
Abstract
Chronic heart diseases have in common an unresolved inflammatory status. In atherosclerosis, myocarditis, myocardial infarction, or atrial fibrillation, mounting evidence suggests that unresolved inflammation contributes to the chronicity, aggravation, and morbidity of the disease. Following cardiac injury or infection, acute inflammation is a normal and required process to repair damaged tissues or eliminate pathogens and promote restoration of normal functions and structures. However, if acute inflammation is not followed by resolution, a chronic and deleterious inflammatory status may occur, characterized by the persistence of inflammatory biomarkers, promoting aggravation of myocardial pathogenesis, abnormal structural remodeling, development of cardiac fibrosis, and loss of function. Although traditional antiinflammatory strategies, including the use of COX-inhibitors, to inhibit the production of inflammation promotors failed to promote homeostasis, mounting evidence suggests that activation of specific endogenous autacoids may promote resolution and perpetuate cardioprotective effects. The recent discovery of the active mechanism of resolution suggests that proresolving signals and cellular processes may help to terminate inflammation and combat the development of its chronic profile in cardiac diseases. This review discussed (I) the preclinical and clinical evidence of inflammation-resolution in cardiac disorders including atrial fibrillation; (II) how and why many traditional antiinflammatory treatments failed to prevent or cure cardiac inflammation and fibrosis; and (III) whether new therapeutic strategies may interact with the resolution machinery to have cardioprotective effects.
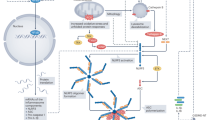
Graphical abstract
RvD D-series resolving, RvE E-series resolving, LXA4 lipoxin A4, MaR1 maresin-1

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Unresolved inflammation is the common denominator of various cardiac disorders including myocarditis, atherosclerosis, congestive heart failure, and atrial arrhythmia [1]. Long-term use of traditional antiinflammatory medications is often responsible for various adverse effects and increased morbidity in patients with a history of cardiac disease [2]. Recent studies suggest that therapeutic strategies promoting the activation of recently described proresolving biological processes may contribute to attenuate the consequences of chronic inflammation in neurodegenerative diseases, pulmonary diseases, obesity, or aging [3]. Few data are available about the role of these specialized proresolving mediators (SPMs) in cardiac diseases.
In the present article, the historical evolution of the concept of resolution from 1907 to 2021 is itemized (Fig. 1) and the recent consensus around the biochemical mechanisms of inflammation-resolution is reported (Figs. 2, 3 and 4).

Concept of resolution: Historical timeline of knowledge. Evolution of the concept of resolution and significant discoveries from 1907 to 2021

Inflammation resolution in cardiac tissue. Schematic of the vascular and tissue responses to inflammation after cardiac infection or injury. During acute inflammation, three consecutive phases aim to promote healing and homeostasis. Initiation: The initiation phase is characterized by increased proinflammatory signaling, PMN recruitment, and proinflammatory-(M1)-macrophage infiltration to phagocytize damaged cells and pathogens. LM-Class switching: Phagocytic PMN and M1-macrophages secrete 12/15 LOX enzyme. This promotes the lipid-mediator class switching, characterized by activation of secretion of proresolving lipids from AA, EPA, and DHA. These signals promote cessation of PMN infiltration and augmentation of anti-inflammatory-(M2)-macrophage phagocytosis of apoptotic PMN. Resolution: Complete resolution is identified by elimination of debris, efferocytosis, clearance, and restoration of normal cardiac function and structure. Chronic Inflammation: Failure in the activation of proresolution signals may promote chronic inflammation, characterized by the persistence of proinflammatory lipid mediators and proinflammatory cytokines, cardiomyocytes hypertrophy and necrosis, cardiac fibrosis, ischemia, and loss of cardiac function

Biochemical machinery of resolution-promoting endogenous molecules. Schematic of biochemical enzymatic reactions from essential fatty acids AA, DHA, and EPA, to the production of their proinflammatory and pro-resolving bioactive lipid metabolites

Resolution-promoting Lipid Mediators and their Receptors. Specialized proresolving mediators (SPM), including RvE1, RvE2, RvD1, RvD2, RvD3, RvD5, LXA4, PDG2, and their known receptors. For each SPM, the molecular structure is provided. For each receptor, the main location and the effects of its interaction with corresponding SPM is described. Picture of SPMs’ molecular structure is provided by PubChem’s free database: https://pubchem.ncbi.nlm.nih.gov/
This review focuses on the impact of proresolution therapy in cardiac disease, and discusses evidence of detection of SPMs receptors in cardiac tissue (Table 1), and the relevance of inflammation resolution therapeutic strategies in cardiac disease (Tables 2 and 3), considering the recent preclinical and clinical studies that have evaluated the role of SPMs in myocarditis, myocardial infarction, atherosclerosis, right heart disease, and atrial fibrillation.
Concept of inflammation resolution
Historical timeline of knowledge
Inflammation is a normal reaction in response to tissue damage or infection [4]. The essential role and physiological purpose of inflammation are to repair the tissue and to restore its homeostatic function.
One of the first reports of resolution of inflammation is from Eugene L. Opie from the Rockefeller Institute of Medical Research of New York in 1907 [5]. In this article, the author described the role of leukocytes’ enzymatic activity in the resolution of fibrinous pleurisy experimentally induced in dogs. He observed that during the first stage (initiation phase) of inflammation from day 0 to day 3, the animals were inert and fluids with polynuclear leukocytes (PNL) were abundant in the pleural cavity. Then a second phase, the resolution of inflammation (from day 3 to day 5), was characterized by degeneration and disappearance of PNL and fluids, persistence of mononuclear cells, and recovery of the animal’s health [5].
In 1970, Taber’s Cyclopedic Medical Dictionary defined resolution as the decomposition, absorption, or breakdown of the products of inflammation, and the termination of inflammation and return to homeostasis. The molecules and/or cells involved in disappearance of inflammation and/or its dispersion were defined as resolvents [6, 7].
During the 1980s, resolution of inflammation was described as well-orchestrated cellular events from the activation of vascular endothelial cells, and recruitment of polymorphonuclear leukocytes (PMN) in the initiation phase [8], to the clearance of cellular debris and dead PMN cells by macrophages in the resolution phase [9]. The endogenous roles of eicosanoids (Prostaglandins [PGs], thromboxanes [TXs], leukotrienes [LTs]), cytokines, and reactive oxygen species (ROS) on proinflammatory response were discovered and associated with initiation and efferocytosis via leukocyte stimuli, cellular chemotaxis and recruitment, phagocytosis, and induced apoptosis [10, 11].
In the ‘90 s, Serhan and collaborators discovered that lipoxins (LXs), could stop PMN recruitment and attenuate fibrosis [12,13,14]. Resolution was then described as an active reaction to terminate acute inflammation, and that chronic inflammation may be a consequence of failed resolution process [15]. Antiinflammatories such as glucocorticoids were shown to inhibit the production of proinflammatory products from arachidonic acid (AA) metabolism by inhibition of phospholipase A2 (PLA2) and COX2 enzymes [16]. COX2 inhibitors were also developed to efficiently inhibit the production of PGs, TXs, and LTs but also LXs, which may explain why their use was associated with complications and side effects [17, 18].
Most recently, during the first decade of the 2000s, Serhan and colleagues discovered that non-toxic endogenous autacoids called resolvins (Rvs) are implicated in the active process of resolution [19]. Serhan and collaborators have described that omega-3 polyunsaturated fatty acids (n-3 PUFAs) are metabolized by enzymes such as COX-2 and 5-LOX to produce these bioactive mediators [20]. Hence, eicosapentaenoic acid (EPA) is a precursor to the E-series resolvins (RvE1, RvE2, RvE3), and eicosahexaenoic acid (DHA) is a precursor to the D-series resolvins (RvD1-6), neuroprotectin D1 (NPD1), and maresins (MaR1) [20]. They have also observed that low-dose aspirin can trigger the biosynthetic production of a variety of analog forms of these lipid mediators, called aspirin-triggered resolvins (AT-Rvs) via EPA and DHA [20].
