
Abstract
Carbonate apatite (CA) is a synthetic derivative of hydroxyapatite, which we have been exploring as a drug delivery nanocarrier in the context of cancer in vitro and in vivo. This nanocarrier showed great potential delivering anti-cancer drugs, plasmids containing tumour suppressor genes and siRNAs against oncogenes in pre-clinical models. We compared here two formulations of CA—the low-Ca2+ CA (made with 4 mM Ca2+) used for in vitro studies in cell lines and the high-Ca2+ CA (made with 40 mM Ca2+) used in mouse models—in terms of protein corona formed with different concentrations of serum in vivo and in vitro. The 10-fold more Ca2+ in high-Ca2+ CA helped produce enough particles in an injectable volume for in vivo delivery of therapeutics. Both formulations made particles of similar size, but their composition differed slightly in terms of Na and Mg content. In serum-containing media, the size of the particles was dependent on the serum concentration. The protein corona around both formulations was almost similar and included albumin, fetuin, haemoglobin, and immunoglobulins. CA was not cytotoxic, and instead an increased expression of ribosomal machinery and glycolytic and cytoskeletal proteins was observed, which promoted translation, growth, and proliferation in cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nanoparticles (NPs) are nanosized structures of 1–100 nm diameter with a high surface area. The ability of NPs to carry cargoes of small molecules to target destinations has made them a popular field of exploration in physical science and medicine in recent years [1]. Along with drug delivery, many recently developed NPs showed promising capabilities of self-reporting and tracking through imaging [2,3,4]. NPs can be lipid-based, carbon-based, polymeric, biomimetic, or inorganic [5]. Therapeutic usage of inorganic NPs is quite challenging as it not only requires functionalisation of NPs with proteins, antibodies, DNA, small molecule drugs but also demands stability and safety in vivo, and toxicity, bioaccumulation and biodistribution of these NPs are still issues to be addressed successfully.
When administered to the bloodstream, serum proteins coat the NPs within minutes. The composition of this coating, which does not substantially change over time, may cause changes to the physicochemical properties of the NPs and contribute to their biodistribution, targeting efficiency, uptake by the immune cells, and clearance from the body [6]. Most of the in vitro studies use a lower serum concentration of about 10%, which does not reflect the condition in vivo [7]. Serum concentration may affect the quality and quantity of the protein corona formed around the particles [7]. A study on the impact of 10% and 100% serum concentrations on pure silica and mesoporous silica nanoparticles (MSNs) showed little impact of the surface chemistry of NPs and incubation time on protein corona compared to that imposed by serum concentration. Adsorption of opsonins such as apolipoproteins increases with serum concentration, which increases the clearance of NPs by the macrophages [7]. Generally, the NP-serum interaction studies use mouse or bovine serum, which is important to take note of as the composition of serum varies among species. A study on polystyrene NPs showed lesser clearance of human serum-coated NPs by macrophages compared to that for the same NP coated with mouse serum [8].
In our lab, we developed an inorganic nanocarrier of carbonate apatite (CA). Until now, CA has successfully delivered anti-cancer drugs, doxorubicin [9] and cyclophosphamide [10], siRNAs against growth factor receptors, anti-apoptotic proteins and cell adhesion molecules [11, 12], and plasmids (p21, p53, and PTEN) [13, 14] to mouse models of colon and breast cancers. Modified CA was also tested for therapeutic delivery to cancer models [15, 16]. A version of CA modified with citrate showed great potential as an imaging agent in an ex-vivo study on human tissue [17].
Interaction of CA, whether in its original or modified form, with serum is very important for its function. In this study, for in vitro experiments, serum is added during formulation, while for in vivo experiments, the CA comes into contact with serum proteins after intravenous administration into the animal model (mouse). Serum proteins spontaneously coat the CA particles and determine the fate of the particles in terms of biodistribution, clearance, and uptake by the tissues. We previously reported the interaction between CA and serum proteins [18]. In this paper, we investigated the impact of serum concentration on CA to extrapolate the protein corona formed in vivo. We also discussed how CA impacts the cellular proteome, with calcium as a major constituent of it.
Methods and materials
Reagent
Dulbecco’s modified Eagle’s medium (DMEM), calcium chloride dihydrate (CaCl2.2H2O), sodium bicarbonate (NaHCO3), dimethyl sulfoxide (DMSO), thiazolyl blue tetrazolium bromide (MTT), tris(hydroxymethyl)aminomethane, EDTA, EGTA, beta-glycerophosphate, phosphatase inhibitor, and EDTA-free protease inhibitor were purchased from Sigma-Aldrich (St Louis, MO, USA). DMEM powder, foetal bovine serum (FBS), trypsin-ethylenediaminetetraacetate (trypsin-EDTA) and penicillin-streptomycin were obtained from Gibco BRL (California, USA). MS Grade Pierce trypsin protease was obtained from Thermo Fisher Scientific (MA, USA). Fluorescence siRNA was purchased from QIAGEN. 4T1, MCF-7, and MDA-MB-231 cells were obtained from ATCC. Six to eight-weeks-old balb/c female mice were bred and maintained at the Animal Facility Unit at Monash University (Ethics no. MARP/2016/126 A).
CA formulation
CA was prepared in DMEM media. 100 ml of DMEM was prepared using 1.35 g of DMEM powder and 0.37 g of sodium bicarbonate [18]. The pH was adjusted to 7.4. Low-Ca2+ CA was prepared with 4 μl of 1 M CaCl2.2H2O (final concentration 4 mm) in 1 ml of DMEM. High-Ca2+ CA was prepared with 40 mM Ca2+ in 100 μl DMEM. The mixture of CaCl2.2H2O and DMEM was incubated at 37 °C for 30 min. After incubation, serum was added to each microcentrifuge tube (concentration mentioned with the respective experiments), except for high-Ca2+ CA prepared to inject into mice. 50% serum was used for protein corona analysis of high-Ca2+ CA, which is described in detail in the “Sample preparation for protein corona (PC) analysis” section.
Microscopic observation of cellular uptake of CA
100,000 MCF-7 cells were seeded per well in a 24-well plate. On the following day, media was replaced with 1 nM fluorescence siRNA-loaded CA or 1 nM free siRNA in DMEM, with or without FBS. After 4 h of incubation at 37 °C, media was aspirated from each well to remove free siRNA or CA-siRNA, which were not taken up by the cells. Each well was then washed with 5 mM EDTA in PBS to remove all CA particles that were not taken up by the cells, followed by washes with only PBS. Images were taken before and after wash using a motorised inverted research microscope (Olympus IX81, Tokyo, Japan).
SEM and EDX
Low-Ca2+ and high-Ca2+ CA were prepared as stated in the “CA formulation” section. Without adding serum, the samples were centrifuged at 13,000 rpm for 15 minutes. The supernatant was discarded, and precipitated particles were washed with 1 ml ultrapure water at the same condition. The supernatant was aspirated carefully, leaving a few microliters of water to dissolve the pellet. 3 μl CA sample was put on a cover slip and let dry at 37 °C for 1 h. The dried sample was subjected to platinum sputtering for 45 s at a current of 30 mA, and a tooling factor of 2.3. The particles were visualised at 10 kV for EDX and 5 kV for SEM. The device used was a Hitachi SU8010 (Tokyo, Japan) [18].
