (5-methyl-cytosine) in CpG sites and can be used to profile genomic DNA methylation from archived formalin-fixed, paraffin-embedded (FFPE) resected tumor material [9, 10, 12, 13]. DNA is extracted from an FFPE tissue block and treated with bisulfite to convert unmethylated cytosines to uracil. Amplification of the DNA replaces uracil with thymidine. Following amplification and fragmentation, the DNA is hybridized to the MethylationEPIC BeadChip, which contains methylation status-specific oligonucleotide probes for over 850,000 methylation sites. Further processing steps and data analysis allow comparison of an individual tumor’s genomic DNA methylation profile to those of known tumors in a reference set using a random forest brain tumor classifier developed by the German Cancer Research Center (DKFZ, www.molecularneuropathology.org/mnp/) [9] and/or by data dimensionality reduction algorithms, e.g., t-distributed Stochastic Neighbor Embedding (tSNE) or Uniform Manifold Approximation and Projection (UMAP) followed by two- or three-dimensional mapping [9,10,11,12,13,14] (Fig. 1).

Unsupervised UMAP dimension reduction analysis map of genomic DNA methylation from the DFKZ CNS tumor reference set. The reference set includes methylation data for 2729 tumors (76 pathological diagnoses) and 72 normal brain tissues. Each group of data points represent a specific normal tissue or tumor diagnosis or closely related diagnoses [9]. The arrow indicates HGNET MN1 tumors (consisting mostly of EET MN1-BEND2)
Early ependymal tumor with MN1-BEND2 fusion
Early ependymal tumors with MN1-BEND2 fusion (EET MN1-BEND2) are pediatric cerebral tumors most often occurring in the parietal or frontal lobes [10], however, a spinal tumor containing the fusion has also been described (Fig. 2, Table 1, Table S1). To date, cases with confirmed MN1-BEND2 fusions have only been well documented in females (n = 19), the vast majority in children (mean and median ages, 9.6 and 9 years, respectively, n = 18) [10] (Table S1). However, larger studies are needed to confirm their sex distribution.

Typical MR findings of EET MN1-BEND2. T1 post-contrast MR images of EET MN1-BEND2 presenting in a 9-year-old girl. T1 post-contrast axial and sagital images show typical well-demarcated complex solid and cystic appearance of EET MN1-BEND2. Like other supratentorial ependymal tumors, they often show a bubbly and/or multinodular appearance [59]. Images courtesy of Dr. Bret Mobley, Vanderbilt University
EET MN1-BEND2 was first identified as a subset of pediatric CNS tumors belonging to a methylation class defined as high-grade neuroepithelial tumors with MN1 alteration (HGNET-MN1). This designation was based on DNA methylation profiling combined with identification of MN1-BEND2 and MN1-CXXC5 fusions by RNA-seq in a small number of cases, and detection of nonspecific MN1 gene rearrangement by break-apart fluorescence in situ hybridization (FISH) [13].
The protein encoded by the meningioma (disrupted in balanced translocation) 1 (MN1) gene at chromosome 22q12.1 acts as a chromatin remodeler and transcriptional coregulator [15]. The function of BEN domain containing 2 encoded by BEND2 at Xp22.13 is unknown. However, other BEN domain-containing DNA-binding proteins are involved in chromatin remodeling [16].
Studies from our group confirmed the presence of MN1-BEND2 fusions in additional tumors within the HGNET-MN1 methylation class and demonstrated that they are typically associated with patient survival of over 10 years [10, 12]. Clearly, many tumors within this methylation class are not clinically high-grade. Therefore, I will henceforth refer to it as neuroepithelial tumors with MN1 alteration (NET-MN1).
In our study, the 5-year (n = 6)Footnote 3 and 10-year (n = 5) survival values for EET MN1-BEND2 were both 100% [12]. Tumor recurrences requiring re-resection were relatively common, however. One-half of patients experienced recurrence after initial resection: one patient at 4.3 years; one at 2, 4 and 4.5 years; and one at 1, 4, 5, 8 and 11 years.
Histologically, EET MN1-BEND2 are characterized by abundant perivascular tumor cell pseudorosettes arranged in a solid and/or loose papillary pattern corresponding to solid and cystic components by imaging, respectively [12, 17] (Figs. 2, 3B and C). EET MN1-BEND2 are generally well circumscribed both radiographically and microscopically and generally do not infiltrate brain parenchyma. Tumor blood vessels are often hyalinized and can appear sclerotic, as may the intervening tumor stroma (Fig. 3C). Mitotic activity and necrosis are frequently present (Fig. 3D). In addition to polygonal, columnar, and sometimes tapered perivascular tumor cells, focal clear or rhabdoid cytomorphology may also occur [10, 17,18,19,20]. Immunohistochemistry reveals that EET MN1-BEND2 usually demonstrate cell membrane and dot-like cytoplasmic epithelial membrane antigen (EMA, aka MUC1) and podoplanin immunoreactivity, and variable patchy or negative glial fibrillary acidic protein (GFAP) immunoreactivity [10, 19, 21, 22] (Fig. 3G and I). These histologic and immunohistochemical features are also found in other supratentorial and spinal ependymal tumors [18, 23,24,25,26,27] (Fig. 3E, H and J).

Histopathology of EET MN1-BEND2. A Perivascular pseudorosette in EET MN1-BEND2, B EET MN1-BEND2 papillary growth pattern. C Vascular and stromal sclerosis in EET MN1-BEND2. D EET MN1-BEND2 showing tumor necrosis. E ZFTA-RELA supratentorial ependymoma pseudorosette. F MAPK astroblastoma pseudorosette demonstrating more elongated cells with prominent nucleoli. A multinucleate cell is indicated by the arrow. G EET MN1-BEND2 EMA immunohistochemical stain demonstrating membrane and dot-like cytoplasmic positivity. H ZFTA-RELA supratentorial ependymoma showing similar EMA immunostaining. I and J Scattered GFAP immunostaining in EET MN1-BEND2 and ZFTA-RELA ependymoma, respectively. K Diffuse GFAP staining in MAPK astroblastoma. L MN1 immunohistochemical stain of EET MN1-BEND2 depicting strong nuclear staining. Such staining was absent in MAPK astroblastomas, however the sensitivity and specificity of MN1 immunohistochemical staining for EET MN1-BEND2 is not yet known [10]
EET MN1-BEND2 were termed astroblastoma, MN1 altered in the 2021 WHO classification of CNS tumors [18] because of their general resemblance to astroblastomas originally described by Percival Bailey, Harvey Cushing, and Paul Bucy [28, 29], that is, their demonstration of numerous, often back-to-back tumor cell perivascular pseudorosettes, sometimes referred to as astroblastic pseudorosettes. The latter are similar to, and at times indistinguishable from, pseudorosettes seen in supratentorial or spinal ependymomas [13, 17, 23, 30] (Fig. 3A, B and E).
Colleagues and I have recently shown that the gene expression profile of EET MN1-BEND2 strongly suggests ependymal differentiation, particularly derivation from an early ependymal precursor, and not astrocytic differentiation or derivation from an astrocyte precursor as “astroblastoma” implies [10]. EET MN1-BEND2 express high mRNA levels of the ependymoma-associated genes FOXJ1, IGF2, CELSR1, RFX3, KCNJ5, TFF3 and YAP1 and relatively low levels of messages of canonical astrocyte marker genes such as OLIG2, GFAP, ALDH1L1, and S100 β [10, 31]. EET MN1-BEND2 are also enriched for homeobox gene expression, e.g., CUX2, SHOX, SOX1, SOX14, IRX2, PAX1, HOXD10, DLX5 and PRRX2, and for HES1, H19, and the ATP binding cassette transporter gene ABCC1 encoding multidrug resistance-associated protein 1. These genes are highly expressed during embryonic and fetal development by the primitive neuroepithelium and/or ventricular zone radial glia (vRG) neural stem cells, the latter of which conventional ependymomas are believed to be derived from [10, 32, 33]. We have thus defined these tumors as early ependymal tumors with MN1-BEND2 fusion due to their expression of both early neural stem/progenitor cell and canonical ependymoma genes (Fig. 4).