The omega-3-derived promotors of resolution are called specialized pro-resolving mediators (SPMs) [21]. Moreover, the discovery of specific receptors of SPMs (i.e., ALX/FPR2 receptor of LXA4 and RvD1; GPR32 receptor of RvD1; ChemR23 receptor of RvE1) supports the concept that resolution is an active process mediated by complex intracellular signaling in response to inflammation, to promote homeostasis and prevent chronic inflammation [22] (Fig. 1).
Consensus: definition and chronology of events from acute inflammation to resolution
Acute inflammation is characterized by the succession of three major phases: the initiation, lipid-mediators class switching, and resolution. Resolution can be defined as the succession of active processes involving endogenous specialized pro-resolving mediators (SPMs), aiming to terminate inflammation and restore normal functions [23].
In response to chemical or physical deleterious stimuli from infection or injury, the attacked tissue releases pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) that interact with pattern recognition receptors, such as toll-like receptors (TLRs), expressed on the affected cell membrane. TLRs activate intracellular pathways including mitogen-activated protein kinases (MAPK) pathways, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). NFkB mediates the activation of expression of various genes involved in the activation of inflammatory processes. Among them, NFkB promotes the assembly and activation of NLRP3-inflammasome and accentuates the secretion of proinflammatory cytokines (IL1β, IL6, IL18), and chemokines (CXCL1/2) involved in the initiation of the inflammatory response [24].
AA is metabolized via CYP450, cyclooxygenase-2 (COX2) and 5-, 12-, and 15-lipoxygenase (LOX) into proinflammatory lipid-mediators including thromboxanes (TXA2: vasoconstriction), prostaglandins (PGs: vasodilation, lymphocytes proliferation), and leukotrienes (LTs: vasopermeability, chemotaxis) (Fig. 2) to install the cardinal signs of initiation of inflammation, namely: warmness, redness, nodule, and ache [25].
Apoptotic neutrophils start to produce endogenous SPMs such as lipoxins (LXA4 and LXB4) [26]. This pro-resolution signal marks the end of the initiation phase of acute inflammation, attenuation of the cardinal signs of inflammation, and the beginning of lipid-mediator-class switching to activate the resolution phase [27] (Figs. 1 and 2).
Circulating essential n-3 PUFAs (EPA and DHA) are accumulated in the damaged tissue via edema to be enzymatically metabolized by COX2 and 5/12/15-LOX and converted into bioactive SPMs. Lipoxins (LXs) inhibit chemotaxis of further PMN and stimulate the nonphlogistic recruitment of monocytes at the site of injury. SPMs, including LXs, Rvs, maresins, and/or protectins are produced and accumulated in the injured tissue. The SPMs stimulate the cessation of neutrophil infiltration in the inflamed area, and they inhibit proinflammatory cytokines-signaling [28]. SPMs promote the differentiation of monocytes into anti-inflammatory (M2) macrophages [29]. SPMs activate efferocytosis and clearance by stimulating M2-macrophage phagocytosis of apoptotic neutrophils and cellular debris. M2-macrophages produce anti-inflammatory cytokines (IL10) and more SPMs to promote resolution. Resolution of inflammation results in tissue healing, relief of pain, remission of fever, and regeneration of normal structure and function [23] (Figs. 1 and 2).
When the tissue fails to activate the lipid-mediator-class switching which promotes SPMs production and resolution, proinflammatory signaling will persist and abnormally stimulate further recruitment of PMN and perpetuation of the inflammatory status. This may promote chronic inflammatory profile, development of fibrosis, and loss of function. However, mechanisms or therapeutic strategies promoting bioavailability and accumulation of SPMs in chronically inflamed organs may activate resolution, wound healing, and restitution of normal function and structure [29, 30] (Figs. 1 and 2).
Specialized pro-resolving mediators’ receptors in cardiac cells
The presence of SPM receptors is well described in lung, brain, kidney, eye, or liver tissue [29]. Few data are available about their expression in the heart. Some studies on murine models of ischemia–reperfusion reported the presence of SPM receptors in ventricle [30, 31]. Recently, ChemR23, GPR32, ALX/FPR2, BLT1, GPR18, and GPR37 were detected in rat atrial tissue [32]. To our knowledge, little is known about the expression of SPM receptors on isolated atrial cardiomyocytes and atrial fibroblasts.
SPM receptors on cardiomyocytes
The expression of SPM receptors has been evaluated on cardiomyocytes from rat, murine and human heart tissue. In a mouse model of autoimmune myocarditis, BML-111, an analog of LXA4, prevented cardiomyocyte death in vitro. This study suggests that under inflamed conditions, proresolving stimuli may promote cardiomyocyte’s expression of ALX/FPR2, the specific receptor of LXA4 [33]. GPR18, the specific receptor of RvD2 was detected on ventricular cardiomyocytes in rats. GPR18 activation ameliorated ventricular function [34]. ChemR23, also known as Chemerin Chemokine-Like Receptor 1, is the receptor of RvE1 and RvE2. ChemR23 was detected on rat ventricular cardiomyocytes [35] and murine atrial cardiomyocytes [36]. ChemR23 activation was shown to mediate cardiomyocyte apoptosis [36] (Table 1 and Figs. 3 and 4).
SPM receptors in cardiac fibroblasts
The activity of SPM on fibroblast’s proliferation and differentiation have been reported on diseases associated with fibrosis-induced malfunctions, including neurodegenerative diseases or aneurysm [29]. In the heart, studies from Ganesh Halade’s group revealed that RvD1 treatment reduced ventricular fibrosis in myocardial infarction [30, 37]. These data suggest that cardiac fibroblasts may express RvD1 receptors GPR32 and/or ALX/FPR2. Consistent with this, in a rat model of monocrotaline-induced right heart disease, RvD1 reduced atrial fibrosis and atrial fibrosis-related markers POSTN, TGFβ3, COL1A1, COL3A1, and ACTA1 as detected by microarray assay, RT-qPCR, and Western blot [32]. In a recent study, ALX/FPR2 and ChemR23 were detected on isolated ventricular fibroblasts. RvD1 treatment prevented lipopolysaccharide (LPS)-induced increase in IL6, MCP-1, and TNFα. The authors observed that RvD1 and RvE1 prevented the adhesion of mononuclear cells to cardiac fibroblasts [38]. Recently GPR18 was detected on cardiomyoblasts from patients with heart failure. GPR18 expression may be increased in response to injury, and its inactivation may promote heart failure chronicity [39] (Table 1, Figs. 3 and 4).
SPM receptors on other cardiac cell types
Coronary vessels (arteries and veins) are essential in cardiac physiology to irrigate the heart. Blood supplies the heart muscle with oxygen, nutrients, and immune cells (in case of injury or infection) via the coronary arteries and is drained via the coronary veins [40]. The coronary vessels are made of an inner thin layer of endothelial cells, the intima; a thicker layer made of several concentric sheets of vascular smooth muscle cells (VSMC), the media; and an external layer comprised of elastin and collagen fibers, the adventitia [41] (Table 1, Figs. 2, 3 and 4).
SPM receptors on cardiac endothelial cells
Cardiac endothelial cells (CEC) play an important role in the initiation and resolution phases of inflammation. CEC are sensitive to chemotaxis stimuli, as they are in direct contact with blood. They can modulate the intima’s permeability to allow or limit immune cells infiltration in the myocardium. Hence, CEC have been shown to express ALX/FRP2 and ChemR23 which signaling are important in the CEC-controlled cessation of PMN recruitment and the activation of non-phlogistic monocyte infiltration during LM-class switching [42]. It has been shown that EC also express GPR18 and GPR32 that are activated to attenuate EC permeability and promote EC adherens-junction integrity during inflammation [43] (Table1).