Size measurement by Zeta sizer
CA NPs were prepared as stated above; dilutions and serum concentrations were mentioned in the result section with each experiment. Malvern Nano Zetasizer (Worcestershire, UK) and accompanying software were used. Samples were prepared and measured in duplicates, and an average value (± SD) was calculated.
Cell viability assay
50,000 4T1 and MDA-MB-231 cells per well were seeded in a 24-well plate and maintained at 37 °C in a CO2 incubator. The next day, cells were treated with low-Ca2+ CA (prepared with 4 mM of Ca2+) or only DMEM (as a control). After 48 h, MTT was added to each well and incubated for 4 h, followed by the addition of DMSO to dissolve the purple formazan crystals. The plates were read at 595 nm with a reference at 630 nm. Cell viability in the treatment group was calculated as a ratio of that in the control group.
Liquid chromatography with tandem mass spectrometry (LCMS/MS) for identification of protein corona (PC) on CA and differential protein expression in CA treated cells
Sample preparation for protein corona (PC) analysis
Respectively, 1 ml and 100 μl of low-Ca2+ and high-Ca2+ CA were prepared. 10% FBS was added to the low-Ca2+ CA. To mimic the in vivo condition of a mouse model, 100 μl of high-Ca2+ was mixed with 1.5 ml of 50% FBS containing DMEM. The protein-bound CA was precipitated through centrifugation at 13,200 rpm for 15 min, followed by two washes with ultrapure water at the same condition. The precipitate was dissolved in 100 μl of 50 mM EDTA. The whole volume of the sample was used for PC analysis.
Sample preparation for differential protein expression
50,000 4T1 cells were seeded per well in a 24-well plate, followed by their treatment with low-Ca2+ CA the next day. A control group was maintained with media only. Cells were maintained in a humidified incubator at 37 °C and 5% CO2 for 48 h. Cells were lysed in 100 μl lysis buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100 (Bio-Rad, CA, USA), 1 mM EDTA, 1 mM EGTA, 50 mM beta-glycerophosphate, phosphatase inhibitor cocktail 2 (Sigma, St Louis, MO, USA), 50 nM NaF, and complete EDTA-free protease inhibitor (Sigma, St Louis, MO, USA). Protein concentrations in cell lysates were determined using the Quick Start Bradford Protein Assay kit (Bio-Rad, CA, USA). From each sample, 30 μg of protein was processed for further analysis.
Tryptic digestion of protein
Both CA samples for PC analysis and cellular protein samples for differential protein expression were processed in the same way. All samples were desalted with a C18 spin column (Thermo Fisher Scientific). Solvents were evaporated in a centrifugal evaporator (EYELA Centrifugal Evaporator CVE 3110). Proteins were subjected to trypsin digestion and prepared for Q-TOF mass spectrometry according to the Agilent in-solution tryptic digestion procedure. Briefly, the protein pellet was resuspended in 100 μl of 0.1% formic acid, followed by reduction with ammonium bicarbonate, TFE (2,2,2-trifluoroethanol), and DTT (dithiothreitol). The reduced protein was alkylated by incubating it with iodoacetamide (IAM) in the dark. Excess IAM was neutralised with DTT, followed by raising pH to 7–9 with ammonium bicarbonate. At this stage, protein was digested with trypsin (1 μg of enzyme for 30 μg of protein) at 37 °C for 18 h. After digestion, enzyme activity was stopped with TFA. The samples were dried in a vacuum concentrator and stored at -20 °C until performing Q-TOF mass spectrometry.
Sample preparation for Q-TOF mass spectrometry
Dry protein digest was dissolved in 30 μl of 0.1% formic acid in water with a brief vortex and sonication. Afterwards, the samples were centrifuged at 13,200 g for 10 min. 1 μl of supernatant was injected into an MS tube and placed on an autosampler for the LC-QTOF analysis.
Nanoflow liquid chromatography electrospray-ionisation coupled with LC-MS/MS
The digested peptides were loaded into an Agilent C18 300 Å large-capacity chip column that was equilibrated with 0.1% formic acid in water (solution A). The peptides were eluted from the column with 90% acetonitrile in water with 0.1% formic acid (solution B) using gradients of 5% solution B over 0–45 min and 70% solution B over 45–55 min. Q-TOF polarity was set to positive, with capillary and fragmentor voltages set at 1900 V and 360 V, respectively, and 5 l/min of gas flow at a temperature of 325 °C. The peptide spectrum was analysed in auto MS mode, ranging from 110–3000 m/z for the MS scan and 50–3000 m/z for the MS/MS scan. Acquisition rates were 2 (spectra/s) for MS and 4 (spectra/s) for MS/MS. The spectrum was then analysed with Agilent MassHunter (Agilent Technologies) data acquisition software and PEAKS 8.0 software (Bioinformatics Solutions Inc., Waterloo, ON, Canada).
Protein identification by automated de novo sequencing (PEAKS Studio 8.0)
Protein identification was performed with PEAKS Studio 8.0 (Bioinformatics Solution Inc.). The Uniprot Mus musculus (Mouse) database (April 2018) was used for protein identification and homology search by comparing the de novo sequence tag. Carbamidomethylation was set as the fixed modification, with maximum mixed cleavages at 3. Parent mass and fragment mass error tolerance were both set to 0.1 Da with monoisotopic mass as the precursor mass search type. Trypsin was selected as the enzyme used for digestion. A false discovery rate of 1% and unique peptides 1 were used for filtering out inaccurate proteins. Only proteins showing high confidence levels (-10lgP > 20) in PEAKS were chosen, as it targets very few decoy matches above that threshold.
Tumour progression analysis in mouse model
Tumour was induced with 4T1 cells in the mammary fat pad of 6–8-week-old balb/c female mice on day 0. When tumour volume reached 13.5 mm3 after about a week, 100 μl CA or normal saline were administered via the tail vein. The second dose was given after two days. Tumour volume was measured on alternate days until day 22, when mice were humanely euthanised and tumours were harvested and measured.
Results
Cellular uptake of CA with and without serum: microscopic visualisation
After 4 h of incubation of cells with free siRNA in serum-containing or serum-free DMEM, fluorescence was not visible in the wells (Fig. 1, rows 1 and 2, before-wash images). This could be due to the degradation of free siRNA by nucleases in the media. However, after removal of the media, a faint signal was noticed from the cells treated with siRNA in serum-containing media (Fig. 1, row 1, after-wash image), while in the case of siRNA in serum-free media, the signal was hardly detectable (Fig. 1, row 2, after-wash image). From the images presented, it was clear that FBS facilitates the cellular entry of free siRNA. In the case of CA-siRNA (low-Ca2+ CA), serum reduced the aggregation of particles (Fig. 1, rows 3 and 4, before-wash images) by coating them with proteins, which is described in later sections. Serum also facilitates CA entry into the cells, which is clear from the after-wash images in rows 3 and 4.