Pediatric supratentorial ependymal tumor genes are highly expressed in the late embryonic/early fetal period and classical astroblastoma associated genes are expressed later during fetal and postnatal gliogenesis. The developmental expression timecourse (in post-conception weeks) of select genes overexpressed or mutated in EET MN1-BEND2 and other pediatric supratentorial ependymomas and MAPK astroblastomas was obtained from the Allen Human Developmental Transcriptome database. Pediatric supratentorial ependymoma and related tumor genes (relatively overexpressed or mutated) are depicted in plain type on the left and MAPK astrocytoma associated genes are in bold on the right. The patient age bar progressive color scheme is arbitrary. Pediatric ependymal tumor associated genes, including MAMLD1, PATZ1, FOXJ1, YAP1, MN1, CXXC5, RELA, EWSR1, BCOR and ZFTA1 are more highly expressed prior to 25 pcw. Some EET MN1-BEND2 associated genes, e.g., CELSR1, DLX5, HES1, FOXJ1, YAP1, SOX1, BCOR and H19, are most highly expressed prior to 10 pcw during the late embryonic/early fetal period. MAPK astrocytoma associated genes are more highly expressed after 25 pcw. Transcript expression is normalized by reads per kilobase of transcript per million mapped reads (RPKM) to compensate for RNA-seq generation of more sequencing reads from longer RNA molecules. Data is from up to 16 brain regions from 42 specimens [Allen Institute for Brain Science. Allen Human Brain Atlas. BrainSpan: Atlas of the Developing Human Brain—Developmental Transcriptome, 2010; https://www.brainspan.org/rnaseq/search/index.html (Accessed 10/31/2022)]
Additional genetic features of EET MN1-BEND2
In addition to MN1 and BEND2, overexpression or mutation of additional genes involved in chromatin remodeling were found in EET MN1-BEND2 tumors, e.g., in the SWI/SNF complex genes SMARCA1, SMARCAD1 and alpha-thalassemia, X-linked mental retardation gene (ATRX), and in lysine methyltransferase 2A (KMT2A, aka MLL1) [10]. The latter is a histone methyltransferase and transcriptional coactivator important in chromatin structural regulation and neural progenitor proliferation [6]. It is a subunit of the MLL1/MLL multiprotein complex that mediates both methylation of histone 3 lysine 4 (H3K4me) and acetylation of histone 4 lysine 16 (H4K16ac) [34]. EET MN1-BEND2 additionally overexpressed CHD3 encoding a component of the Mi-2/NuRD histone deacetylase complex. The mismatch repair gene MSH3 (at chromosome 5q14.1) was also frequently mutated in EET MN1-BEND2 [10].
Chromosomal copy number variations (CNVs) found in EET MN1-BEND2 include loss of portions of chromosomes 6, 8, 9, 10, 14, 16, 18, 22q and X, and gains of 6p, 9p and X, however, the most frequent CNVs observed were losses of chromosomes 14, 16, 22q and X [12]. Notably, 22q loss is the most common chromosomal abnormality in ependymomas [35].
Additional genes highly expressed in EET MN1-BEND2 include the ventricular zone radial glia-enriched gene H19, which is implicated as a tumor suppressor in the pediatric neoplasm Wilms tumor. The H19 gene product is a long noncoding RNA required for the recruitment of methyl-CpG-binding domain protein 1 (MBD1) and thus histone deacetylase to methylated sites on the nearby insulin growth factor 2 (IGF2) gene, resulting in its decreased transcription. miR483, also overexpressed in Wilms tumor and EET MN1-BEND2, enhances transcription of IGF2 and IGF2 antisense (IGF2-AS) genes. FAM3B, whose protein is involved in insulin secretion and apoptosis of insulin secreting cells, was also overexpressed. Like others, we found that MUM1, which facilitates DNA damage repair-associated chromatin changes, appears highly overexpressed in EET MN1-BEND2, however this was not the case when compared to normal brain controls [10, 36].
MAPK pathway activated classical astroblastomas
Other tumors also traditionally called astroblastomas more highly express OLIG2, GFAP, ALDH1L1, and S100 β astrocyte genes and exhibit histomorphologic and patient demographic characteristics more closely matching original descriptions of astroblastoma [10, 28]. These astrocyte-like astroblastomas are associated with intermediate-grade biological behavior and occur in male children, and young to middle-aged adults (rarely older adults) of both sexes [10, 12]. They demonstrate genomic methylation patterns similar, but not identical, to those of pleomorphic xanthoastrocytoma (PXA). Like PXA and other astrocytomas, they highly express and frequently harbor mutations in mitogen activated protein kinase (MAPK) pathway genes, e.g., BRAF, MAP3K5, MAP4K4 and NF1. BRAF V600E mutation especially occurred in young adult female patients [10, 12, 37].
MAP3K1, however, exhibited deletion mutations more often in EET MN1-BEND2 and other supratentorial ependymal tumors including ZFTA-RELA ependymomas and papillary tumors of the pineal region (PTPR). The latter share other genetic features with ependymomas and are considered to be ependymal tumors by some authors [10, 24, 38]. MAP3K1 is a cell survival/apoptosis regulator involved in ERK and JNK MAPK pathways, and in NF-κB and p53 signaling. It was also mutant in an EET MN1-BEND2 case reported by others and its gene promoter is hypermethylated in pediatric supratentorial and spinal ependymomas [21, 39].
A large subset of MAPK astroblastomas also showed PI3K/AKT/mTOR pathway alterations, including phosphoinositide-3-kinase (PI3K) subunit (PI3KCA, PI3KC3, PI3KR1, and PI3KR3) overexpression or mutations, AKT2, TSC2, RABEP1 and PTEN mutations, and TERT fusions not found in EET MN1-BEND2 [10]. They also harbor mutations in AHNAK (an actin binding protein related to phospholipase C signaling and cell migration), and more frequent mutations in WNT pathway genes, and show TCF4 and FAM107A overexpression [10]. MAPK astroblastomas also demonstrate mutations in multiple chromatin regulatory genes, e.g., SMARCA1, SMARCA2, SMARCAD1, SMARCD3, ATRX, and in histone methyltransferases, e.g., KMT2A, KMTA2C, KMT2D, and KMT2E. Additionally, MAPK pathway-associated astroblastomas show greater gene expression overlap with cerebral astrocyte precursor cells, i.e., outer radial glia (oRG) and truncated radial glia (tRG), than do EET MN1-BEND2 [10, 40].
Histologically, MAPK astroblastomas more often demonstrate elongate, tapering perivascular cells and more prominent nucleoli than do EET MN1-BEND2 (Fig. 3F). These types of rosettes would more appropriately be described as “astroblastic” and morphologically resemble tRG. They also more frequently show multinucleate tumor cells, may exhibit eosinophilic granular bodies [10, 12, 17] and demonstrate invasion of neighboring brain parenchyma [17]. These histomorphologic features were also noted in the original descriptions of astroblastomas by Bailey and Cushing, and Bailey and Bucy [28, 29]. Unlike generally seen in ependymal tumors, MAPK astroblastomas are diffusely GFAP positive and EMA negative by immunohistochemical staining consistent with astrocyte differentiation (Fig. 3K). We suggest that the term astroblastoma should be reserved for these MAPK pathway dominant tumors (astroblastoma, MAPK type) [10].
Other NET-MN1 methylation class tumors
The NET-MN1 methylation class contains two additional, closely associated subclasses of tumors characterized by MN1-CXXC5 or EWSR1-BEND2 fusions [13, 41, 42] (Table 1). EWSR1 encodes EWS RNA binding protein 1, a transcriptional activator whose gene at 22q12.2 is also the first component of fusions found in the pediatric tumor Ewing sarcoma. EWSR1-BEND2 harboring tumors tend to present in children to young adults [41] (Table 1, Table S1). Radiologic findings are similar to EET MN1-BEND2 [42]. Four cases presenting in the lower medulla and/or upper spinal cord have been reported in males: an infant aged 3 months (medulla to C4), a 20-year-old (brainstem NOS), a 38-year-old (lower medulla) and a 36-year-old (T3–T5) [41,42,43,44]. One cervicospinal and two frontal tumors with EWSR1-BEND2 have been reported in females, aged 6, and 6 and 26 years, respectively [41]. The mean and median ages of reported cases with documented fusions are 19 and 20 years (n = 7), respectively. The EWSR1-BEND2 fusion was also reported in a tumor described only as a spinal ependymoma [45]. Additionally, a cervicothoracic tumor with EWSR1 rearrangement by FISH was reported in a 6-year-old girl [46] and a multiply recurrent cervicomedullary tumor matching the NET-MN1 methylation class, but not otherwise molecularly characterized, was reported in a woman who presented at approximately 16 years of age [47]. A pontomedullary tumor exhibiting vascular and stromal sclerosis and MN1 rearrangement by FISH was reported in an 11-year-old male, possibly representing an EET MN1-BEND2 or MN1-CXXC5 lesion [48].