SPM receptors on cardiac vascular smooth muscle cells
Intimal hyperplasia is a major clinical problem causing coronary artery bypass graft failure due to proliferation and migration of VSMC in the intima of coronary vessels, leading to their progressive obstruction. In a recent study involving a rat model of intima hyperplasia, ChemR23 was detected on VSMC. ChemR23 deletion was associated with progression of intimal hyperplasia and alteration of VSMC phenotype [44]. Consistent with these results, patients with coronary artery disease (CAD) treated with EPA and DHA had higher plasma levels of RvE1 and lower levels of LTB4 compared to untreated CAD patients. This was associated with coronary artery plaque regression [45]. The sensitivity of VSMC to LTB4 suggests that these cells may express its specific receptor BLT1 [46]. VSMC have also been shown to express ALX/FPR2. VSMC modulates the permeabilization of the artery wall during initiation and resolution of inflammation via activation of ALX/FPR2 which promotes vasorelaxation, cessation of PMN recruitment and augmentation of monocyte infiltration across the coronary vessels wall [46] (Table 1, Figs. 2, 3 and 4).
SPM receptors on immune cells in heart tissue
SPMs’ receptors are well described on immune cells [29]. In this section, the review focuses on reports that assessed the expression of SPMs’ receptors on immune cells in the context of cardiac tissue only. In a murine model of myocardial infarction, ALX/FPR2 was expressed on PMN and macrophages. ALX/FPR2 activation limited myocardial scar after myocardial infarction [30, 37]. In patients with CAD, ChemR23 was detected on monocytes and macrophages [47]. BLT1 was shown to be expressed on the surface of PMN, and pro-inflammatory macrophages (M1-macrophages). The authors observed that BLT1 inhibition was associated with amelioration of sepsis-induced myocardial injury [48]. GPR18 was shown to be expressed on PMN, monocytes, lymphocytes, M1- and anti-inflammatory (M2)-macrophages. GPR18 activation promoted inhibition of PMN infiltration, M2-polarization and phagocytosis, and M1-macrophage apoptosis [34]. GPR32 has been proven to be expressed on human PMN and macrophages to promote M2-macrophage polarization and phagocytosis of apoptotic PMN. GPR32 activity via RvD1 is associated with clearance, efferocytosis, and resolution [49, 50] (Table 1 and Figs. 3 and 4).
Evidence of specialized pro-resolving mediators in cardiac diseases
Myocarditis and cardiac inflammation
Myocarditis is a progressive inflammation status of the myocardium in response to infection, cardiotoxic insults, or autoimmune disease [1]. Myocarditis is characterized by an unresolved inflammatory profile, cardiac cell death, and oxidative stress, which can lead to severe dilated cardiomyopathy and heart failure [1].
The role of LXA4 analog BML-111 has been evaluated on a murine model of experimental autoimmune myocarditis (EAM). Myocarditis was induced in 8-week-old female BALB/c mice by subcutaneous immunization with 350 µg of cardiac myosin in a 1:1 emulsion with Complete Freund's Adjuvant containing 5 mg/mL of Mycobacterium tuberculosis H37RA. Control animals received an equivalent volume of saline. BML-111 (1 mg/kg/d) was injected from day 7 to day 21. All animals were sacrificed at day 21. Compared to control, EAM was associated with statistically significant increased ventricular myocardium expression of inflammation related genes Il1b, Il6, and Tnfa; fibrosis-related genes Tgfb1, Col1a1, and Col3a1; and oxidative stress markers 8-OHdG and Keap1. BML-111 treatment significantly decreased and normalized the level of expression of these genes (except Il6 and Col3a1). The LXA4 analog also prevented EAM-induced cardiomyocyte apoptosis, LV hypertrophy, and reduction of LV ejection fraction and fraction shortening. The authors proposed that BLM-111 may play its cardioprotective effects by activating the CaMKK2/AMPK axis to promote activation of antioxidant Nrf2 pathway [37] (Table 2).
Myocardial infarction and cardiac fibrosis
Myocardial infarction (MI) is characterized by LV ischemia and persistent irreversible collagen deposition promoting LV loss of function and progression of heart failure [51]. The role of RvE1 was evaluated in an in vivo model of LV ischemia/reperfusion (I/R). Male Sprague–Dawley rats were subjected or not (sham), to the ligation of the left anterior descending coronary artery (LAD). At 28 min post-ligation, the animals received a single intravenous injection of vehicle or RvE1 at 0.03, 0.1, or 0.3 mg/kg. At 30 min post-ligation, ischemia was stopped by releasing the coronary artery snare to allow reperfusion for 4 h. Compared to rats with I/R-alone; RvE1 treatment did not improve the mean blood pressure post-I/R. However, the infarct size was significantly smaller when treated with RvE1 at 0.1 and 0.3 mg/kg. The mean leukocytes infiltration was reduced in the post-ischemic area in RvE1-treated rats (0.3 mg/kg) compared to I/R-alone. Myocardial levels of Ser473 phospho-Akt, Thr308 phospho-Akt, and phospho-ERK1/2 were significantly increased in I/R animals compared to sham. RvE1 treatment dose-dependently increased these levels compared to I/R-alone without affecting total Akt or total ERK1/2 levels. Moreover, although caspase-3 was significantly increased in I/R rats compared to sham, RvE1 significantly decrease its level of expression in the infarct area when injected at 0.1 and 0.3 mg/kg, 28 min post-I/R [52].
The inflammation resolving response of RvD1 treatment was studied in a mice model of myocardial infarction (MI) using permanent LAD. Infarcted C57BL/6 J mice were treated or not with RvD1 (3 µg/kg/d) 3 h post-MI. The animals were sacrificed 1 day or 5 days post-MI. RvD1 prevented MI-induced LV hypertrophy and attenuated MI-induced reduction of fraction shortening. RvD1 treatment significantly enhanced the expression of ALX/FPR2 in the infarcted area at day 5 post-MI. RvD1 reduced M1-macrophages density, oriented M2-macrophage polarization, and promoted pro-resolution clearance in the LV at day 5 post-MI as shown by decreased expression of Tnfα, Il6, Ccl2, and Il1β and increased expression of Mrc-1, Arg1, and Ym-1. Moreover, RvD1 decreased collagen deposition and ECM gene expression thereby improved LV healing and function post-MI [30] (Table 2).
Atherosclerosis and cardiac ischemia
Chronic unresolved inflammation participates in the development and aggravation of atherosclerosis. Recent reports have attempted to understand the mechanisms by which failed resolution may promote atherosclerosis. It has been shown that in human, 5-LOX-derived SPM levels were lower in vulnerable atherosclerotic plaque compared to stable lesions. The same study reported that administration of RvD1 reduced necrosis and collagenase while promoting efferocytosis, in a murine model of chronic atherosclerosis [53]. Atherosclerosis progression was prevented by RvD2 and MaR1 repetitive treatment in mice [54]. Pirault and Bäck reviewed the role of four SPMs receptors (ALX/FRP2, GPR32, GPR18, and ChemR23) in atherosclerosis, aortic aneurysm, and ischemia/reperfusion. They reported that activation of these receptors may promote cardiovascular protection via activation of P-ERK1/2 and eNOS activity. Inhibition of these specific SPMs’ receptors provoked inhibition of p38 MAPK signaling and reduction of proinflammatory cytokines production in PMN, macrophages, vascular smooth muscle cells, and cardiomyocytes [42].
These data suggest that atherosclerosis is characterized by an abnormal decrease in SPM/[proinflammatory LTs + PGs] ratio. Proresolving treatment may promote resolution and prevent atheroprogression (Table 2 and Fig. 2).
Valvulopathy and cardiac calcification
Aortic valve stenosis (AVS) is the most common valve disease characterized by thickening and calcification of the aortic valve leaflets (AVL) causing progressive narrowing and malfunction of the aortic valve (AV) [55].