Cellular uptake of free or CA-bound fluorescence siRNA or CA-siRNA (AllStars Neg. siRNA Alexa Fluor 488). 1 × 105 MCF-7 cells were seeded in a 24-well plate. On the following day, the cells were treated with free or CA-bound siRNA, with or without FBS. After 4 hours, images were taken and presented in columns 1 and 2. Then each well was washed with 5 mM EDTA in PBS, submerged in 100 µl PBS, and another set of images was taken (column 3 and 4). Scale was 50 µm
High Ca2+ and low Ca2+ CA: SEM and EDX
As mentioned before, in our lab, we use two slightly different formulations of CA for in vitro and in vivo usage, for which the molecular structure of apatite was confirmed by FTIR in previous studies [18]. The reaction conditions were the same for both formulations, except for the concentration of exogenously added Ca2+ (Table 1). For in vitro experiments, we prepared low-Ca2+ CA in 1 ml DMEM with 4 mM of Ca2+, while for systemic delivery in a mouse model, high-Ca2+ CA was prepared in only 100 μl DMEM with 40 mM of Ca2+. We used scanning electron microscopy (SEM) and energy dispersive X-ray analysis (EDX) to explore how this variation impacted the size and composition of these two CA formulations.
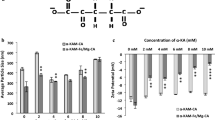
The SEM images showed that low-Ca2+ CA had a particle population of varying sizes between a range of 100 and 600 nm, with a high number of particles of diameter around 200 nm. The high-Ca2+ CA had mostly particles of diameter 200 nm, with a few large particles of 450-700 nm. The surface looked rougher for the low-Ca2+ CA (Fig. 2). Though the high-Ca2+ CA had 10 times higher Ca2+ in the reaction milieu, the similar sizes of both NPs indicated that particle formation was controlled by the low yet fixed concentration of PO43−(Table 1).

SEM images of low-Ca2+ and high-Ca2+ formulations of CA. Bar diagrams show particle size distributions from the SEM images. A. CA: low-Ca2+ formulation (4 mM exogenous Ca2+); B. high-Ca2+ formulation (40 mM exogenous Ca2+)
The EDX analysis of both formulations exhibited an almost similar proportion of Ca, C, P, and Cl. The low-Ca2+ CA contained a higher amount of O and Mg; however, Na content was higher in the high-Ca2+ CA (listed in Table 2). The Ca/P ratio was 2.2–2.4 and 1.9–2 for low-Ca2+ and high-Ca2+ formulations, respectively. As we confirmed the structure of CA with FTIR previously, we attribute the deviation of the Ca/P ratio from that for pure apatite (Ca/P ratio 1.67) [19] to the presence of many organic molecules coming from DMEM.
Effect of serum on particle size
We observed aggregation and precipitation of CA in serum-free media, while in serum-containing media, the particles looked quite homogenously dispersed under a microscope. We assumed that proteins in serum stabilised the particles and prevented further particle formation and their aggregation. In accordance with our speculation, particles’ size, measured via the DLS method by the particle size analyser, zetasizer, reduced with increasing concentration of serum in the media. For the same formulation of CA (low-Ca2+ CA made with 4 μl Ca2+ in 1 ml DMEM), a size of 1249 nm was recorded in serum-free media, which gradually decreased to only 24 nm in 20% serum-containing media (Fig. 3).

Effect of serum concentration on particle size. 5 sets of CA were prepared with 4 mM of exogenous Ca2+, followed by the addition of different concentrations of serum. Particle size was measured with a zetasizer. Experiments were performed in duplicates, and all data was significant at a p value of 0.05
From the above experiment, we deduced that serum concentration had a role in particle size. In an attempt to explore if the volume of diluent also mattered, we prepared a stock of high-Ca2+ CA suspension with 40 mM of Ca2+ in 1 ml of DMEM and did not add serum after incubation to stop the reaction. Immediately afterwards, 50, 100, or 200 μl of NP suspension were taken out of the stock and topped up to 1 ml with serum-free or serum-containing DMEM, which resulted in 20-fold, 10-fold, and 5-fold dilutions, respectively (illustrated in Fig. 4.A).

Effect of media volume or dilution on particle size. A. experiment design. B. Change in particle size upon different dilutions of CA in serum-free and serum-containing DMEM. Each measurement was done in duplicate, and all data was significant at a p value 0.05
In this experiment, serum-free DMEM was used as a control diluent to compare with DMEM with various concentrations of serum. For the controls, the 2 μm size of high-Ca2+ CA interestingly increased with 5- and 10-fold dilutions while decreasing slightly with a 20-fold dilution (Fig. 4.B). We assume this was a random behaviour by the particles, as there was no uniform pattern and CA NPs are prone to interact among themselves in the absence of serum, giving rise to particles of various sizes. However, when the same dilutions were performed in 10% serum containing DMEM, the size of particles decreased from 2 μm to < 500 nm (Fig. 4.B). However, there was no statistically significant difference among the sizes in three dilutions of 10% serum containing DMEM. We can conclude that it’s the concentration of serum that controls particle size, not the volume of media or diluent.
Variation of protein corona formed around high-Ca2+ and low-Ca2+ CA
The high-Ca2+ CA for in vivo delivery of therapeutics comes into contact with approximately 50% plasma in the bloodstream after intravenous delivery into animals. In this study, the volume of high-Ca2+ CA injected intravenously into a mouse model was 100 μl. The average volume of blood in a 25-g adult mouse is around 1.5 ml, and mouse blood contains 52% of plasma. To mimic this condition, a single dosage of high-Ca2+ CA was prepared and incubated for 30 min in 1.5 ml of 50% mouse plasma containing DMEM. Simultaneously, the low-Ca2+ CA was prepared with 10% mouse plasma. Both CA samples, along with the bound proteins, were precipitated and digested with trypsin by the Agilent in-solution tryptic digestion method, followed by detection of the proteins via LC-MS/MS.
Table 3 lists all the detected proteins in the protein corona (PC) of low-Ca2+ CA and high-Ca2+ CA. Both formulations’ PCs included albumin, protease inhibitors, haemoglobin subunits, complement factor, and immunoglobulin subunits. The high-Ca2+ CA also bound apolipoproteins, while pentaxin, the cytochrome P450 subunit, and a few proteins related to ubiquitin-mediated degradation were found only in the PC of the low-Ca2+ CA. As the most abundant protein in plasma, with a concentration of 35–50 μg/μl, the presence of albumin in the PC was anticipated [20]. Iron-binding proteins may bind to the CA NPs due to their affinity for divalent Ca2+ ions. Compared to the in vitro formulation with only 10% plasma, the in vivo formulation with 50% plasma was exposed to a larger quantity of proteins; however, the PC of both CAs was approximately the same. This could be due to the masking effect of more abundant proteins. The PC of high-Ca2+and low-Ca2+ CA was discussed in detail in the discussion section.
Differential protein expression in untreated and CA-treated cells
Compared to untreated cells, the CA-treated cells expressed higher levels of proteins related to growth processes such as transcription, translation, stress management, glycolysis, movement, and recycling. Specifically, proteins with significant differences between the two groups were transcription factor high mobility group box 1, ribosomal subunits and elongation factors, chaperones (heat shock proteins), proteins facilitating disulfide bond formation for folding, glycolytic enzymes, cytoskeletal proteins, albumin, annexin, and associated EF-hand proteins, and proteins of the ubiquitin system (Fig. 5).