The histologic features of EWSR1-BEND2 tumors may be identical to those of EET MN1-BEND2, i.e., abundant perivascular pseudorosettes, including perivascular and stromal sclerosis in some cases, conspicuous mitotic activity, focal necrosis, patchy variable GFAP immunoreactivity, and often diffuse EMA positivity [41]. Overall survival of patients with EWSR1-BEND2 tumors is less favorable than for EET MN1-BEND2, at approximately 60% at 5 years and likely attributable to their frequent medullary and upper spinal cord locations.
A lumbospinal tumor with MAMLD1-BEND2 fusion matching the NET-MN1 methylation class, was reported in a 3-year-old girl [49]. MAMLD1, located at Xq28, encodes a developmentally important transcriptional coactivator [50]. YAP1 at 11q22.1, encoding for a DNA-binding Hippo pathway regulatory protein, and MAMLD1 fusions (YAP1-MAMLD1) appear to drive oncogenesis in a subset of supratentorial ependymomas occurring mostly in female infants [23, 51].
CXXC5 at 5q31.2 codes for a protein that binds unmethylated CpG sites and promotes chromatin structural changes, thereby modulating expression of multiple proliferation, cell cycle arrest and cancer related genes [52]. Fewer MN1-CXXC5 harboring tumors have been well described. One case, originally diagnosed as anaplastic ependymoma, presented in the temporal lobe of a 3-year-old boy [53], and another case in the parietal lobe of a 16-year-old male [13]. An additional case showing more poorly differentiated, tumor architecture lacking prominent pseudorosettes, occurred in the parietal lobe of a 36-year-old woman [54].
The extent in which MN1-CXXC5, EWSR1-BEND2 and MAMLD1-BEND2 containing tumors are biologically similar to EET MN1-BEND2 is not currently known. The histology of MN1-CXXC5 tumors may vary compared to EET MN1-BEND2 and EWSR1-BEND2 tumors, and indeed MN1-CXXC5 tumors appear to form a slightly distant satellite cluster of the NET-MN1 methylation class [54]. The common denominator of BEND2 as the downstream gene in MN1-BEND2, EWSR1-BEND2 and MAMLD1-BEND2 fusion harboring tumors has led to speculation that BEND2 is the more biologically important overexpressed gene function in these histologically similar lesions [10, 41, 49]. Perhaps the NET-MN1 methylation class should be renamed NET-BEND2 altered.
High-grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication
High-grade neuroepithelial tumors with BCOR exon 15 internal tandem duplication (HGNET BCOR ex15 ITD) are rare pediatric tumors belonging to a discrete methylation class [13, 55]. BCOR (BCL6 co-repressor) at Xp11.4 represses gene transcription through interaction with the DNA binding protein BCL-6 [56] and may recruit a histone deacetylase. BCOR mutation, however, results in methylation of histone 3 lysines 4 and 36 leading to reactivated transcription of silenced genes [57].
HGNET BCOR ex15 ITD are predominantly cerebral tumors, but have also rarely occurred in the basal ganglia, cerebellum, and pons. From their series and literature review, Ferris et al. reported that they occur nearly equally in males and females (n = 35) in patients ranging from 0 to 22 years with a median patient age of 3.5 years [55]. Imaging shows large, well‐circumscribed, heterogeneous tumors demonstrating variable enhancement, often with central necrosis or hemorrhage, and restricted diffusion indicative of a highly cellular lesion [55].
The largest reported series of HGNET BCOR ex15 ITD describes them as being histologically heterogenous, but they typically contain more classical ependymoma-like pseudorosettes demonstrating a fibrillary perivascular anuclear zone, palisading necrosis, and an absence of microvascular proliferation [55, 58]. Homer Wright rosettes were additionally seen in some cases, which are not characteristic of ependymal tumors, but occur in more primitive embryonal tumors [58]. Although generally well circumscribed, some cases can be infiltrative. They are reportedly mostly GFAP-negative and lack ependymoma-like EMA immunoreactivity, however most cases demonstrate NeuN and BCOR nuclear immunoreactivity. EET MN1-BEND2 also highly express BCOR [10, 19]. HGNET BCOR ex15 ITD are truly high-grade tumors. Their prognosis appears to be significantly worse than that of EET MN1-BEND2, however, some long-term survivors are reported [55, 59]. Nosologically, they may be best considered an anaplastic early ependymal tumor or an embryonal tumor (Fig. 4).
Neuroepithelial tumors with PATZ1 fusions
Neuroepithelial tumors with PATZ1 fusions (NET-PATZ1) are a diverse group of mostly pediatric tumors harboring fusions between nearby chromosome 22q12 region genes, i.e., MN1-PATZ1 or EWSR1-PATZ1 [60] (Table 1). PATZ1 at 22q12.2, like MN1 at 22q12.1, encodes a chromatin remodeler and transcriptional coregulator. NET-PATZ1 tumors are relatively heterogenous histologically. Most were originally diagnosed as glioblastoma or high-grade astrocytoma, followed by anaplastic ependymoma. A cerebral tumor in a 13-year-old girl demonstrating a perivascular pseudorosette pattern harbored both EWSR1-PATZ1 and MN1-GTSE1 fusions [61]. GTSE1 at 22q13.31 encodes a cell cycle regulatory protein that binds p53 and shuttles it out of the nucleus in response to DNA damage. In a separate report, a tumor with a EWSR1-PATZ1 fusion was described as a ganglioglioma [62].
NET-PATZ1 frequently show necrosis, but like EET MN1-BEND2 and HGNET BCOR ex15 ITD, generally lack microvascular proliferationFootnote 4. NET-PATZ1 appear to occur equally in male and female patients in multiple CNS locations (cerebrum, cerebellum, spinal cord), however most are supratentorial. Although, the demographic and anatomic data of NET-PATZ1 were not described individually for MN1 and EWSR1 fusion tumors [60]. tSNE analysis of their tumor methylation profiles reveals that both fusion types form a methylation class grouping, or perhaps separate, but closely associated subgroups [60]. Some NET-PATZ1, presumably those originally diagnosed as anaplastic ependymoma, show histology very similar to EET MN1-BEND2 [60]. NET-PATZ1 are predicted to show intermediate biological behavior. However, the latter is not established, and their clinical aggressiveness could be variable because of their overall heterogeneity.
Diagnosis of EET BEND2 and related tumors
Although some features may be more common in one tumor type versus another within the extended NET-MN1 methylation class, HGNET BCOR ex15 ITD, other ependymal or astroblastoma-like tumors, and MAPK astroblastomas, these tumors cannot be reliably distinguished by histology alone [12]. Because it encompasses three or more distinct pathological entities, prior studies of cases assigned to the HGNET-MN1 methylation class should be interpreted with caution [59, 63,64,65].
Pathological diagnosis requires ancillary testing. Immunohistochemistry should be performed for EMA or podoplanin, GFAP, BCOR, and p65-RELA or L1CAM for ZFTA-RELA ependymomas [26, 66, 67]. Unlike MAPK astroblastoma and many astrocytomas, which tend to be diffusely GFAP immunoreactive, EET MN1-BEND2 shows variable, but usually only focal GFAP immunoreactivity, but like ependymoma, when present tends to be positive in perivascular pseudorosettes [10, 68, 69]. Like other ependymal tumors [70], NET-MN1 class lesions may occasionally show immunoreactivity for neuronal markers [59].
Currently, molecular studies are necessary to evaluate for characteristic fusions by FISH, PCR, RNA or DNA sequencing, or Nanostring technology [12, 21, 67, 71]. Genomic DNA methylation analysis will likely become increasingly helpful to establish a precise diagnosis of many pediatric CNS tumors [10, 11, 19]. FISH for MN1-BEND2 using fusion probes rather than relatively nonspecific break apart analysis may be preferable [21]. More than one molecular diagnostic modality may be required, for example to confirm FISH or genomic DNA methylation results.
DNA methylation can be reliably performed on FFPE tissue, however, is currently only available at a limited number of academic clinical centers and is not yet FDA approved. Immunohistochemistry for MN1 is a promising cost-efficient procedure to help identify EET MN1-BEND2 tumors that can be easily performed in most medical centers (Fig. 3L). However, further studies are needed to establish its sensitivity and specificity, as it may be positive in other tumors highly expressing MN1, perhaps especially rare tumors with alternate MN1 fusions.
Treatment of EET MN1-BEND2 and related tumors
Treatment for EET MN1-BEND2 is primarily surgical. Complete resection should be pursued whenever possible, as it may be curative, offer the potential for very long-term patient survival, reduce morbidity and/or negate the need for adjuvant cytotoxic chemotherapy or potentially biotransformative radiation therapy [12, 18, 21].