The role of n-3 PUFAs and their derived specialized proresolving mediators have been evaluated on the development of AVS in human tricuspid aortic valves from patients who underwent AV replacement surgery, and in transgenic mice with AVS [56]. It has been shown that EPA and DHA, as well as RvE1 and RvD3 were decreased, whereas LTB4 was increased, in calcified compared to non-calcified human AV. RvE1 reduced phosphate-induced calcification in human valvular interstitial cells (VICs) in vitro. In transgenic mice, targeted deletion of ChemR23 induced increased thickening and malfunction of the aortic valve after aortic valve wire injury. The activation of the n-3 PUFA/RvE1/ChemR23 axis promoted M2 macrophage polarization, inflammation-resolution, and AV cusp motion while preventing AVL thickening and calcification [56] (Table 2).
Right heart disease
Various inflammatory diseases such as congenital cardiac disease, chronic obstructive pulmonary disease, pulmonary hypertension, are associated with right heart remodeling characterized by RV hypertrophy, RH dilation, RV systolic pressure, tricuspid regurgitation, and atrial dilation [57, 58]. The impact of inflammation resolution on the incidence of these diseases is suggested, but little is known on the underlying pathophysiological mechanism. Recently the role of inflammation resolution has been tested in a rat model of right heart disease (RHD)-induced AF. RHD has been induced in Wistar rats by a single injection of 60 mg/kg of monocrotaline i.p. Control animals received an equivalent dose of vehicle. Starting one day before MCT administration, the intervention group received daily doses of 2 µg/kg/d of RvD1 for 3 weeks. RvD1 significantly prevented RHD-induced increase in P wave duration, RR and QT intervals, and tricuspid annulus plane systolic excursion [32]. Previously, the role of MAG-DPA (docosapentaenoic acid monoacylglyceride), a synthetic metabolic precursor of EPA and DHA has been tested in an MCT model of PH-induced RHD in rats. MAG-DPA was administrated at 231 mg/kg/d during 3-weeks. MCT-induced increases in RV weight and wall thickness were prevented by MAG-DPA treatment. MAG-DPA stimulated metabolic production of RvD5 in cardiac tissue [59]. These data suggest that proresolving treatment may prevent accentuation of inflammation-induced cardiac structural and functional remodeling in RHD (Table 2 and Fig. 2).
Congestive heart failure
Congestive heart failure (CHF) is characterized by a typical inflammation profile [60]. A recent report supports the concept that alteration of resolution of inflammation is associated with chronicity of heart failure [61]. This study involved 50 patients: 27 had CHF and 23 were healthy. CHF patients showed lower levels of leukocytic 15LOX activity, D-series resolvins, and RvD1 receptor GPR32 compared to healthy subjects. The authors concluded that altered proresolution mediators may promote chronicity of inflammation and progression of CHF [61] (Table 2 and Fig. 2).
Atrial arrhythmia
Mounting evidence suggest that unresolved inflammation may promote atrial fibrosis and development of atrial arrhythmogenic substrate leading to enhanced AF susceptibility [62]. The role of RvD1 has been tested in AF prevention. In a rat model of RHD, RvD1 daily treatment attenuated RHD-induced RA fibrosis. RvD1 promoted reduction of density in M1-macrophages and increased M2-macrophages in RA. RvD1 prevented RHD-induced overexpression of proinflammatory markers (CXCL1/2), NLRP3-inflammasome components (ASC, CASP1, IL1β), and fibrosis-related agents (COL3A1, TGFβ3). Although no changes were observed in ALX/FPR2 expression, RvD1 increased ChemR23 expression in RA compared to RHD-only and control animals. RvD1 also prevented RHD-induced RA electrical conduction abnormalities. These right atrial effects of RvD1 were associated with significant prevention of RHD-induced increase in atrial tachyarrhythmia susceptibility [32, 63] (Table 2 and Fig. 2).
SPMs vs traditional antiinflamatory treatments in cardiac diseases
Non-steroidal antiinflammatory drugs and SPMs in cardiac diseases and AF
COX-inhibitors are resolution-toxic
The normal process of acute inflammation is regulated by bioactive lipid mediators produced from the well-orchestrated sequence of enzymatic interactions of COX1/2 and 5/15/15LOX with AA, EPA, DHA [29]. The highly selective COX2 inhibitors are efficient to block the production of PGs, LTs, and TXs from AA. However, they can cause serious cardiovascular adverse effects [64]. The inhibition of COX2 by COX2-inhibitors is nonselective. Hence, important metabolite products involved in the resolution process (PGE2, PGD2) are also blocked. In a randomized trial involving 2035 patients, use of celecoxib was associated with a dose-dependent augmentation of mortality from cardiovascular causes, myocardial infarction, heart failure and stroke [65]. Celecoxib has been shown to significantly increase P-wave duration in patients with inflammatory arthritis suggesting that this medication caused atrial remodeling that predisposed these patients to AF [66]. In a 9-year retrospective study on 32,602 patients, the use of COX2 inhibitors was associated with an increased risk of AF and atrial flutter [67] (Table 3 and Figs. 3 and 4).
Potential aspirin-triggered resolution
Aspirin is a traditional non-steroidal antiinflammatory drug (NSAID) that acetylates COX2 and stops the production of PGs and LTs from AA. In this manner, aspirin attenuates the LM-class switching and some resolution process mediated by PGE2 and PGD2. Paradoxically, aspirin-induced acetylated COX2 remains active for enzymatic interactions with EPA and DHA leading to production of aspirin-triggered (AT) resolvins (AT-Rvs). It is unclear whether aspirin-triggered production of SPMs homologs and complete inhibition of PGs would have beneficial outcomes or not for the host. In cardiovascular disease, low-dose aspirin (80 mg and under) is recommended for secondary prevention, when the patient is between 40 to 70 years of age, at high risk of cardiovascular disease and at low risk of bleeding, with a history of coronary disease, stroke, or heart attack. However, aspirin is not recommended as a primary preventive approach for healthy patients [68]. Aspirin has been used in AF treatment as an anticoagulant in some patients [69], but chronic use of aspirin was not associated with diminution of AF incidence [70] (Table 3 and Figs. 3 and 4).
Impact of glucocorticoids on resolution in cardiac diseases and AF
Glucocorticoids may appear as ideal medications in the treatment of inflammation, particularly cardiac inflammation, as they promote reduction of proinflammatory biomarkers (initiation phase) and stimulate nonphlogistic monocyte polarization into M2-macrophage, efferocytosis, and clearance (resolution phase) [71,72,73]. In a prospective study, 138 patients that underwent catheter ablation were randomly assigned to two groups: control and steroid treatment. Short-term steroid therapy decreased the incidence of early recurrence of AF compared to control. No efficacity was observed on late recurrence of AF [74]. In a clinical trial involving 4494 patients; dexamethasone did not prevent postoperative AF [75]. Glucocorticoids are known to be associated with various adverse effects and risk of complications [76]. A recent review suggests that their use may increase the risk of atherosclerosis, and atrial arrhythmias [77] (Table 3).
Other anti-inflammatory targets in resolution of cardiac diseases and AF
Colchicine
Colchicine is a well-known antiinflammatory drug with beneficial effects in pericarditis. The mode of action of colchicine suggests beneficial effects to promote resolution of inflammation. Colchicine inhibits NLRP3 inflammasome activation, inactivates NFkB expression, decreases TNFα expression, reduces the release of the pro-inflammatory cytokines IL-1β and IL-18, prevents PMN chemotaxis, and promotes cessation of inflammation [78]. In a retrospective study on 1412 patients, colchicine administration significantly reduced AF incidence after cardiac surgery [79]. Recently, in the COLchicine Cardiovascular Outcomes Trial (COLCOT), low dose colchicine (0.5 mg daily) has been demonstrated to significantly reduce the risk of ischemic cardiovascular events in 2366 MI patients compared to 2379 MI patients treated with placebo [80]. In the Low-dose colchicine trial (LoDoCo) involving 532 patients with chronic coronary disease, daily administration of 0.5 mg colchicine decreased the risk acute cardiovascular events compared to patients treated with placebo [81]. In the LoDoCo2 trial, recently published in 2020, 5522 patients were randomized to receive either colchicine (2762) or placebo (2760). The incidence of spontaneous myocardial infarction, cardiovascular death or ischemia-driven coronary revascularization were significantly lower in patients treated with colchicine compared to placebo [82] (Table 3).