Heatmap of proteins differentially expressed in untreated and CA-treated 4T1 cell line. Each test was run in triplicate. PEAKS 8.0 software and the Uniprot Mus musculus (Mouse) database were used for protein identification and homology search
Overall, the proteome analysis shows a proliferative role for CA, which is anticipated due to the stimulation of Ca pathways by the Ca2+ ions in CA. Like the in vitro proteome data on cell lines, tumours in CA-treated mice grew similarly to the ones in untreated mice (Figs. 6B and 6C). However, one interesting observation was that in the cell viability assay, treatment with CA showed ~ 20% cell death (compared to untreated cells) (Fig. 6A), even though a higher level of expression of proteins positively associated with proliferation was observed in the crude protein sample collected from the CA-treated cells as stated above. We infer that the cell death occurred at an early stage of treatment when the number of cells was low and an excessive number of CA crystals surrounding them played a toxic role.

A. Viability of 4T1 cells after 48 h of treatment with low-Ca2+ CA, data significant at p value 0.05. B. Breast tumour growth over a 22-day period in untreated and high-Ca2+ CA-treated mice: difference between the two groups was not statistically significant. C. Weight of tumour harvested from the sacrificed mice: difference between the two groups was not statistically significant. D. Level of pyruvate kinase M (PKM) enzyme in CA-treated and CA-PKM2 siRNA-treated 4T1 cells. The relative level of PKM expression was determined with label-free quantification of proteins via LC-MS/MS
As a drug delivery vehicle, CA (low- and high-Ca2+) plays a growth-promoting role, which to some extent undermines its acceptability as a nanocarrier in cancer. To explore how an siRNA works in the context of our nanocarriers’ growth-promoting nature, we delivered siRNA against pyruvate kinase, a glycolytic enzyme upregulated in CA-treated cells. We observed in two breast cancer cell lines, 4T1 and MDA-MB-231, that CA-siRNA reduced PKM expression significantly (Fig. 6D).
Discussion
Microscopic imaging
Microscopic images showed strong fluorescence from CA-siRNA-treated cells, with very little signal from the free siRNA-treated ones. It was also evident that serum facilitated the cellular uptake of free siRNA and CA-siRNA. siRNA’s large size, negative charge, and hydrophilicity negatively impact its diffusion via the plasma membrane. However, once siRNA makes its way into the cell, it can bind to and be protected by the RNA-induced silencing complex (RISC) [21]. Systemic delivery of free siRNA is a challenge, as in systemic circulation, unmodified free siRNA is quickly degraded. Hickerson et al. showed that human serum degrades siRNA partially, leaving it still functional, while bovine serum destroys 70–90% of unmodified siRNA in just 5 h. [22]. Though serum, for the most part, does not support free siRNA in systemic circulation, argonaute 2, a protein of RISC inside cells, was detected in circulation too and reported to carry 90% of extracellular miRNA [23]. Research showed that HDL and LDL also transported foreign miRNAs in vivo [24]. We assume that structural and functional similarities between siRNA and miRNA enabled their binding and transportation to the cells. In fact, an in vivo study on mice showed increased delivery of HDL-conjugated siRNA to hepatic cells [25].
While both CA-siRNA with and without FBS showed noticeably enhanced delivery of siRNA, the cells treated with FBS looked healthier than those treated without FBS. For this experiment, cells were treated with free siRNA or CA-siRNA the day after seeding when the cells were attached to the wells, and the images were taken after 4 h of treatment. At that time, cells were not growing much and frequent washes with EDTA and PBS might cause detachment of some cells. However, as these were the common procedures for the whole experiment, we assume that bigger particles formed by aggregation in serum-free media damaged the cells, whereas CA in serum-containing media was more soluble and thus less toxic to the cells.
SEM and EDX
The inorganic phase of biological apatite was at first identified as being composed of calcium, hydroxide, and phosphate with the proposed formula Ca10(PO4)6OH2. Afterwards, extensive research by LeGeros and his colleagues reported the incorporation of other radicals and elements in hydroxyapatite, with a corrected formula (Ca,Mg,Na,X)10(PO4,CO3,HPO4)6(OH,Cl,F)2, where X stood for other calcium substituents. Varying proportions of the substituents in biological apatites influence the mechanical strength, function, interaction with proteins, and acid solubility of different biological apatites. We infer that it is the proportion of the elements in CA NPs that attributes to its circular shape, pH dependence and other properties, compared to the ribbon or needle-shaped hard apatite in our bone or enamel [19].
The NP fabricated in our lab was confirmed to be CA by the presence of CO32− and PO43− in FT-IR [18]. However, its Ca/P ratio was 1.9–2.4 (Table 2), unlike 1.67 in biological samples, accompanied by a much higher proportion of Mg, Na, and CO32−. For synthetic apatites, the substitution and thereby composition depend on reaction conditions. Usually precipitation at low temperatures causes a B type substitution of CO32− for PO43−[26]. This CO32− for PO43− substitution was reported to be coupled with a Na for Ca substitution [27]. A smaller size and higher solubility of CA may be attributed to a higher amount of CO32− and Mg2+ [19]. Our NP had slightly higher Cl, which could be due to Cl− coming from not only the inorganic salts in media but also the hydrochloride forms of amino acids and vitamins in DMEM.
Between the low-Ca2+ and high-Ca2+ formulations, the latter had a 10-fold higher Ca2+ concentration in the reaction milieu while the rest of the reactants were the same in both. Among the five fields explored during EDX, Ca content varied from 9.4–13.4%, which means Ca incorporation was not uniform in the crystals. Though Ca2+ and CO32− incorporations were not significantly different between low-Ca2+ and high-Ca2+ formulations, P content was higher in the latter. In the high-Ca2+ formulation, Na incorporation was almost doubled, and it replaced a portion of Mg. Simultaneously, O content was 6% less in the high-Ca2+ formulation. We assume it was due to the loss of OH ions to balance the positive charges from the metal ions.
Effect of serum
We noticed that in the absence of serum, particle formation continued even at room temperature, and the already formulated particles remained aggregated. In contrast, serum stopped particle formation by coating them with proteins. This coating of proteins, or PC around the CA particles is a dynamic feature, with particle size changing proportionately with increasing serum concentration (Fig. 3). The role of protein concentration to reduce particle size was also evident from Fig. 4B, where CA NPs came down to similar sizes in 10% serum-containing media regardless of the dilution factor. Figure 4B also showed that once particles were formed, DMEM could not impact their structure; it is only the serum proteins that control the size of CA.
This phenomenon of size reduction is unlike that of other inorganic NPs such as iron oxide and gold NPs, which increase in size in contact with serum proteins [28, 29]. This incident can be explained by CA particles’ structural similarity to bone hydroxyapatite. For bone tissues, demineralization and remineralization are regular processes. Undesired precipitation of bone minerals is prevented by matrix proteins, which keep bones ‘soluble’. It is evident from the protein corona that it is in fact the proteins related to bone metabolism that bind around CA particles and thus impart size reduction [30].
Two facts have been established from this experiment. First, once the particles are formed, the volume of media plays no role in disintegrating it and second, the particle size depends on serum concentration and not on media volume. Based on these findings, we can conclude that the size of CA would be the same in bigger animals as in mice, as long as their plasma/serum composition is similar [31].