Intraoperative pathological diagnosis or pre-resection stereotactic biopsy should be able to confirm ependymal histology and guide the surgical approach. A recent study demonstrated the feasibility of intraoperative tumor DNA methylation analysis, which could portend the future of intraoperative pathological diagnosis [72]. Fluorescence-guided resection using 5-aminolevulic acid (5-ALA) may be helpful to achieve gross total resection (GTR) [22].
As recurrence is common, long-term surveillance is necessary for EET MN1-BEND2 patients. Adjuvant therapy should be considered for patients with multiple recurrences and/or whose tumors are not completely resectable. For pediatric supratentorial ependymomas, GTR is associated with improved progression free survival, but not necessarily overall survival [69]. Conformational radiation (CRT) increases 5-year event free survival in ependymoma [73]. It would therefore be rational to treat EET-MN1 BEND2 and other new ependymal tumor entities with CRT if GTR is not possible. Complete resection may be hampered by the multinodular/multicystic nature of EET MN1-BEND2 and EWSR1-BEND2 tumors. In brainstem or spinal cord tumors GTR may not be possible, therefore adjuvant therapy appears indicated [42, 74].
Confirmed and probable EET MN1-BEND2 cases have been treated with radiation or radiation and temozolomide with unclear benefits due to the variable, but overall indolent natural history of this entity [21, 22, 64]. Medullary and spinal cord related-tumors (e.g., EWSR1-BEND2 lesions) that are not completely resectable have been successfully treated with radiation and temozolomide [42]. Yamada et al. [74] report a T1–T2 spinal cord astroblastoma-like tumor, with an apparent MN1 tandem duplication by FISH, in a 20-year-old woman who exhibited dramatic functional improvement and tumor shrinkage in response to radiation, temozolomide, and bevacizumab. Because of EET MN1-BEND2’s overexpression of IGF2 pathway components and ABCC1, agents directed at these targets could be therapeutic candidates, perhaps in combination with radiation and temozolomide [10].
One EET MN1-BEND2 case presenting in a 6-year-old girl recurred multiple times over ten years and appeared to undergo malignant transformation with acquired mutations in NF-κB signaling proteins and increased expression of p65-RELA [21]. The patient was treated with radiation therapy and temozolomide after a second resection, and combined CCNU/temozolomide following a third. Therefore, transformation may have theoretically been treatment related.
Summary
Astroblastoma has been a controversial entity. Some have argued they were of ependymal differentiation; others favored astrocytic derivation, while some have opined that astroblastoma histomorphology simply represents a nonspecific pattern [24]. We used the terminology early ependymal tumors with MN1-BEND2 because of expression of early neural stem/progenitor cell and ependymoma-associated genes in this new tumor entity. They might also be appropriately called ependymoma with MN1-BEND2. Their histological features, immunohistochemical profile and generally noninvasive behavior overlap with established supratentorial ependymomas, as do those of related EWSR1-BEND2 harboring tumors. These and other newly described pediatric astroblastoma- or ependymoma-like tumors should therefore probably be considered ependymal tumors.
Perhaps the most compelling reason for their classification as ependymal is that their current treatment and prognosis is more similar to that of other ependymal tumors than to that of astrocytic tumors. Neuroepithelial tumor is too broad a term as it can be used to describe any tumor ultimately derived from the primitive neuroepithelium, essentially all primary CNS tumors, and has thus become a “wastebasket” term [75]. Astroblastoma is similarly becoming a wastebasket description for several new tumor entities. Creation of additional tumor categories based on unique genetic features, e.g., specific gene fusions, that do not correlate with a truly novel histology or clinical behavior is not helpful, and medicine may be better served by considering such lesions subtypes of established lineages if they share similar overall genetics and biological behavior.
EET-MN1 patients may have very long-term survival despite the presence of intermediate to high-grade tumor histological features, i.e., mitotic activity, and necrosis, in many examples [12]. Similar to other supratentorial ependymal tumors, EET MN1-BEND2 tend to recur and may require multiple re-resections. Like ependymoma their defining histologic feature is perivascular pseudorosettes and a tendency for discrete borders with uninvolved brain tissue and only local tumor cell invasion if any. The latter likely contributes to their relatively indolent biological behavior. Indeed, many cases of EET MN1-BEND2 and other NET MN1 methylation class tumors were originally diagnosed as ependymoma or anaplastic ependymoma [25, 45, 46, 66]. ZFTA fusion-positive supratentorial ependymomas with alternate (non-RELA) fusion partners form satellite subclusters of the RELA Ependymoma methylation class and include tumors demonstrating astroblastoma-like histologic features, further supporting that the latter are within the spectrum of ependymal differentiation [26].
Many genes involved in fusions or mutated in the ependymal astroblastoma-like tumors discussed in this review are chromatin remodelers and/or transcriptional regulators affecting DNA methylation and gene expression. This suggests perturbations effecting DNA methylation and downstream chromatin and transcriptional regulation are important factors in pediatric CNS tumorigenesis, perhaps particularly ependymomagenesis. DNA damage causing double strand breaks repaired by error prone non-homologous and alternative end joining [76], particularly involving chromosomes X, 11, and 22, may lead to gene translocations in ependymomagenesis.
Fusions between chromosome 22.12 to 22.13 genes in NET-PATZ1 may be generated by a type of genomic instability called chromothripsis: a process in which catastrophic chromosomal instability leads to clustered deletions and rearrangements within a particular chromosome. Chromothripsis may also be responsible for generating chromosome 22q fusions in rare supratentorial astroblastoma-like tumors lacking MN1 alterations [67] and chromosome 11q13.1 gene fusions in ZFTA-RELA harboring supratentorial ependymomas [26]. Chromosome X chromothripsis may possibly facilitate MN1-BEND2 fusion in some cases of EET MN1-BEND2 [12, 65]. Characteristic gene fusions in such tumors may drive their oncogenic phenotypes. Chromothripsis itself may be initiated by mutations in SWI/SNF chromatin remodeling proteins or mismatch repair proteins [77].
Mutations in histone modifying proteins may also be important in pediatric ependymomagenesis. DNA methylation and histone deacetylation are intimately linked. In addition to methyl-CpG binding proteins, DNA methyltransferase 1 (DNMT1), which maintains genomic DNA methylation, also recruits histone deacetylase [78]. SWI/SNF remodeling proteins recognize acetylated or methylated histones and alter nucleosome structure to allow transcription [79]. In astrocytomas, a hypermethylated genomic DNA state (CpG island methylator phenotype or CIMP) in isocitrate dehydrogenase (IDH1/2) mutant tumors correlates with increased histone methylation, altered gene expression and improved patient survival [80, 81]. Mutant IDH1/2 causes elevated levels of 2-hydroxyglutarate, which inhibits histone demethylases and the TET family of 5-methlycytosine hydroxylases leading to increased histone and DNA methylation, respectively [82]. IDH-mutant tumors, thus, have a better prognosis than IDH1/2 wildtype astrocytomas.
Inappropriate hypomethylation of growth factor genes such as IGF2 and other imprinted genes may be an important factor in driving EET MN1-BEND2 tumorigenesis [10]. Altered gene promoter methylation could possibly be secondary to chromatin regulatory gene mutation resulting in chromatin structural changes that effect the activity of DNA methyltransferases [10, 83, 84]. Thus, chromatin structural regulation including by DNA methylation and post-translational modifications of histone proteins may be particularly important in pediatric CNS tumorigenesis.
Change history
30 January 2023
The original version of this article has been revised: The missing color shading has been added to Table 1, and a reference to Fig. 3L in the last paragraph of the section 'Diagnosis of EET BEND2 and related tumors'. Also, some typos have been corrected.
Notes
Not to be confused with DNA methylation.
Because the N-terminal methionine is cleaved from histone proteins, H3K28 has historically been referred to as H3K27, with clinical usage of H3K28 occurring only recently [3].
Updated data.