Omega-3 PUFAs
During the last two decades, EPA and DHA were central subjects of controversy. Their antinflammatory and antiarrhythmogenic properties have been demonstrated, but no clinical trials confirmed their efficacy to prevent AF [83]. Recently, in the multicenter study REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial), Deepak L. Bhatt and colleagues observed that daily administration of icosapent ethyl (IPE) could decrease the risk of ischemia, myocardial infarction, and cardiovascular death [84]. These results were opposed to the STRENGTH trial results in which combination of EPA and DHA did not show significant benefits [85]. In terms of arrhythmia, these studies suggested that AF risk was increased as 2.2% patients in the omega-3 group vs. 1.3% in the placebo group induced AF in the STRENGTH trial, while 5.3% patients in the IPE group vs. 3.9% in the placebo group induced AF in the REDUCE-IT trial. Regarding the biochemical system involving EPA and DHA in the resolution process, it is suspected that most of trials involving these n-3 PUFAs did not consider promoting their enzymatic metabolism into bioactive compounds [29, 86]. The present review supports the idea that a new paradigm must be adopted in omega-3 treatments considering their level of degradation after administration, their optimized interaction with specific enzymes (5/12/15LOX) and the successful production of their bioactive proresolving metabolites (Table 3 and Fig. 3). Supporting the hypothesis that therapy promoting optimization of SPM production must be prioritized instead of simply administrating Omega-3 PUFAS or inhibiting inflammatory compound productions, the STABILITY trial (STabilization of Atherosclerotic plaque By Initiation of darapLadIb TherapY) involving 15,828 patients revealed that compared to placebo, darapladib (an inhibitor PLA2) did not prevent cardiovascular events in patients with coronary heart disease [87]. As previously described, PLA2 is at the top of the metabolic cascade of production of either proinflammatory biomarkers from AA and proresolution mediators from EPA and DHA. This review suggests that inhibition of PLA2 may prevent production of inflammatory signals but also SPM, which may explain no beneficial effects against cardiovascular events in the STABILITY study (Figs. 2 and 3).
NLRP3-inflammasome, IL1 and IL6 INHIBITION
Various chronic cardiac inflammatory diseases, including AF, have in common the overexpression of NLRP3 inflammasome-related genes and components [88,89,90,91,92]. The NLRP3 inflammasome senses PAMPs and DAMPs signals to promote proinflammatory response to infection or injury by stimulating secretion of proinflammatory interleukins such as IL1β, IL6, and IL18 [93, 94]. Genetic deficiency of NLRP3 was associated with reduced expression of proinflammatory cytokines (TNFα, IL1β, IL6), reduced secretion of proinflammatory PGE2 and LTB4, and increased expression of LXA4 and LXB4, which suggests that presence of NLRP3 may negatively influence the LM-class switching and prevent resolution process during acute inflammation [95]. In the CANTOS (Canakinumab ANtiinflammatory Thrombosis Outcome Study) trial involving 10,061 patients with myocardial infarction and high-sensitivity C-reactive protein level of ≥ 2 mg per liter, specific inhibition of IL1β by the monoclonal antibody canakinumab resulted in a significant reduction in the risk of cardiovascular events compared to patients treated with placebo [96]. In the randomized RESCUE trial, IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk was associated with reduction of circulating levels of biomarkers of thrombosis and inflammation commonly related to atherosclerosis [97]. Recently the role of SPMs in the inhibition of NLRP3 inflammasome to promote resolution has been tested [63]. The authors observed that RvD2, via its specific receptor GPR18, suppressed expression of pro-IL1β, reduced secretion of IL1β, attenuated ASC oligomerization, and decreased CASP1 activity [98]. In the heart, murine models of inhibition of NLRP3 by MCC950 or specific knockdown showed reduction of cardiac fibrosis, and expression levels of IL1β and IL18 [99, 100]. In a rat model of right heart disease, RvD1 treatment decreased arrhythmogenic atrial remodeling-associated increases in right atrial expression of NLRP3, ASC, CASP1, and AF inducibility [32, 63] (Table 3).
Reported limits about SPM’s activity
In a recent study from Giorgio Mottola and collaborators involving a model of rat carotid angioplasty, periprocedural oral administration of RvD1 led to the increased plasmatic concentration of RvD1 3 h post-gavage, which prevented arterial inflammation but did not reduce intimal hyperplasia [101]. In a rat model of right heart disease, daily treatment with RvD1 during 21 days attenuated cardiac fibrosis and inflammation but did not prevent cardiac dilation in response to induced right-sided chronic overload [32]. As previously described in Fig. 3, SPMs are generated via fragile metabolic processes, including oxidation and epoxidation, meaning that SPMs can themselves be metabolized shortly after their production. This character confers an extremely short half-life to the SPMs of about 1 h to 5 h depending on the study [103]. The short half-life of SPMs versus their potential strong pro-resolution effects suggest that strategies to optimize their bioavailability are required [102, 103].
Summary
Acute inflammation is governed by a complex biochemical system involving bioactive proinflammatory and proresolving lipid mediators. In optimal conditions, the initiation of inflammation must be terminated to optimize the activation of resolution signals promoting clearance and homeostasis. In chronic inflammatory cardiovascular diseases including AF, resolution may have failed to be activated. This review suggests that future therapeutic strategies may target resolution via the maximization of the production of bioactive proresolving mediators and the inhibition of proinflammatory biomarkers, in a spatiotemporal-dependent manner, to prevent or cure cardiac inflammation and AF. In a larger spectrum, identifying the right moment and the right place to tackle inflammation is a significant challenge in the development of new therapies targeting diseases characterized by chronic inflammatory status.
Abbreviations
- AA:
-
Arachidonic acid
- AF:
-
Atrial Fibrillation
- COX:
-
Cyclooxygenase
- IL:
-
Interleukin
- LOX:
-
Lipoxygenase
- MI:
-
Myocardial Infarction
- NLRP3:
-
NOD-like receptor family, pyrin domain containing 3
- RHD:
-
Right Heart Disease
- Rvs:
-
Resolvin
- SPM:
-
Specialized Pro-resolvin mediator
References
Cooper LT Jr (2009) Myocarditis. N Engl J Med 360:1526–1538. https://doi.org/10.1056/NEJMra0800028
Varga Z, Sabzwari SRA, Vargova V (2017) Cardiovascular risk of nonsteroidal anti-inflammatory drugs: an under-recognized public health issue. Cureus 9:e1144. https://doi.org/10.7759/cureus.1144
Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510:92–101. https://doi.org/10.1038/nature13479
Punchard NA, Whelan CJ, Adcock I (2004) The Journal of Inflammation. J Inflamm (London, England) 1:1. https://doi.org/10.1186/1476-9255-1-1
Opie EL (1907) Experimental pleurisy-resolution of a fibrinous exudate. J Exp Med 9:391–413
Taber CW (1970) Taber's Cyclopedic Medical Dictionary, 11th Ed., F. A. Davis, Philadelphia, 1405
Grob D (1946) Proteolytic Enzymes: II. The physiological significance of the control of their activity, especially with respect to bacterial growth. J Gen Physiol 29:249–255
Weissmann G, Smolen JE, Korchak HM (1980) Release of inflammatory mediators from stimulated neutrophils. N Engl J Med 303:27–34. https://doi.org/10.1056/NEJM198007033030109
Savill JS, Henson PM, Haslett C (1989) Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Invest 84:1518–1527. https://doi.org/10.1172/jci114328
Houck JC (1979) Chemical Messengers of the Inflammatory Process, Elsevier/North-Holland Biomedical Press Amsterdam, ed 1–420.