Variation in protein corona formed around high-Ca2+ and low-Ca2+ CA
As soon as the NPs are administered into the bloodstream, serum proteins start coating them; however, the types of coating proteins depend on the nanoparticles’ chemical composition, charge, size, and shape. In many cases, protein corona is a dynamic feature that changes with time [28, 32]. As CA NP shares compositional similarity with hydroxyapatite, predictably, we found the protein corona on CA was composed of mostly the same proteins involved in hard tissue metabolism or bone remodelling. In fact, the PC on CA was very similar to the proteins bound to natural calcium phosphate crystals in our body [33]. Primarily, these proteins bind via the electrostatic interaction between carboxylate anions and Ca cations on proteins and NP, respectively [34].
Bone remodelling is a continuous, lifelong process in our body. This process releases ions—mainly calcium and phosphates—in the bloodstream and eventually in extracellular fluids [30, 35]. To maintain their optimum concentration in circulation and simultaneously prevent unwanted ectopic calcification, homeostasis of these ions is tightly regulated by calcitropic hormones and phosphatonins, with bone, kidneys, and intestine—the major organs involved in the undertaking [36, 37]. Several proteins regulated by the two above-mentioned hormones, work as calcification inhibitors, forming soluble colloidal mineral-protein complexes with these ions [30]. Calcification inhibitor proteins are generally acidic and may contain stretches of polyaspartic acid, substantially phosphorylated serine/threonine, or γ-carboxyglutamic acid [38].
We start the discussion on the list of proteins forming PC around CA with albumin. In vitro studies showed that one-half of the mineralisation-inhibitory effect of serum is attributed to fetuins or alpha-2-HS-glycoprotein, with the rest coming from albumin [38, 39]. Albumin is the most abundant serum protein, constituting 50% of total protein content [40]. A few interesting features of this carrier protein make it a potential facilitator of tumour-targeted delivery of drugs and NPs. Firstly, it binds foreign particles and increases their circulation time by reducing clearance by the mononuclear phagocytic system. Secondly, albumin is taken up by tumour tissue as a source of amino acids and energy [41]. Thirdly, albumin can enter tumour cells via either macropinocytosis or gp60 and caveolin-1-mediated mechanisms [42]. Once inside, it remains there due to the enhanced permeability and retention (EPR) effect. In addition, in some cancers, affinity for overexpressed Secreted Protein Acidic and Rich in Cysteine (SPARC) keeps albumin inside the tumour tissue [43]. Pre-coating of NPs, such as polystyrene nanosphere and gold NP, with albumin reduced opsonin binding and increased their circulation time [44, 45]. In line with our findings, a study by Gernett and Dieppe showed that serum decreased the growth of synthetic hydroxyapatite crystals, with half of the inhibitory effect attributed to albumin alone [39]. The interaction occurred between the cationic Ca ion and the anionic carboxylate of the protein, with the adsorption reinforced by divalent Ca and Mg ions [46].
The second group of proteins in our PC are serine-phosphorylated and cystatin-domain containing cysteine-type endopeptidase inhibitors— alpha-2-HS-glycoprotein/fetuin and SPP24. These proteins regulate bone remodelling by inhibiting cysteine proteases such as cathepsins and are important in bone remodelling and, therefore, hydroxyapatite metabolism [47]. Produced in the liver, alpha-2-HS-glycoprotein/fetuin is found in high concentrations in serum and mineralised bone. It comprises a major fraction of non-collagenous proteins of bone and teeth and binds calcium phosphate on the N-terminus site [36, 38, 48]. Besides bone remodelling, it forms calciprotein particles with calcium phosphate in ectopic organs, which are cleared via Kupffer cells and marginal macrophages of the reticuloendothelial system [36, 48]. Fetuin entry in osteoblast, chondrocyte, and vascular smooth muscle cells was found to be mediated by annexin calcium channels in the presence of extracellular calcium. This process is involved in endocytosis and plays a role in calcium influx. A similar pattern of fetuin uptake in a calcium-dependent manner via annexins II and IV was observed in breast carcinoma. This fetuin-calcium entry into cells happens via fluid phase endocytosis and not receptor-mediated [49]. The other cysteine protease inhibitor forming PC on CA, secreted phosphoprotein 24, is also an important influencer of mineral calcification. It stabilises the hydroxyapatite interaction with the bone matrix. Discovered first as a non-collagenous bone matrix protein, it comprises a minor part in the fetuin-mineral complex (FMC), which also involves minor amounts of matrix Gla protein, serum amyloid P component, and prothrombin [50]. FMC prevents unwanted calcification by inhibiting the precipitation of calcium phosphate.
Haemoglobin subunits and cytochrome P450 (containing heme cofactor) detected in the PC of CA refer to the heme group’s affinity towards hydroxyapatite, a phenomenon previously reported in other studies. For example, a hydroxyapatite column is used to isolate acidic (pI value 6.8) haemoglobin through HPLC during the production of a blood substitute [51, 52]. Zhao et al. used haemoglobin as a soft template to attach HAP microspheres to a 3-D nanosheet [34]. Similar to the case of haemoglobin, immunoglobulin’s affinity for hydroxyapatite was exploited in the chromatographic isolation of immunoglobulin G. Both proteins’ interactions with both Ca and phosphate sites of hydroxyapatite are reinforced by other divalent ions [46, 53]. Along with immunoglobulins (Ig M), we also detected a few complement activators, such as properdin and pentaxin, which interact with the complement component C1q complex in the classical pathway [54]. In vitro studies reported the adsorption of complement activators to hydroxyapatite implants. Thus, osteoclasts can phagocytose hydroxyapatite to create porosity [55]. The apolipoprotein coating on CA reduces neutrophil activation, thus working as opsonin to clear the particles from the body.
Proteosomes and neuralized-like proteins are parts of ubiquitin-proteasome pathway for protein catabolism. The proteasome has a natural affinity for hydroxyapatite, a characteristic taken advantage of to purify this protein through hydroxyapatite chromatography [56].
Differential protein expression in untreated and CA-treated cells
Ca is a ubiquitous and universal intracellular second messenger essential to a wide range of processes such as fertilisation, development, growth, learning and memory, contraction and secretion, stress tolerance, and apoptosis—almost every process from the beginning to the end of a cell [57]. A high calcium gradient is maintained across the cellular membrane with a 104-fold higher extracellular concentration. Even though the cytoplasmic concentration of calcium is around 100 nM, intracellular organelles such as the ER and lysosome store calcium at a milimolar level [58, 59]. Strictly regulated extracellular and intracellular calcium levels, along with those in the organelles and their speed of change, amplitude, and spatio-temporal patterning, are important for proliferation, survival, and apoptosis. Intracellular increases in Ca2+ induce the expression of Ca2+ dependent transcription factors for proliferation. During cytoplasmic increases of Ca2+, Ca2+ enters mitochondria via the transporters and activates the dehydrogenases that lead to ATP synthesis [60]. However, in healthy cells, prolonged intracellular Ca2+ can induce apoptosis by releasing mitochondrial cytochrome C and activating caspases. Anomalies in Ca2+ homeostasis are linked to cancer, Alzheimer's, diabetes, heart disease, etc. In a review, Varghese et al. described that aberrant function of voltage-gated calcium channel family proteins was associated with 19 different cancers, including breast cancer. Modified cross-talk between ER and mitochondria evades apoptosis and promotes malignancy [61].