References
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38:23–38. https://doi.org/10.1038/npp.2012.112
El-Osta A, Wolffe AP (2000) DNA methylation and histone deacetylation in the control of gene expression: basic biochemistry to human development and disease. Gene Expr 9:63–75. https://doi.org/10.3727/000000001783992731
Leske H, Rushing E, Budka H, Niehusmann P, Pahnke J, Panagopoulos I (2018) K27/G34 versus K28/G35 in histone H3-mutant gliomas: A note of caution. Acta Neuropathol 136:175–176. https://doi.org/10.1007/s00401-018-1867-2
Rice JC, Allis CD (2001) Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol 13:263–273. https://doi.org/10.1016/S0955-0674(00)00208-8
Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, Fischle W, Bonaldi T, Pasini D (2014) Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell 53:49–62. https://doi.org/10.1016/j.molcel.2013.10.030
Huang YC, Shih HY, Lin SJ, Chiu CC, Ma TL, Yeh TH, Cheng YC (2015) The epigenetic factor Kmt2a/Mll1 regulates neural progenitor proliferation and neuronal and glial differentiation. Dev Neurobiol 75:452–462. https://doi.org/10.1002/dneu.22235
Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E (2002) ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10:1119–1128. https://doi.org/10.1016/s1097-2765(02)00740-2
Harutyunyan AS, Chen H, Lu T, Horth C, Nikbakht H, Krug B, Russo C, Bareke E, Marchione DM, Coradin M, Garcia BA, Jabado N, Majewski J (2020) H3K27M in gliomas causes a one-step decrease in H3K27 methylation and reduced spreading within the constraints of H3K36 methylation. Cell Rep 33:108390. https://doi.org/10.1016/j.celrep.2020.108390
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, Kratz A, Wefers AK, Huang K, Pajtler KW, Schweizer L, Stichel D, Olar A, Engel NW, Lindenberg K, Harter PN, Braczynski AK, Plate KH, Dohmen H, Garvalov BK, Coras R, Holsken A, Hewer E, Bewerunge-Hudler M, Schick M, Fischer R, Beschorner R, Schittenhelm J, Staszewski O, Wani K, Varlet P, Pages M, Temming P, Lohmann D, Selt F, Witt H, Milde T, Witt O, Aronica E, Giangaspero F, Rushing E, Scheurlen W, Geisenberger C, Rodriguez FJ, Becker A, Preusser M, Haberler C, Bjerkvig R, Cryan J, Farrell M, Deckert M, Hench J, Frank S, Serrano J, Kannan K, Tsirigos A, Bruck W, Hofer S, Brehmer S, Seiz-Rosenhagen M, Hanggi D, Hans V, Rozsnoki S, Hansford JR, Kohlhof P, Kristensen BW, Lechner M, Lopes B, Mawrin C, Ketter R, Kulozik A, Khatib Z, Heppner F, Koch A, Jouvet A, Keohane C, Muhleisen H, Mueller W, Pohl U, Prinz M, Benner A, Zapatka M, Gottardo NG, Driever PH, Kramm CM, Muller HL, Rutkowski S, von Hoff K, Fruhwald MC, Gnekow A, Fleischhack G, Tippelt S, Calaminus G, Monoranu CM, Perry A, Jones C, Jacques TS, Radlwimmer B, Gessi M, Pietsch T, Schramm J, Schackert G, Westphal M, Reifenberger G, Wesseling P, Weller M, Collins VP, Blumcke I, Bendszus M, Debus J, Huang A, Jabado N, Northcott PA, Paulus W, Gajjar A, Robinson GW, Taylor MD, Jaunmuktane Z, Ryzhova M, Platten M, Unterberg A, Wick W, Karajannis MA, Mittelbronn M, Acker T, Hartmann C, Aldape K, Schuller U, Buslei R, Lichter P, Kool M, Herold-Mende C, Ellison DW, Hasselblatt M, Snuderl M, Brandner S, Korshunov A, von Deimling A, Pfister SM (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Lehman NL, Spassky N, Sak M, Webb A, Zumbar CT, Usubalieva A, Alkhateeb KJ, McElroy JP, Maclean KH, Fadda P, Liu T, Gangalapudi V, Carver J, Abdullaev Z, Timmers C, Parker JR, Pierson CR, Mobley BC, Gokden M, Hattab EM, Parrett T, Cooke RX, Lehman TD, Costinean S, Parwani A, Williams BJ, Jensen RL, Aldape K, Mistry AM (2022) Astroblastomas exhibit radial glia stem cell lineages and differential expression of imprinted and X-inactivation escape genes. Nat Commun 13:2083. https://doi.org/10.1038/s41467-022-29302-8
Perez E, Capper D (2020) Invited review: DNA methylation-based classification of paediatric brain tumours. Neuropathol Appl Neurobiol 46:28–47. https://doi.org/10.1111/nan.12598
Lehman NL, Usubalieva A, Lin T, Allen SJ, Tran QT, Mobley BC, McLendon RE, Schniederjan MJ, Georgescu MM, Couce M, Dulai MS, Raisanen JM, Al Abbadi M, Palmer CA, Hattab EM, Orr BA (2019) Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7:42. https://doi.org/10.1186/s40478-019-0689-3
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I, Ryzhova M, Koelsche C, Pfaff E, Allen SJ, Balasubramanian G, Worst BC, Pajtler KW, Brabetz S, Johann PD, Sahm F, Reimand J, Mackay A, Carvalho DM, Remke M, Phillips JJ, Perry A, Cowdrey C, Drissi R, Fouladi M, Giangaspero F, Lastowska M, Grajkowska W, Scheurlen W, Pietsch T, Hagel C, Gojo J, Lotsch D, Berger W, Slavc I, Haberler C, Jouvet A, Holm S, Hofer S, Prinz M, Keohane C, Fried I, Mawrin C, Scheie D, Mobley BC, Schniederjan MJ, Santi M, Buccoliero AM, Dahiya S, Kramm CM, von Bueren AO, von Hoff K, Rutkowski S, Herold-Mende C, Fruhwald MC, Milde T, Hasselblatt M, Wesseling P, Rossler J, Schuller U, Ebinger M, Schittenhelm J, Frank S, Grobholz R, Vajtai I, Hans V, Schneppenheim R, Zitterbart K, Collins VP, Aronica E, Varlet P, Puget S, Dufour C, Grill J, Figarella-Branger D, Wolter M, Schuhmann MU, Shalaby T, Grotzer M, van Meter T, Monoranu CM, Felsberg J, Reifenberger G, Snuderl M, Forrester LA, Koster J, Versteeg R, Volckmann R, van Sluis P, Wolf S, Mikkelsen T, Gajjar A, Aldape K, Moore AS, Taylor MD, Jones C, Jabado N, Karajannis MA, Eils R, Schlesner M, Lichter P, von Deimling A, Pfister SM, Ellison DW, Korshunov A, Kool M (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164:1060–1072. https://doi.org/10.1016/j.cell.2016.01.015
Terry M, Wakeman K, Williams BJ, Miller DM, Sak M, Abdullaev Z, Pacheco MC, Aldape K, Lehman NL (2022) Malignant melanotic nerve sheath tumor with PRKAR1A, KMT2C and GNAQ mutations. Free Neuropathol 3:21. https://doi.org/10.17879/freeneuropathology-2022-3864
van Wely KH, Molijn AC, Buijs A, Meester-Smoor MA, Aarnoudse AJ, Hellemons A, den Besten P, Grosveld GC, Zwarthoff EC (2003) The MN1 oncoprotein synergizes with coactivators RAC3 and p300 in RAR-RXR-mediated transcription. Oncogene 22:699–709. https://doi.org/10.1038/sj.onc.1206124
Dai Q, Ren A, Westholm JO, Serganov AA, Patel DJ, Lai EC (2013) The BEN domain is a novel sequence-specific DNA-binding domain conserved in neural transcriptional repressors. Genes Dev 27:602–614. https://doi.org/10.1101/gad.213314.113
Lehman NL, Hattab EM, Mobley BC, Usubalieva A, Schniederjan MJ, McLendon RE, Paulus W, Rushing EJ, Georgescu MM, Couce M, Dulai MS, Cohen ML, Pierson CR, Raisanen JM, Martin SE, Lehman TD, Lipp ES, Bonnin JM, Al-Abbadi MA, Kenworthy K, Zhao K, Mohamed N, Zhang G, Zhao W (2017) Morphological and molecular features of astroblastoma, including BRAFV600E mutations, suggest an ontological relationship to other cortical-based gliomas of children and young adults. Neuro Oncol 19:31–42. https://doi.org/10.1093/neuonc/now118
WHO Classification of Tumours Editorial Board (2021) Central Nervous System Tumours. International Agency for Research on Cancer, Lyon (France)