Samuelsson B (1982) From studies of biochemical mechanisms to novel biological mediators: prostaglandin endoperoxides, thromboxanes and leukotrienes. Biosci Rep 3:791–813. https://doi.org/10.1007/BF01133779
Serhan CN, Fiore S, Brezinski DA, Lynch S (1993) Lipoxin A4 metabolism by differentiated HL-60 cells and human monocytes: conversion to novel 15-oxo and dihydro products. Biochemistry 32:6313–6319. https://doi.org/10.1021/bi00076a002
Takano T, Clish CB, Gronert K, Serhan CL (1998) Neutrophil-mediated changes in vascular permeability are inhibited by topical application of aspirin-triggered 15-epi-lipoxin A4 and novel lipoxin B4 stable analogues. J Clin Invest 101:819–826. https://doi.org/10.1172/JCI1578
Serhan CN (1994) Lipoxin biosynthesis and its impact in inflammatory and vascular events. Biochim Biophys Acta 1212:1–25. https://doi.org/10.1016/0005-2760(94)90185-6
Serhan CN (2002) Endogenous chemical mediators in anti-inflammation and pro-resolution. Curr Med Chem Anti-Inflamm Anti-Allergy Agents 1:177–192. https://doi.org/10.2174/1568014023355881
Barnes PJ, Pedersen S, Busse WW (1998) Efficacy and safety of inhaled corticosteroids New developments. Am J Respir Crit Care Med 157(3):1–53. https://doi.org/10.1164/ajrccm.157.3.157315
Adelizzi RA (1999) COX-1 and COX-2 in health and disease. J Am Osteopath Assoc 99:S7–S12
Fiorucci S, de Lima OM, Palazzetti B, Distrutti E, McKnight W, Dicay M, Ma L, Romano M, Morelli A, Wallace JL (2002) Cyclooxygenase-2-derived lipoxin A4 increases gastric resistance to aspirin-induced damage. Gastroenterology 123:1598–1606. https://doi.org/10.1053/gast.2002.36558
Serhan CN, Chiang N (2004) Novel endogenous small molecules as the checkpoint controllers in inflammation and resolution: entrée for resoleomics. Rheum Dis Clin North Am 30:69–95. https://doi.org/10.1016/S0889-857X(03)00117-0
Serhan CN, Gotlinger K, Hong S, Arita M (2004) Resolvins, docosatrienes, and neuroprotectins, novel omega-3-derived mediators, and their aspirin-triggered endogenous epimers: an overview of their protective roles in catabasis. Prostaglandins Other Lipid Mediat 73:155–172. https://doi.org/10.1016/j.prostaglandins.2004.03.005
Fredman G, Serhan CN (2011) Specialized proresolving mediator targets for RvE1 and RvD1 in peripheral blood and mechanisms of resolution. Biochem J 437:185–197. https://doi.org/10.1042/BJ20110327
Serhan CN, Krishnamoorthy S, Recchiuti A, Chiang N (2011) Novel anti-inflammatory–pro-resolving mediators and their receptors. Curr Top Med Chem 11:629–647. https://doi.org/10.2174/1568026611109060629
Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LJA, Perretti M, Rossi AG, Wallace JL (2007) Resolution of inflammation: state of the art, definitions and terms. FASEB J 21:325–332. https://doi.org/10.1096/fj.06-7227rev
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820. https://doi.org/10.1016/j.cell.2010.01.022
Ricciotti E, FitzGerald GA (2011) Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31:986–1000. https://doi.org/10.1161/ATVBAHA.110.207449
Fiore S, Brezinski ME, Sheppard KA, Serhan CN (1991) The lipoxin biosynthetic circuit and their actions with human neutrophils. Adv Exp Med Biol 314:109–132. https://doi.org/10.1007/978-1-4684-6024-7_7
Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN (2001) Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2:612–619. https://doi.org/10.1038/89759
Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggström JZ, Petasis NA, Serhan CN (2013) The novel 13S, 14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. Faseb J 27:2573–2583. https://doi.org/10.1096/fj.13-227728
Serhan CN, Gupta SK, Perretti M, Godson C, Brennan E, Li Y, Soehnlein O, Shimizu T, Werz O, Chiurchiù V, Azzi A, Dubourdeau M, Gupta SS, Schopohl P, Hoch M, Gjorgevikj D, Khan FM, Brauer D, Tripathi A, Cesnulevicius K, Lescheid D, Schultz M, Särndahl E, Repsilber D, Kruse R, Sala A, Haeggström JZ, Levy BD, Filep JG, Wolkenhauer O (2020) The Atlas of Inflammation Resolution (AIR). Mol Aspects Med 74:100894. https://doi.org/10.1016/j.mam.2020.100894
Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, Joshi M, Halade GV (2015) Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol 84:24–35. https://doi.org/10.1016/j.yjmcc.2015.04.003
Kain V, Prabhu SD, Halade GV (2014) Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic Res Cardiol 109(6):444. https://doi.org/10.1007/s00395-014-0444-7
Hiram R, Xiong F, Naud P, Xiao J, Sirois M, Tanguay J-F, Tardif J-C, Nattel S (2021) The inflammation-resolution promoting molecule resolvin-D1 prevents atrial proarrhythmic remodelling in experimental right heart disease. Cardiovasc Res 117:1776–1789. https://doi.org/10.1093/cvr/cvaa186
Jaén RI, Fernández-Velasco M, Terrón V, Sánchez-García S, Zaragoza C, Canales-Bueno N, Val-Blasco A, Vallejo-Cremades MT, Boscá L, Prieto P (2020) BML-111 treatment prevents cardiac apoptosis and oxidative stress in a mouse model of autoimmune myocarditis. FASEB J 34:10531–10546. https://doi.org/10.3390/antiox9121259
Matouk AI, Taye A, El-Moselhy MA, Heeba GH, Abdel-Rahman AA (2017) The effect of chronic activation of the novel endocannabinoid receptor GPR18 on myocardial function and blood pressure in conscious rats. J Cardiovasc Pharmacol 69:23–33. https://doi.org/10.1097/FJC.0000000000000438
Zhang R, Liu S, Guo B, Chang L, Li Y (2014) Chemerin induces insulin resistance in rat cardiomyocytes in part through the ERK1/2 signaling pathway. Pharmacology 94:259–264. https://doi.org/10.1159/000369171
Rodríguez-Penas D, Feijóo-Bandín S, García-Rúa V, Mosquera-Leal A, Durán D, Varela A, Portolésm M, Roselló-Lletí E, Rivera M, Diéguez C, Gualillo O, González-Juanatey JR, Lago F (2015) The adipokine chemerin induces apoptosis in cardiomyocytes. Cell Physiol Biochem 37:176–192. https://doi.org/10.1159/000430343
Halade GV, Kain V, Serhan CN (2018) Immune responsive resolvin D1 programs myocardial infarction-induced cardiorenal syndrome in heart failure. FASEB J 32:3717–3729. https://doi.org/10.1096/fj.201701173RR
Salas-Hernández A, Espinoza-Pérez C, Vivar R, Espitia-Corredor J, Lillo J, Parra-Flores P, Sánchez-Ferrer CF, Peiró C, Díaz-Araya G (2021) Resolvin D1 and E1 promote resolution of inflammation in rat cardiac fibroblast in vitro. Mol Biol Rep 48:57–66. https://doi.org/10.1007/s11033-020-06133-8
Li J, Su H, Zhu Y, Cao Y, Ma X (2020) ETS2 and microRNA-155 regulate the pathogenesis of heart failure through targeting and regulating GPR18 expression. Exp Ther Med 19:3469–3478. https://doi.org/10.3892/etm.2020.8642
Malakar AK, Choudhury D, Halder B, Paul P, Uddin A, Chakraborty S (2019) A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol 234:16812–16823. https://doi.org/10.1002/jcp.28350
Tsai S, Vega GL (2020) Coronary and peripheral artery plaques: do differences in plaque characteristics translate to differences in lipid management? J Investig Med 68:1141–1151. https://doi.org/10.1136/jim-2019-001252