Calcium is taken up by the nucleus to support transcription during proliferation. A study on mouse 3T3 cells showed a positive link between Ca2+ influx and DNA replication and cell cycle progression [61]. An early study by K.V. Chin et al. on hepatocytes showed that sequestration of Ca2+ by the ER rather than mobilisation of it from the ER enhances translation. Their study on multiple cancer and non-cancer cell lines proved that Ca2+ supports the assembly of ribosomal subunits into polysomes and enhances translation initiation [62]. In our study, we observed that treating cells with CA increased the transcription regulator high mobility group box 1, ribosomal subunits and other translation-associated proteins by up to 2.8 fold.
CA also increased the level of chaperones such as heat shock proteins, 84b, and 90 alpha. As the name suggests, heat shock proteins render protection from stress caused by heat, UV, and oxidation among others. Besides, they work as chaperones and are involved in the maturation and regulation of client proteins. Their expression is often high in many cancers to correct the increased number of misfolded proteins [63]. HSP90 has a larger range of client proteins, some of which are especially important in cancer, such as epidermal growth factor receptor-2 (HER-2), insulin-like growth factor-IR (IGF-1R), Raf-1, androgen and oestrogen receptors, and hypoxia-inducible factor-1α [64]. HSP70, reduced slightly in CA-treated cells, attenuates calcium and contributes to the inhibition of the mitochondrial apoptosis pathway [64]. Interestingly, it may also give an ‘eat me’ signal to the immune system [65]. Another protein increased in CA-treated cells was peptidyl-prolyl cis-trans isomerase, which contributes to not only the folding of extracellular matrix proteins, such as collagen and hensin but also calcium handling in gene expression [66, 67]. This protein catalyses Ca2+ uptake by sarco(endo)plasmic reticulum calcium ATPase (SERCA2a) and Na+/Ca2+ exchanger 1 proteins [68].
We noticed an increase in glycolytic enzymes in CA-treated cells, which could be caused by the Ca2+ from CA fed into the mitochondria. In cancer cells, higher levels of cytoplasmic Ca2+ are buffered by Ca2+-dependent proteins such as the calcium-dependent mitochondrial carrier for adenine nucleotides, SCaMC-1. Overexpressed in many cancer cells, it can potentially help them withstand a higher Ca2+ load, which otherwise would trigger cell death by opening the mitochondrial permeability transition pore [60]. Also, the voltage-dependent anion channel (VDAC) on the outer membrane and the mitochondrial calcium uniporter (MCU) on the inner membrane allow calcium entry into the mitochondria and are associated with malignancy and metastasis. Overexpressed in many cancers, these proteins promote glycolysis [69, 70].
The cytoskeleton, such as tubulin, was detected to be significantly different between the two groups. However, we also detected actin alpha and gamma (at -10lgP < 20, data not reported). The Ca/calmodulin switch controls numerous effectors, one of which is actin the cytoskeleton machinery [59]. Actin alpha and gamma are involved in cell migration. Recent research showed that actin remodelling helps breast cancer cells evade natural killer cells [71]. Tubulin contributes to the Warburg effect, first by blocking VDAC and secondly by regulating glycolytic flux during metabolic stress [72].
Annexins, a family of Ca2+ effector proteins, can interact with the EGF receptor, formyl peptide receptor, selectin, actin, and integrin A4 [73]. They create membrane-cytoskeleton linkage in their Ca2+-bound conformation and play roles in exocytosis and endocytosis [59, 74]. We detected a small rise in ANXA1 in CA-treated cells. Besides its regular role in endosome and actin reorganisation, in breast cancer, this protein positively regulates NF-kB and c-myc and is found to be associated with proliferation and metastasis [75]. A few calcium-binding EF-hand proteins were detected to increase a little. EF-hand proteins of the S100 family interact with annexins [74]. S100A4 is a metastatic factor [76].
We observed a slight increase in KxDL motif-containing protein, a part of the BORC complex that works to move and localise lysosomes to the periphery. Lysosomes positively contribute to energy metabolism, growth, movement, and immune escape in tumour cells. It recycles cellular nutrients via autophagy to cope with a limited supply. During aerobic glycolysis, it helps maintain intracellular pH by taking in extra protons released by the lactic acid produced. Cathepsin released by the lysosome also remodels the ECM and promotes metastasis [77].
We found a 2.6-fold increase in the E3 ubiquitin-protein ligase RNF213, a part of the ubiquitination system. Ubiquitin tags damaged proteins for degradation by the proteasome. The proteasome breaks proteins into small peptides, which are further broken down to amino acids by peptidases for reuse. The protein misfolding rate is high in cancer cells. If it goes beyond the repair ability of the chaperones, proteasomal action takes over [78]. 26S proteosome and proteosomal activity increased in breast, colon, and blood cancers [79]. E3 ligase regulates proliferation, cell cycle arrest, and apoptosis. The E3 family follows protein kinases for their importance in cancer [80]. The effect of calcium on ubiquitination and proteasomes can be direct or indirect via calmodulin or calmodulin-dependent kinases [81].
We measured a sharp 3.4-fold increase in cellular albumin in CA-treated cells. Cancer cells take up albumin for amino acid and energy [82]. As CA also binds albumin, its entrance into the cell may contribute to increasing intracellular albumin too.
Macrophage migration inhibitory factor (MIF), an inflammatory cytokine that promotes tumour progression and metastasis by modulating the immunosuppressive tumour microenvironment, increased by two folds. [65]. This protein also interacts with calcium signalling [83].
Conclusion
This study established three facts. First, low-Ca2+ and high-Ca2+ particles are almost similar in size and composition. Second, CA particles are much smaller in vivo than they are in crystalline solid form, and the particle size of CA is controlled by the serum proteins; therefore, it can be manipulated with specific serum proteins as required. Third, the PC formed around the particles is the one involved in hard tissue metabolism, making CA safer for in vivo use. And fourth, CA itself is growth-promoting to the cancer cells. However, this growth-promoting role of CA could be outweighed by the anti-cancer effect of the delivered therapeutic drugs and genetic materials such as siRNA and plasmids, which was evident from our previous studies and the data reported in Fig. 6D. In the future, our research will focus on safely increasing the retention time of CA in mouse by modifying the classic composition of CA.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article.