Jamshidi P, McCord M, Horbinski C, Jennings L, Santana dos Santos L, Fudyma IA, DeCuypere M, Yap KL, Rathbun P, Wadhwani N (2022) Methylation profiling improves the care of pediatric brain tumor patients. AJSP: Rev dReports. https://doi.org/10.1097/pcr.0000000000000493
Mhatre R, Sugur HS, Nandeesh BN, Chickabasaviah Y, Saini J, Santosh V (2019) MN1 rearrangement in astroblastoma: study of eight cases and review of literature. Brain Tumor Pathol 36:112–120. https://doi.org/10.1007/s10014-019-00346-x
Burford A, Mackay A, Popov S, Vinci M, Carvalho D, Clarke M, Izquierdo E, Avery A, Jacques TS, Ingram WJ, Moore AS, Frawley K, Hassall TE, Robertson T, Jones C (2018) The ten-year evolutionary trajectory of a highly recurrent paediatric high grade neuroepithelial tumour with MN1:BEND2 fusion. Sci Rep 8:1032. https://doi.org/10.1038/s41598-018-19389-9
Fudaba H, Momii Y, Kawasaki Y, Goto H, Nobusawa S, Fujiki M (2020) Well-differentiated astroblastoma with both focal anaplastic features and a meningioma 1 gene alteration. NMC Case Rep J 7:205–210. https://doi.org/10.2176/nmccrj.cr.2020-0028
Andreiuolo F, Varlet P, Tauziede-Espariat A, Junger ST, Dorner E, Dreschmann V, Kuchelmeister K, Waha A, Haberler C, Slavc I, Corbacioglu S, Riemenschneider MJ, Leipold A, Rudiger T, Korholz D, Acker T, Russo A, Faber J, Sommer C, Armbrust S, Rose M, Erdlenbruch B, Hans VH, Bernbeck B, Schneider D, Lorenzen J, Ebinger M, Handgretinger R, Neumann M, van Buiren M, Prinz M, Roganovic J, Jakovcevic A, Park SH, Grill J, Puget S, Messing-Junger M, Reinhard H, Bergmann M, Hattingen E, Pietsch T (2019) Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol 29:205–216. https://doi.org/10.1111/bpa.12659
Lehman NL (2008) Central nervous system tumors with ependymal features: a broadened spectrum of primarily ependymal differentiation? J Neuropathol Exp Neurol 67:177–188. https://doi.org/10.1097/NEN.0b013e31816543a6
Neumann JE, Spohn M, Obrecht D, Mynarek M, Thomas C, Hasselblatt M, Dorostkar MM, Wefers AK, Frank S, Monoranu CM, Koch A, Witt H, Kool M, Pajtler KW, Rutkowski S, Glatzel M, Schuller U (2020) Molecular characterization of histopathological ependymoma variants. Acta Neuropathol 139:305–318. https://doi.org/10.1007/s00401-019-02090-0
Tauziede-Espariat A, Siegfried A, Nicaise Y, Kergrohen T, Sievers P, Vasiljevic A, Roux A, Dezamis E, Benevello C, Machet MC, Michalak S, Puiseux C, Llamas-Gutierrez F, Leblond P, Bourdeaut F, Grill J, Dufour C, Guerrini-Rousseau L, Abbou S, Dangouloff-Ros V, Boddaert N, Saffroy R, Hasty L, Wahler E, Pages M, Andreiuolo F, Lechapt E, Chretien F, Blauwblomme T, Beccaria K, Pallud J, Puget S, Uro-Coste E, Varlet P, Renoclip-Loc tBc, (2021) Supratentorial non-RELA, ZFTA-fused ependymomas: a comprehensive phenotype genotype correlation highlighting the number of zinc fingers in ZFTA-NCOA1/2 fusions. Acta Neuropathol Commun 9:135. https://doi.org/10.1186/s40478-021-01238-y
Ishizawa K, Komori T, Shimada S, Hirose T (2009) Podoplanin is a potential marker for the diagnosis of ependymoma: a comparative study with epithelial membrane antigen (EMA). Clin Neuropathol 28:373–378
Bailey P, Bucy PC (1930) Astroblastomas of the brain. Acta Psychiatr Scand 5:439–461. https://doi.org/10.1111/j.1600-0447.1930.tb08230.x
Bailey P, Cushing H (1926) A Classification of the Tumors of the Glioma Group on a Histogenetic Basis with a Correlated Study of Prognosis. JB Lippincott Company, Philadelphia, PA
Dulai MS, Caccamo DV, Briley AL, Edwards MS, Fisher PG, Lehman NL (2010) Intramedullary papillary ependymoma with choroid plexus differentiation and cerebrospinal fluid dissemination to the brain. J Neurosurg Pediatr 5:511–517. https://doi.org/10.3171/2009.12.PEDS09130
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. https://doi.org/10.1523/JNEUROSCI.4178-07.2008
Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, Macdonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ (2005) Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8:323–335. https://doi.org/10.1016/j.ccr.2005.09.001
Spassky N, Merkle FT, Flames N, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A (2005) Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci 25:10–18. https://doi.org/10.1523/JNEUROSCI.1108-04.2005
Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG (2005) Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121:873–885. https://doi.org/10.1016/j.cell.2005.04.031
Carter M, Nicholson J, Ross F, Crolla J, Allibone R, Balaji V, Perry R, Walker D, Gilbertson R, Ellison DW (2002) Genetic abnormalities detected in ependymomas by comparative genomic hybridisation. Br J Cancer 86:929–939. https://doi.org/10.1038/sj.bjc.6600180
Lastowska M, Trubicka J, Sobocinska A, Wojtas B, Niemira M, Szalkowska A, Kretowski A, Karkucinska-Wieckowska A, Kaleta M, Ejmont M, Perek-Polnik M, Dembowska-Baginska B, Grajkowska W, Matyja E (2020) Molecular identification of CNS NB-FOXR2, CNS EFT-CIC, CNS HGNET-MN1 and CNS HGNET-BCOR pediatric brain tumors using tumor-specific signature genes. Acta Neuropathol Commun 8:105. https://doi.org/10.1186/s40478-020-00984-9
Boisseau W, Euskirchen P, Mokhtari K, Dehais C, Touat M, Hoang-Xuan K, Sanson M, Capelle L, Nouet A, Karachi C, Bielle F, Guegan J, Marie Y, Martin-Duverneuil N, Taillandier L, Rousseau A, Delattre JY, Idbaih A (2019) Molecular profiling reclassifies adult astroblastoma into known and clinically distinct tumor entities with frequent mitogen-activated protein kinase pathway alterations. Oncologist 24:1584–1592. https://doi.org/10.1634/theoncologist.2019-0223
Coy S, Dubuc AM, Dahiya S, Ligon KL, Vasiljevic A, Santagata S (2017) Nuclear CRX and FOXJ1 expression differentiates non-germ cell pineal region tumors and supports the ependymal differentiation of papillary tumor of the pineal region. Am J Surg Pathol 41:1410–1421. https://doi.org/10.1097/PAS.0000000000000903
Rogers HA, Kilday JP, Mayne C, Ward J, Adamowicz-Brice M, Schwalbe EC, Clifford SC, Coyle B, Grundy RG (2012) Supratentorial and spinal pediatric ependymomas display a hypermethylated phenotype which includes the loss of tumor suppressor genes involved in the control of cell growth and death. Acta Neuropathol 123:711–725. https://doi.org/10.1007/s00401-011-0904-1
Allen DE, Donohue KC, Cadwell CR, Shin D, Keefe MG, Sohal VS, Nowakowski TJ (2022) Fate mapping of neural stem cell niches reveals distinct origins of human cortical astrocytes. Science 376:1441–1446. https://doi.org/10.1126/science.abm5224
Lucas CG, Gupta R, Wu J, Shah K, Ravindranathan A, Barreto J, Gener M, Ginn KF, Prall OWJ, Xu H, Kee D, Ko HS, Yaqoob N, Zia N, Florez A, Cha S, Perry A, Clarke JL, Chang SM, Berger MS, Solomon DA (2022) EWSR1-BEND2 fusion defines an epigenetically distinct subtype of astroblastoma. Acta Neuropathol 143:109–113. https://doi.org/10.1007/s00401-021-02388-y
Smith-Cohn M, Abdullaev Z, Aldape K, Quezado M, Rosenblum M, Vanderbilt C, Rodriguez F, Laterra J, Eberhart C (2021) Molecular clarification of brainstem astroblastoma with EWSR1-BEND2 fusion in a 38-year-old man. Free Neuropathol. https://doi.org/10.17879/freeneuropathology-2021-3334