Pirault J, Bäck M (2018) Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front Pharmacol 9:1273. https://doi.org/10.3389/fphar.2018.01273
Chattopadhyay R, Raghavan S, Rao GN (2017) Resolvin D1 via prevention of ROS-mediated SHP2 inactivation protects endothelial adherens junction integrity and barrier function. Redox Biol 12:438–455. https://doi.org/10.1016/j.redox.2017.02.023
Artiach G, Carracedo M, Clària J, Laguna-Fernandez A, Bäck M (2018) Opposing effects on vascular smooth muscle cell proliferation and macrophage-induced inflammation reveal a protective role for the proresolving lipid mediator receptor chemR23 in intimal hyperplasia. Front Pharmacol 9:1327. https://doi.org/10.3389/fphar.2018.01327
Welty FK, Schulte F, Alfaddagh A, Elajami TK, Bistrian BR, Hardt M (2021) Regression of human coronary artery plaque is associated with a high ratio of (18-hydroxy-eicosapentaenoic acid + resolvin E1) to leukotriene B4. FASEB J 35:e21448. https://doi.org/10.1096/fj.202002471R
Petri MH, Laguna-Fernandez A, Tseng CN, Hedin U, Perretti M, Bäck M (2015) Aspirin-triggered 15-epi-lipoxin A4 signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int J Cardiol 179:370–372. https://doi.org/10.1016/j.ijcard.2014.11.010
Sato K, Yoshizawa H, Seki T, Shirai R, Yamashita T, Okano T, Shibata K, Wakamatsu MJ, Mori Y, Morita T, Matsuyama T-A, Ishibashi-Ueda H, Hirano T, Watanabe T (2019) Chemerin-9, a potent agonist of chemerin receptor (ChemR23), prevents atherogenesis. Clin Sci (Lond) 133:1779–1796. https://doi.org/10.1042/CS20190336
Xie S, Qi X, Wu Q, Wei L, Zhang M, Xing Y, Shi W, Chen S, Zeng X, Wang S, Guo H, Deng W (2021) Inhibition of 5-lipoxygenase is associated with downregulation of the leukotriene B4 receptor 1/ Interleukin-12p35 pathway and ameliorates sepsis-induced myocardial injury. Free Radic Biol Med 166:348–357. https://doi.org/10.1016/j.freeradbiomed.2021.02.034
Halade GV, Tourki B (2019) Specialized pro-resolving mediators directs cardiac healing and repair with activation of inflammation and resolution program in heart failure. Adv Exp Med Biol 1161:45–64. https://doi.org/10.1007/978-3-030-21735-8_6
Schmid M, Gemperle C, Rimann N, Hersberger M (2016) Resolvin D1 polarizes primary human macrophages toward a proresolution phenotype through GPR32. J Immunol 196:3429–3437. https://doi.org/10.4049/jimmunol.1501701
Braunwald E (2012) The treatment of acute myocardial infarction: the Past, the Present, and the Future. Eur Heart J Acute Cardiovasc Care 1:9–12. https://doi.org/10.1177/2048872612438026
Keyes KT, Ye Y, Lin Y, Perez-Polo JR, Gjorstrup P, Birnbaum Y (2010) Resolvin E1 protects the rat heart against reperfusion injury. Am J Physiol Heart Circ Physiol 299:H153–H164. https://doi.org/10.1152/ajpheart.01057.2009
Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I (2016) An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun 23(7):12859. https://doi.org/10.1038/ncomms12859
Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre-Roig C, Dittmar G, Döring Y, Drechsler M, Weber C, Zimmer R, Cenac N, Soehnlein O (2016) Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res 119:1030–1038. https://doi.org/10.1161/CIRCRESAHA.116.309492
Grimard BH, Safford RE, Burns EL (2016) Aortic stenosis: diagnosis and treatment. Am Fam Physician 93:371–378
Artiach G, Carracedo M, Plunde O, Wheelock CE, Thul S, Sjövall P, Franco-Cereceda A, Laguna-Fernandez A, Arnardottir H, Bäck M (2020) Omega-3 polyunsaturated fatty acids decrease aortic valve disease through the resolvin E1 and ChemR23 axis. Circulation 142:776–789. https://doi.org/10.1161/CIRCULATIONAHA.119.041868
Frangogiannis NG (2017) Fibroblasts and the extracellular matrix in right ventricular disease. Cardiovasc Res 113:1453–1464. https://doi.org/10.1093/cvr/cvx146
Hiram R, Provencher S (2021) Pulmonary disease, pulmonary hypertension and atrial fibrillation. Card Electrophysiol Clin 13:141–153. https://doi.org/10.1016/j.ccep.2020.10.001
Morin C, Hiram R, Rousseau E, Blier PU, Fortin S (2014) Docosapentaenoic acid monoacylglyceride reduces inflammation and vascular remodeling in experimental pulmonary hypertension. Am J Physiol Heart Circ Physiol 307:H574–H586. https://doi.org/10.1152/ajpheart.00814.2013
Hajar R (2019) Congestive heart failure: a history. Heart Views 20:129–132. https://doi.org/10.4103/HEARTVIEWS.HEARTVIEWS_77_19
Chiurchiù V, Leuti A, Saracini S, Fontana D, Finamore P, Giua R, Padovini L, Incalzi RA, Maccarrone M (2019) Resolution of inflammation is altered in chronic heart failure and entails a dysfunctional responsiveness of T lymphocytes. FASEB J 33:909–916. https://doi.org/10.1096/fj.201801017R
Harada M, Nattel S (2021) Implications of inflammation and fibrosis in atrial fibrillation pathophysiology. Card Electrophysiol Clin 13:25–35. https://doi.org/10.1016/j.ccep.2020.11.002
Yarmohammadi F, Hayes AW, Karimi G (2021) Possible protective effect of resolvin D1 on inflammation in atrial fibrillation: involvement of ER stress mediated the NLRP3 inflammasome pathway. Naunyn Schmiedebergs Arch Pharmacol 394(8):1613–1619. https://doi.org/10.1007/s00210-021-02115-0
Cairns JA (2007) The coxibs and traditional nonsteroidal anti-inflammatory drugs: a current perspective on cardiovascular risks. Can J Cardiol 23:125–131. https://doi.org/10.1016/s0828-282x(07)70732-8
Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk H, Bertagnolli M (2005) Adenoma prevention with celecoxib (APC) study investigators. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 352:1071–1080. https://doi.org/10.1056/NEJMoa050405
Pizzuto K, Averns HL, Baranchuk A, Abdollah H, Michael KA, Simpson C, Redfearn DP (2014) Celecoxib-induced change in atrial electrophysiologic substrate in arthritis patients. Ann Noninvasive Electrocardiol 19:50–56. https://doi.org/10.1111/anec.12097
Schmidt M, Christiansen CF, Mehnert F, Rothman KJ, Sørensen HT (2011) Non-steroidal anti-inflammatory drug use and risk of atrial fibrillation or flutter: population-based case-control study. BMJ 343:3450. https://doi.org/10.1136/bmj.d3450
Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, Himmelfarb CD, Khera A, Lloyd-Jones D, McEvoy JW, Michos ED, Miedema MD, Muñoz D, SmithVirani SCSS, WilliamsYeboah KAJ, Ziaeian B (2020) 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circulation 140(11):e596–e646. https://doi.org/10.1161/CIR.0000000000000678
Miller VT, Rothrock JF, Pearce LA, Feinberg WM, Hart RG, Anderson DC (1993) Ischemic stroke in patients with atrial fibrillation: effect of aspirin according to stroke mechanism. Stroke prevention in atrial fibrillation investigators. Neurology 43:32–36. https://doi.org/10.1212/wnl.43.1_part_1.32
Ofman P, Petrone AB, Peralta A, Hoffmeister P, Albert CM, Djousse L, Gaziano JM, Rahilly-Tierney C (2014) Aspirin use and risk of atrial fibrillation in the Physicians’ Health Study. J Am Heart Assoc 3:e000763. https://doi.org/10.1161/JAHA.113.000763