References
Khan I, Saeed K, Khan I (2019) Nanoparticles: properties, applications and toxicities. Arab J Chem 12(7):908–931
Cristina Zavaleta DH, Chung EJ (2018) Theranostic nanoparticles for tracking and monitoring disease state. SLAS Technol 23(3):281–293
Robin Bofinger GW, Evans R, Glaser M, Sander K, Allan H, Hochhauser D, Kalber TL, Årstad E, Hailes HC, Ng T, Tabor AB (2021) Drug delivery, biodistribution and anti-EGFR activity: theragnostic nanoparticles for simultaneous in vivo delivery of tyrosine kinase inhibitors and kinase activity biosensors. Nanoscale 13(44):18520–18535
Giner-Casares JJ et al (2016) Inorganic nanoparticles for biomedicine: where materials scientists meet medical research. Mater Today 19(1):19–28
Hui Xu SL, Liu Y-S (2022) Nanoparticles in the diagnosis and treatment of vascular aging and related diseases. Signal Transduct Target Ther 7(1)
Francesca Giulimondi EV, Digiacomo L, Giuli MV, Mancusi A, Capriotti AL, Laganà A, Cerrato A, Chiozzi RZ, Nicoletti C, Amenitsch H, Cardarelli F, Masuelli L, Bei R, Screpanti I, Pozzi D, Zingoni A, Checquolo S, Caracciolo G (2022) Opsonin-deficient nucleoproteic corona endows UnPEGylated liposomes with stealth properties in vivo. ACS Nano 16(2):2088–2100
Lin CY, Yang CM, Lindén M (2019) Influence of serum concentration and surface functionalization on the protein adsorption to mesoporous silica nanoparticles. RSC Adv 9(58):33912–33921
Müller LK, Simon J, Rosenauer C, Mailänder V, Morsbach S, Landfester K (2018) The transferability from animal models to humans: challenges regarding aggregation and protein corona formation of nanoparticles. Biomacromolecules 12(2):374–385
Sharif Hossain HY, Chowdhury EH, Xin W, Hirose H, Amranul Haque YD, Mori M, Akaike T (2013) Fabrication and intracellular delivery of doxorubicin/ carbonate apatite nanocomposites: effect on growth retardation of established colon tumor. PLoS One 8(4):e60428
SnigdhaTiash IO, Rosli R, Chowdhury EH (2014) Methotrexate- and cyclophosphamide-embedded pure and strontium- substituted carbonate apatite nanoparticles for augmentation of Che- motherapeutic activities in breast cancer cells. Curr Drug Deliv 11:214–222
Kamaruzman NI et al (2018) siRNAs targeting growth factor receptor and anti-apoptotic genes synergistically kill breast cancer cells through inhibition of MAPK and PI-3 kinase pathways. Biomedicines 6(3):72
Ashaie MA et al (2019) Targeting cell adhesion molecules via carbonate apatite-mediated delivery of specific siRNAs to breast cancer cells in vitro and in vivo. Pharmaceutics 11(7):309
Ibnat N et al (2019) Transfection with p21 and p53 tumor suppressor plasmids suppressed breast tumor growth in syngeneic mouse model. Gene 701:32–40
Ibnat N, Islam RA, Chowdhury EH (2021) Inhibition of breast tumour growth with intravenously administered PRKCA siRNA- and PTEN tumour suppressor gene-loaded carbonate apatite nanoparticles. Appl Sci 11(17):8133
Mozar FS, Chowdhury EH (2018) PEGylation of carbonate apatite nanoparticles prevents opsonin binding and enhances tumor accumulation of gemcitabine. J Pharm Sci 107(9):2497–2508
Haque ST et al (2020) Fe/Mg-modified carbonate apatite with uniform particle size and unique transport protein-related protein corona efficiently delivers doxorubicin into breast cancer cells. Nanomaterials (Basel) 10(5):834
Ahmed S et al (2020) Molecular endoscopic imaging for the detection of Barrett’s metaplasia using biodegradable inorganic nanoparticles: an ex-vivo pilot study on human tissue. PLoS One 15(10):e0239814–e0239814
Islam RA et al (2020) Carbonate apatite and hydroxyapatite formulated with minimal ingredients to deliver SiRNA into breast cancer cells in vitro and in vivo. J Funct Biomater 11(3):63
Daculsi G, Bouler JM, LeGeros RZ (1997) Adaptive crystal formation in normal and pathological calcifications in synthetic calcium phosphate and related biomaterials. Int Rev Cytol 172:129–191
Kima SJ, Han SY (2014) Gel electrophoresis analysis of the hard coronas of Human Serum Albumin on silica nanoparticles: size dependence of corona formation. Bull Korean Chem Soc 35(9):2621–2624
Kanasty RL et al (2012) Action and reaction: the biological response to siRNA and its delivery vehicles. Molecular therapy: the journal of the American Society of Gene Therapy 20(3):513–524
Hickerson RP et al (2008) Stability study of unmodified siRNA and relevance to clinical use. Oligonucleotides 18(4):345–354
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, Tait JF, Tewari M (2011) Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci 108(12):6
Pottash AE et al (2019) Protein-based vehicles for biomimetic RNAi delivery. J Biol Eng 13(1):19
Wolfrum C et al (2007) Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol 25(10):1149–1157
Madupalli H, Pavan B, Tecklenburg MMJ (2017) Carbonate substitution in the mineral component of bone: discriminating the structural changes, simultaneously imposed by carbonate in A and B sites of apatite. J Solid State Chem 255:27–35
El Feki H et al (2000) Sodium and carbonate distribution in substituted calcium hydroxyapatite. Solid State Sci 2(5):577–586
Casals E et al (2010) Time evolution of the nanoparticle protein corona. ACS Nano 4(7):3623–3632
McConnell KI et al (2016) Reduced cationic nanoparticle cytotoxicity based on serum masking of surface potential. J Biomed Nanotechnol 12(1):154–164
Heiss A et al (2008) Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J Biol Chem 283(21):14815–14825
Tiash S, Chowdhury EH (2016) Passive targeting of cyclophosphamide-loaded carbonate apatite nanoparticles to liver impedes breast tumor growth in a syngeneic model. Curr Pharm Des 22:8
Chowdhury EH (2016) NANOTHERAPEUTICS From Laboratory to Clinic. CRC Press
Terkeltaub RA et al (1988) Serum and plasma inhibit neutrophil stimulation by hydroxyapatite crystals. Evidence that serum alpha 2-HS glycoprotein is a potent and specific crystal-bound inhibitor. Arthritis Rheum 31(9):1081–9
Zhao XY et al (2014) Hydroxyapatite nanosheet-assembled microspheres: hemoglobin-templated synthesis and adsorption for heavy metal ions. J Colloid Interface Sci 416:11–18
Hadjidakis DJ, Androulakis II (2006) Bone remodeling. Ann N Y Acad Sci 1092:385–96
Herrmann M et al (2012) Clearance of Fetuin-A–containing calciprotein particles is mediated by scavenger receptor-A. Circ Res 111(5):575–584
Lee J et al (2017) Effects of phospho- and calciotropic hormones on electrolyte transport in the proximal tubule [version 1; peer review: 2 approved]. F1000Research 6(1797)
Schinke T et al (1996) The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J Biol Chem 271(34):20789–96
Garnett J, Dieppe P (1990) The effects of serum and human albumin on calcium hydroxyapatite crystal growth. Biochem J 266(3):863–868
Farrugia A (2010) Albumin usage in clinical medicine: tradition or therapeutic? Transfus Med Rev 24(1):53–63
Stehle G et al (1997) Plasma protein (albumin) catabolism by the tumor itself–implications for tumor metabolism and the genesis of cachexia. Crit Rev Oncol Hematol 26(2):77–100
Hoogenboezem EN, Duvall CL (2018) Harnessing albumin as a carrier for cancer therapies. Adv Drug Deliv Rev 130:73–89