Tsutsui T, Arakawa Y, Makino Y, Kataoka H, Mineharu Y, Naito K, Minamiguchi S, Hirose T, Nobusawa S, Nakano Y, Ichimura K, Haga H, Miyamoto S (2021) Spinal cord astroblastoma with EWSR1-BEND2 fusion classified as HGNET-MN1 by methylation classification: a case report. Brain Tumor Pathol 38:283–289. https://doi.org/10.1007/s10014-021-00412-3
Yamasaki K, Nakano Y, Nobusawa S, Okuhiro Y, Fukushima H, Inoue T, Murakami C, Hirato J, Kunihiro N, Matsusaka Y, Honda-Kitahara M, Ozawa T, Shiraishi K, Kohno T, Ichimura K, Hara J (2020) Spinal cord astroblastoma with an EWSR1-BEND2 fusion classified as a high-grade neuroepithelial tumour with MN1 alteration. Neuropathol Appl Neurobiol 46:190–193. https://doi.org/10.1111/nan.12593
Ramkissoon SH, Bandopadhayay P, Hwang J, Ramkissoon LA, Greenwald NF, Schumacher SE, O’Rourke R, Pinches N, Ho P, Malkin H, Sinai C, Filbin M, Plant A, Bi WL, Chang MS, Yang E, Wright KD, Manley PE, Ducar M, Alexandrescu S, Lidov H, Delalle I, Goumnerova LC, Church AJ, Janeway KA, Harris MH, MacConaill LE, Folkerth RD, Lindeman NI, Stiles CD, Kieran MW, Ligon AH, Santagata S, Dubuc AM, Chi SN, Beroukhim R, Ligon KL (2017) Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: precision medicine analysis of 203 pediatric brain tumors. Neuro Oncol 19:986–996. https://doi.org/10.1093/neuonc/now294
Lubieniecki F, Vazquez V, Lamas GS, Camarero S, Nuñez FJ, Baroni L, Schüller U, Alderete D (2022) The spectrum of morphological findings in pediatric central nervous system MN1-fusionpositive neuroepithelial tumors. Childs Nerv Syst. https://doi.org/10.1007/s00381-022-05741-y
Gopakumar S, McDonald MF, Sharma H, Tatsui CE, Fuller GN, Rao G (2022) Recurrent HGNET-MN1 altered (astroblastoma MN1-altered) of the foramen magnum: case report and molecular classification. Surg Neurol Int 13:139. https://doi.org/10.25259/SNI_1208_2021
Shin SA, Ahn B, Kim SK, Kang HJ, Nobusawa S, Komori T, Park SH (2018) Brainstem astroblastoma with MN1 translocation. Neuropathology 38:631–637. https://doi.org/10.1111/neup.12514
Rossi S, Barresi S, Colafati GS, Giovannoni I, Miele E, Alesi V, Cacchione A, Diomedi-Camassei F, Macari G, Antonelli M, Carboni A, Carai A, Mastronuzzi A, Giangaspero F, Gessi M, Alaggio R (2022) Paediatric astroblastoma-like neuroepithelial tumour of the spinal cord with a MAMLD1-BEND2 rearrangement. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12814
Fukami M, Wada Y, Okada M, Kato F, Katsumata N, Baba T, Morohashi K, Laporte J, Kitagawa M, Ogata T (2008) Mastermind-like domain-containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J Biol Chem 283:5525–5532. https://doi.org/10.1074/jbc.M703289200
Pajtler KW, Wei Y, Okonechnikov K, Silva PBG, Vouri M, Zhang L, Brabetz S, Sieber L, Gulley M, Mauermann M, Wedig T, Mack N, Imamura Kawasawa Y, Sharma T, Zuckermann M, Andreiuolo F, Holland E, Maass K, Korkel-Qu H, Liu HK, Sahm F, Capper D, Bunt J, Richards LJ, Jones DTW, Korshunov A, Chavez L, Lichter P, Hoshino M, Pfister SM, Kool M, Li W, Kawauchi D (2019) YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat Commun 10:3914. https://doi.org/10.1038/s41467-019-11884-5
Ayaz G, Razizadeh N, Yasar P, Kars G, Kahraman DC, Saatci O, Sahin O, Cetin-Atalay R, Muyan M (2020) CXXC5 as an unmethylated CpG dinucleotide binding protein contributes to estrogen-mediated cellular proliferation. Sci Rep 10:5971. https://doi.org/10.1038/s41598-020-62912-0
Lake JA, Donson AM, Prince E, Davies KD, Nellan A, Green AL, Mulcahy Levy J, Dorris K, Vibhakar R, Hankinson TC, Foreman NK, Ewalt MD, Kleinschmidt-DeMasters BK, Hoffman LM, Gilani A (2020) Targeted fusion analysis can aid in the classification and treatment of pediatric glioma, ependymoma, and glioneuronal tumors. Pediatr Blood Cancer. https://doi.org/10.1002/pbc.28028
Wallace GC, Macaulay RJB, Etame AB, Aldape K, Pina Y (2022) Histopathologically atypical astroblastoma with MN1-CXXC5 fusion transcript diagnosed by methylation classifier. Arch Community Med Public Health 8:113–117. https://doi.org/10.17352/2455-5479.000185
Ferris SP, Velazquez Vega J, Aboian M, Lee JC, Van Ziffle J, Onodera C, Grenert JP, Saunders T, Chen YY, Banerjee A, Kline CN, Gupta N, Raffel C, Samuel D, Ruiz-Diaz I, Magaki S, Wilson D, Neltner J, Al-Hajri Z, Phillips JJ, Pekmezci M, Bollen AW, Tihan T, Schniederjan M, Cha S, Perry A, Solomon DA (2020) High-grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication-a comprehensive clinical, radiographic, pathologic, and genomic analysis. Brain Pathol 30:46–62. https://doi.org/10.1111/bpa.12747
Huynh KD, Fischle W, Verdin E, Bardwell VJ (2000) BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev 14:1810–1823
Fan Z, Yamaza T, Lee JS, Yu J, Wang S, Fan G, Shi S, Wang CY (2009) BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol 11:1002–1009. https://doi.org/10.1038/ncb1913
Lehman NL (2014) Rosettes and pseudorosettes. In: Aminoff MJ, Daroff RB (eds) Encyclopedia of the neurological sciences, 2nd edn. Academic Press, Oxford, pp 69–73
Tauziede-Espariat A, Pages M, Roux A, Siegfried A, Uro-Coste E, Nicaise Y, Sevely A, Gambart M, Boetto S, Dupuy M, Richard P, Perbet R, Vinchon M, Caron S, Andreiuolo F, Gareton A, Lechapt E, Chretien F, Puget S, Grill J, Boddaert N, Varlet P, Renoclip LOC (2019) Pediatric methylation class HGNET-MN1: unresolved issues with terminology and grading. Acta Neuropathol Commun 7:176. https://doi.org/10.1186/s40478-019-0834-z
Alhalabi KT, Stichel D, Sievers P, Peterziel H, Sommerkamp AC, Sturm D, Wittmann A, Sill M, Jager N, Beck P, Pajtler KW, Snuderl M, Jour G, Delorenzo M, Martin AM, Levy A, Dalvi N, Hansford JR, Gottardo NG, Uro-Coste E, Maurage CA, Godfraind C, Vandenbos F, Pietsch T, Kramm C, Filippidou M, Kattamis A, Jones C, Ora I, Mikkelsen TS, Zapotocky M, Sumerauer D, Scheie D, McCabe M, Wesseling P, Tops BBJ, Kranendonk MEG, Karajannis MA, Bouvier N, Papaemmanuil E, Dohmen H, Acker T, von Hoff K, Schmid S, Miele E, Filipski K, Kitanovski L, Krskova L, Gojo J, Haberler C, Alvaro F, Ecker J, Selt F, Milde T, Witt O, Oehme I, Kool M, von Deimling A, Korshunov A, Pfister SM, Sahm F, Jones DTW (2021) PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol 142:841–857. https://doi.org/10.1007/s00401-021-02354-8
Chadda KR, Holland K, Scoffings D, Dean A, Pickles JC, Behjati S, Jacques TS, Trotman J, Tarpey P, Allinson K, Murray MJ, Genomics England Research C (2021) A rare case of paediatric astroblastoma with concomitant MN1-GTSE1 and EWSR1-PATZ1 gene fusions altering management. Neuropathol Appl Neurobiol 47:882–888. https://doi.org/10.1111/nan.12701
Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, Tang B, Haupfear K, Punchihewa C, Easton J, Mulder H, Boggs K, Shao Y, Rusch M, Becksfort J, Gupta P, Wang S, Lee RP, Brat D, Peter Collins V, Dahiya S, George D, Konomos W, Kurian KM, McFadden K, Serafini LN, Nickols H, Perry A, Shurtleff S, Gajjar A, Boop FA, Klimo PD Jr, Mardis ER, Wilson RK, Baker SJ, Zhang J, Wu G, Downing JR, Tatevossian RG, Ellison DW (2016) Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131:833–845. https://doi.org/10.1007/s00401-016-1539-z