Hyun H et al (2018) Surface modification of polymer nanoparticles with native albumin for enhancing drug delivery to solid tumors. Biomaterials 180:206–224
Ogawara K et al (2004) Pre-coating with serum albumin reduces receptor-mediated hepatic disposition of polystyrene nanosphere: implications for rational design of nanoparticles. J Control Release 100(3):451–455
Bolaños K, Kogan MJ, Araya E (2019) Capping gold nanoparticles with albumin to improve their biomedical properties. Int J Nanomed 14:6387–6406
Fargues C, Bailly M, Grevillot G (1998) Adsorption of BSA and hemoglobin on hydroxyapatite support: equilibria and multicomponent dynamic adsorption. Adsorption 4(1):5–16
Ramage S (2007) The role of secreted phosphoprotein-24 in osteoblast differentiation and matrix mineralization. In Department of Biochemistry and Molecular Biology, Virginia Commonwealth University: Richmond
Szweras M et al (2002) alpha 2-HS glycoprotein/fetuin, a transforming growth factor-beta/bone morphogenetic protein antagonist, regulates postnatal bone growth and remodeling. J Biol Chem 277(22):19991–19997
Chen NX et al (2007) Fetuin-A uptake in bovine vascular smooth muscle cells is calcium dependent and mediated by annexins. Am J Physiol Renal Physiol 292(2):F599–F606
Price PA, Nguyen TM, Williamson MK (2003) Biochemical characterization of the serum fetuin-mineral complex. J Biol Chem 278(24):22153–22160
Curtis E, Wertz MSG (1997) Separation of polymerized hemoglobin from unpolymerized hemoglobin on hydroxyapatite using HPLC, U.S. Patent, Editor. BiopureCorporation, Cambridge, Mass
White HL, Monaghan BR (1935) The isoelectric point of adsorbed hemmoglobin. J Biol Chem 113:371–374
Ng PK, He J, Gagnon P (2007) Mechanistic model for adsorption of immunoglobulin on hydroxyapatite. J Chromatogr A 1142(1):13–18
Merle NS et al (2015) Complement system Part I - molecular mechanisms of activation and regulation. Front Immunol 6:262–262
Albrektsson T (1998) Hydroxyapatite-coated implants: a case against their use. J Oral Maxillofac Surg 56(11):1312–1326
Kanayama HO et al (1992) Demonstration that a human 26S proteolytic complex consists of a proteasome and multiple associated protein components and hydrolyzes ATP and ubiquitin-ligated proteins by closely linked mechanisms. Eur J Biochem 206(2):567–578
Roy A et al (2020) Calcium signaling is involved in diverse cellular processes in fungi. Mycology 12(1):10–24
Stewart TA, Yapa KT, Monteith GR (2015) Altered calcium signaling in cancer cell. Biochimica et Biophysica Acta (BBA) - Biomembranes 1848(10, Part B):2502–2511
Kramer IM (2016) Chapter 6 - Intracellular calcium. In: Kramer IM (ed) Signal transduction (Third Edition). Academic Press, Boston, pp 381–439
Tarasov AI, Griffiths EJ, Rutter GA (2012) Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 52(1):28–35
Varghese E et al (2019) Anti-cancer agents in proliferation and cell death: the calcium connection. Int J Mol Sci 20(12):3017
Chin KV et al (1987) Calcium-dependent regulation of protein synthesis at translational initiation in eukaryotic cells. J Biol Chem 262(34):16509–16514
Chatterjee S, Burns TF (2017) Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci 18(9):1978
Dudeja V, Vickers SM, Saluja AK (2009) The role of heat shock proteins in gastrointestinal diseases. Gut 58(7):1000–1009
Balogh KN, Templeton DJ, Cross JV (2018) Macrophage migration inhibitory factor protects cancer cells from immunogenic cell death and impairs anti-tumor immune responses. PLoS One 13(6):e0197702–e0197702
Lin W et al (2019) Peptidyl prolyl cis/trans isomerase activity on the cell surface correlates with extracellular matrix development. Commun Biol 2(1):58
Perrucci GL et al (2015) Peptidyl-prolyl isomerases: a full cast of critical actors in cardiovascular diseases. Cardiovasc Res 106(3):353–364
Sacchi V et al (2017) Peptidyl-Prolyl Isomerase 1 regulates Ca<sup>2+</sup> handling by modulating sarco(Endo)plasmic reticulum calcium ATPase and Na<sup>2+</sup>/Ca<sup>2+</sup> exchanger 1 protein levels and function. J Am Heart Assoc 6(10):e006837
Chakraborty PK et al (2017) MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun 8:14634–14634
Bustos G et al (2017) Endoplasmic reticulum-mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: a novel potential target. Front Oncol 7:199–199
Al Absi A et al (2018) Actin cytoskeleton remodeling drives breast cancer cell escape from natural killer-mediated cytotoxicity. Can Res 78(19):5631
Parker AL, Kavallaris M, McCarroll JA (2014) Microtubules and their role in cellular stress in cancer. Front Oncol 4:153
Moss SE, Morgan RO (2004) The annexins. Genome Biol 5(4):219–219
Gerke V, Creutz CE, Moss SE (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol 6(6):449–461
Moraes LA, Ampomah PB, Lim LHK (2018) Annexin A1 in inflammation and breast cancer: a new axis in the tumor microenvironment. Cell Adh Migr 12(5):417–423
Mishra SK, Siddique HR, Saleem M (2012) S100A4 calcium-binding protein is key player in tumor progression and metastasis: preclinical and clinical evidence. Cancer Metastasis Rev 31(1–2):163–172
Tang T et al (2020) The role of lysosomes in cancer development and progression. Cell Biosci 10(1):131
Jang HH (2018) Regulation of protein degradation by proteasomes in cancer. J Cancer Prev 23(4):153–161
Soave CL et al (2017) Targeting the ubiquitin-proteasome system for cancer treatment: discovering novel inhibitors from nature and drug repurposing. Cancer Metastasis Rev 36(4):717–736
Shi D, Grossman SR (2010) Ubiquitin becomes ubiquitous in cancer: emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol Ther 10(8):737–747
Mukherjee R et al (2017) Calcium dependent regulation of protein ubiquitination – Interplay between E3 ligases and calcium binding protein. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1864(7):1227–1235
Frei E (2011) Albumin binding ligands and albumin conjugate uptake by cancer cells. Diabetol Metab Syndr 3(1):11
Wennemuth G et al (2000) Macrophage migration inhibitory factor-induced Ca(2+) response in rat testicular peritubular cells. Biol Reprod 62(6):1632–1639
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions The project has been supported by FRGS/1/2018/STG05.
Author information
Authors and Affiliations
Contributions
Conceptualization: Rowshan Ara Islam and Ezharul Hoque Chowdhury; Methodology: Rowshan Ara Islam, Nabilah Ibnat, Maeirah Afzal Ashaie, Syafiq Asnawi Zainal Abidin; Formal analysis and investigation: Rowshan Ara Islam and Ezharul Hoque Chowdhury; Writing—original draft preparation: Rowshan Ara Islam; Writing—review and editing: Ezharul Hoque Chowdhury; Funding acquisition: Ezharul Hoque Chowdhury; Resources: Ezharul Hoque Chowdhury; Supervision: Ezharul Hoque Chowdhury.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Islam, R.A., Ibnat, N., Ashaie, M.A. et al. Carbonate apatite: effect of serum and impact on the cellular proteome. J Nanopart Res 25, 196 (2023). https://doi.org/10.1007/s11051-023-05838-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-023-05838-8