Chen W, Soon YY, Pratiseyo PD, Sutanto R, Hendriansyah L, Kuick CH, Chang KTE, Tan CL (2020) Central nervous system neuroepithelial tumors with MN1-alteration: an individual patient data meta-analysis of 73 cases. Brain Tumor Pathol 37:145–153. https://doi.org/10.1007/s10014-020-00372-0
Baroni LV, Rugilo C, Lubieniecki F, Sampor C, Freytes C, Nobre L, Hansford JR, Malalasekera VS, Zapotocky M, Dodgshun A, Martinez OC, La Madrid AM, Lavarino C, Sunol M, Rutkowski S, Schuller U, Bouffet E, Ramaswamy V, Alderete D (2020) Treatment response of CNS high-grade neuroepithelial tumors with MN1 alteration. Pediatr Blood Cancer. https://doi.org/10.1002/pbc.28627
Hirose T, Nobusawa S, Sugiyama K, Amatya VJ, Fujimoto N, Sasaki A, Mikami Y, Kakita A, Tanaka S, Yokoo H (2018) Astroblastoma: a distinct tumor entity characterized by alterations of the X chromosome and MN1 rearrangement. Brain Pathol 28:684–694. https://doi.org/10.1111/bpa.12565
Pages M, Pajtler KW, Puget S, Castel D, Boddaert N, Tauziede-Espariat A, Picot S, Debily MA, Kool M, Capper D, Sainte-Rose C, Chretien F, Pfister SM, Pietsch T, Grill J, Varlet P, Andreiuolo F (2019) Diagnostics of pediatric supratentorial RELA ependymomas: integration of information from histopathology, genetics, DNA methylation and imaging. Brain Pathol 29:325–335. https://doi.org/10.1111/bpa.12664
Zschernack V, Junger ST, Mynarek M, Rutkowski S, Garre ML, Ebinger M, Neu M, Faber J, Erdlenbruch B, Claviez A, Bielack S, Brozou T, Fruhwald MC, Dorner E, Dreschmann V, Stock A, Solymosi L, Hench J, Frank S, Vokuhl C, Waha A, Andreiuolo F, Pietsch T (2021) Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol 141:455–466. https://doi.org/10.1007/s00401-020-02260-5
Hammas N, Senhaji N, Alaoui Lamrani MY, Bennis S, Chaoui EM, El Fatemi H, Chbani L (2018) Astroblastoma—a rare and challenging tumor: a case report and review of the literature. J Med Case Rep 12:102. https://doi.org/10.1186/s13256-018-1623-1
Lillard JC, Venable GT, Khan NR, Tatevossian RG, Dalton J, Vaughn BN, Klimo P (2019) Pediatric supratentorial ependymoma: surgical, clinical, and molecular analysis. Neurosurgery 85:41–49. https://doi.org/10.1093/neuros/nyy239
Andreiuolo F, Puget S, Peyre M, Dantas-Barbosa C, Boddaert N, Philippe C, Mauguen A, Grill J, Varlet P (2010) Neuronal differentiation distinguishes supratentorial and infratentorial childhood ependymomas. Neuro Oncol 12:1126–1134. https://doi.org/10.1093/neuonc/noq074
Fukuoka K, Kanemura Y, Shofuda T, Fukushima S, Yamashita S, Narushima D, Kato M, Honda-Kitahara M, Ichikawa H, Kohno T, Sasaki A, Hirato J, Hirose T, Komori T, Satomi K, Yoshida A, Yamasaki K, Nakano Y, Takada A, Nakamura T, Takami H, Matsushita Y, Suzuki T, Nakamura H, Makino K, Sonoda Y, Saito R, Tominaga T, Matsusaka Y, Kobayashi K, Nagane M, Furuta T, Nakada M, Narita Y, Hirose Y, Ohba S, Wada A, Shimizu K, Kurozumi K, Date I, Fukai J, Miyairi Y, Kagawa N, Kawamura A, Yoshida M, Nishida N, Wataya T, Yamaoka M, Tsuyuguchi N, Uda T, Takahashi M, Nakano Y, Akai T, Izumoto S, Nonaka M, Yoshifuji K, Kodama Y, Mano M, Ozawa T, Ramaswamy V, Taylor MD, Ushijima T, Shibui S, Yamasaki M, Arai H, Sakamoto H, Nishikawa R, Ichimura K, Japan Pediatric Molecular Neuro-Oncology G (2018) Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6:134. https://doi.org/10.1186/s40478-018-0630-1
Djirackor L, Halldorsson S, Niehusmann P, Leske H, Capper D, Kuschel LP, Pahnke J, Due-Tonnessen BJ, Langmoen IA, Sandberg CJ, Euskirchen P, Vik-Mo EO (2021) Intraoperative DNA methylation classification of brain tumors impacts neurosurgical strategy. Neurooncol Adv. https://doi.org/10.1093/noajnl/vdab149
Merchant TE, Bendel AE, Sabin ND, Burger PC, Shaw DW, Chang E, Wu S, Zhou T, Eisenstat DD, Foreman NK, Fuller CE, Anderson ET, Hukin J, Lau CC, Pollack IF, Laningham FH, Lustig RH, Armstrong FD, Handler MH, Williams-Hughes C, Kessel S, Kocak M, Ellison DW, Ramaswamy V (2019) Conformal Radiation therapy for pediatric ependymoma, chemotherapy for incompletely resected ependymoma, and observation for completely resected, supratentorial ependymoma. J Clin Oncol 37:974–983. https://doi.org/10.1200/JCO.18.01765
Yamada SM, Tomita Y, Shibui S, Takahashi M, Kawamoto M, Nobusawa S, Hirato J (2018) Primary spinal cord astroblastoma: case report. J Neurosurg Spine 28:642–646. https://doi.org/10.3171/2017.9.SPINE161302
Perry A (2020) The “neuroepithelial tumor”: exchanging our trash can for an industrial size dumpster? Free Neuropathol 1:2. https://doi.org/10.17879/fnp-2020-2611
Iliakis G, Murmann T, Soni A (2015) Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen 793:166–175. https://doi.org/10.1016/j.mrgentox.2015.07.001
Voronina N, Wong JKL, Hubschmann D, Hlevnjak M, Uhrig S, Heilig CE, Horak P, Kreutzfeldt S, Mock A, Stenzinger A, Hutter B, Frohlich M, Brors B, Jahn A, Klink B, Gieldon L, Sieverling L, Feuerbach L, Chudasama P, Beck K, Kroiss M, Heining C, Mohrmann L, Fischer A, Schrock E, Glimm H, Zapatka M, Lichter P, Frohling S, Ernst A (2020) The landscape of chromothripsis across adult cancer types. Nat Commun 11:2320. https://doi.org/10.1038/s41467-020-16134-7
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 24:88–91. https://doi.org/10.1038/71750
Tang L, Nogales E, Ciferri C (2010) Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog Biophys Mol Biol 102:122–128. https://doi.org/10.1016/j.pbiomolbio.2010.05.001
Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483. https://doi.org/10.1038/nature10866
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, Cancer Genome Atlas Research N (2010) Identification of a CpG Island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. https://doi.org/10.1016/j.ccr.2010.03.017
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30. https://doi.org/10.1016/j.ccr.2010.12.014
Kinney SR, Pradhan S (2011) Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Prog Mol Biol Transl Sci 101:311–333. https://doi.org/10.1016/B978-0-12-387685-0.00009-3
Robertson AK, Geiman TM, Sankpal UT, Hager GL, Robertson KD (2004) Effects of chromatin structure on the enzymatic and DNA binding functions of DNA methyltransferases DNMT1 and Dnmt3a in vitro. Biochem Biophys Res Commun 322:110–118. https://doi.org/10.1016/j.bbrc.2004.07.083
Acknowledgements
The author thanks Dr. Daniel Mais of the University of Louisville and Dr. Zied Abdullaev of the National Cancer Institute for helpful comments.
Author information
Authors and Affiliations
Contributions
NL Lehman wrote the manuscript and prepared the figures.
Corresponding author
Ethics declarations
Conflict of interest
The author has nothing to disclose.
Data availability
All data is contained within the article and cited references.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lehman, N.L. Early ependymal tumor with MN1-BEND2 fusion: a mostly cerebral tumor of female children with a good prognosis that is distinct from classical astroblastoma. J Neurooncol 161, 425–439 (2023). https://doi.org/10.1007/s11060-022-04222-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-022-04222-1