Birnbaum Y, Ye Y, Lin Y, Freeberg SY, Nishi SP, Martinez JD, Huang M-H, Uretsky BF, Perez-Polo JR (2006) Augmentation of myocardial production of 15-epi-lipoxin-a4 by pioglitazone and atorvastatin in the rat. Circulation 114:929–935. https://doi.org/10.1161/circulationaha.106.629907
Giles KM, Ross K, Rossi AG, Haslett C, Dransfield I (2001) Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol 167:976–986. https://doi.org/10.4049/jimmunol.167.2.976
Lucas CD, Dorward DA, Tait MA, Fox S, Marwick JA, Allen KC, Robb CT, Hirani N, Haslett C, Duffin R, Rossi AG (2014) Downregulation of Mcl-1 has anti-inflammatory pro-resolution effects and enhances bacterial clearance from the lung. Mucosal Immunol 7:857–868. https://doi.org/10.1038/mi.2013.102
Kim YR, Nam GB, Han S, Kim S-H, Kim K-H, Lee S, Kim J, Choi K-J, Kim Y-H (2015) Effect of short-term steroid therapy on early recurrence during the blanking period after catheter ablation of atrial fibrillation. Circ Arrhythm Electrophysiol 8:1366–1372. https://doi.org/10.1161/CIRCEP.115.002957
van Osch D, Dieleman JM, Nathoe HM, Boasson MP, Kluin J, Bunge JJH, Nierich AP, Rosseel PM, van der Maaten JM, Hofland J, Diephuis JC, de Lange F, Boer C, van Dijk D (2015) Dexamethasone for cardiac surgery study group. intraoperative high-dose dexamethasone in cardiac surgery and the risk of rethoracotomy. Ann Thorac Surg 100:2237–2242. https://doi.org/10.1016/j.athoracsur.2015.06.025
Schäcke H, Döcke WD, Asadullah K (2002) Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96:23–43. https://doi.org/10.1016/s0163-7258(02)00297-8
Stroeder J, Evans C, Mansell H (2015) Corticosteroid-induced bradycardia: Case report and review of the literature. Can Pharm J (Ott) 148:235–240. https://doi.org/10.1177/1715163515597451
Dalbeth N, Lauterio TJ, Wolfe HR (2014) Mechanism of action of colchicine in the treatment of gout. Clin Ther 36:1465–1479. https://doi.org/10.1016/j.clinthera.2014.07.017
Lennerz C, Barman M, Tantawy M, Sopher M, Whittaker P (2017) Colchicine for primary prevention of atrial fibrillation after open-heart surgery: systematic review and meta-analysis. Int J Cardiol 249:127–137. https://doi.org/10.1016/j.ijcard.2017.08.039
Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim I, Gamra H, Kiwan GS, Berry C, López-Sendón J, Ostadal P, Koenig W, Angoulvant D, Grégoire JC, Lavoie M-A, Dubé M-P, Rhainds D, Provencher M, Blondeau L, Orfanos A, L’Allier PL, Guertin M-C, Roubille F (2019) Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 381:2497–2505. https://doi.org/10.1056/NEJMoa1912388
Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL (2013) Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol 61:404–410. https://doi.org/10.1016/j.jacc.2012.10.027
Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, The SHK, Xu XF, Ireland MA, Lenderink T, Latchem D, Hoogslag P, Jerzewski A, Nierop P, Whelan A, Hendriks R, Swart H, Schaap J, Kuijper AFM, van Hessen MWF, Saklani P, Tan I, Thompson AG, Morton A, Judkins C, Bax WA, Dirksen M, Alings M, Hankey GJ, Budgeon CA, Tijssen JGP, Cornel GJ, Thompson PL (2020) LoDoCo2 trial investigators. colchicine in patients with chronic coronary disease. N Engl J Med 383:1838–1847. https://doi.org/10.1056/NEJMoa2021372
Christou GA, Christou KA, Korantzopoulos P, Rizos EC, Nikas DN, Goudevenos JA (2015) The current role of omega-3 fatty acids in the management of atrial fibrillation. Int J Mol Sci 16:22870–22887. https://doi.org/10.3390/ijms160922870
Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, DoyleJuliano RTRA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM (2019) REDUCE-IT investigators. cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med 380:11–22. https://doi.org/10.1056/NEJMoa1812792
Nicholls SJ, Lincoff AM, Garcia M, Bash D, Ballantyne CM, Barter PJ, Davidson MH, Kastelein JJP, Koenig W, McGuire DK, Mozaffarian D, Ridker PM, Ray KK, Katona BG, Himmelmann A, Loss LE, Rensfeldt M, Lundström T, Agrawal R, Menon V, Wolski K, Nissen SE (2020) Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the strength randomized clinical trial. JAMA 324:2268–2280. https://doi.org/10.1001/jama.2020.22258
Fussbroich D, Colas RA, Eickmeier O, Trischler J, Jerkic SP, Zimmermann K, Göpel A, Schwenger T, Schaible A, Henrich D, Baer P, Zielen S, Dalli J, Beermann C, Schubert R (2020) A combination of LCPUFA ameliorates airway inflammation in asthmatic mice by promoting pro-resolving effects and reducing adverse effects of EPA. Mucosal Immunol 13:481–492. https://doi.org/10.1038/s41385-019-0245-2
Wallentin L, Held C, Armstrong PW, Cannon CP, Davies RY, Hagström GCB (2016) Lipoprotein-associated phospholipase A2 activity is a marker of risk but not a useful target for treatment in patients with stable coronary heart disease. J Am Heart Assoc 5:e003407. https://doi.org/10.1161/JAHA.116.003407
Fender AC, Kleeschulte S, Stolte S, Leineweber K, Kamler M, Bode J, Li N, Dobrev D (2020) Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res Cardiol 115(2):10. https://doi.org/10.1007/s00395-019-0771-9
Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, Wang Q, Abu-Taha IH, Gorka M, Künzel S, El-Armouche A, Reichenspurner H, Kamler M, Nikolaev V, Ravens U, Li N, Nattel S, Wehrens X, Dobrev D (2020) Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ Res 127(8):1036–1055. https://doi.org/10.1161/CIRCRESAHA.120.316710
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20:3328. https://doi.org/10.3390/ijms20133328
Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha Dr I, Ghezelbash S, Reynolds CL, Shen YH, LeMaire SA, Schmitz W, Müller FU, El-Armouche A, Eissa NT, Beeton C, Nattel S, Wehrens XHT, Dobrev D, Li N (2018) Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 138:2227–2242. https://doi.org/10.1161/CIRCULATIONAHA.118.035202
Chen G, Chelu MG, Dobrev D, Li N (2018) Cardiomyocyte inflammasome signaling in cardiomyopathies and atrial fibrillation: mechanisms and potential therapeutic implications. Front Physiol 9:1115. https://doi.org/10.3389/fphys.2018.01115
Hiram R (2021) Cardiac cytokine therapy? Relevance of targeting Inflammatory mediators to combat cardiac arrhythmogenic remodeling. IJC Heart Vasc 37:100918. https://doi.org/10.1016/j.ijcha.2021.100918
Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21:677–687. https://doi.org/10.1038/nm.3893
Lee S, Nakahira K, Dalli J, Siempos II, Norris PC, Colas RA, Moon J-S, Shinohara M, Hisata S, Howrylak JA, Suh G-Y, Ryter SW, Serhan CN, Choi AMK (2017) NLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B4 synthesis. Am J Respir Crit Care Med 196:713–726. https://doi.org/10.1164/rccm.201604-0892OC
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, CANTOS Trial Group (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377:1119–1131. https://doi.org/10.1056/NEJMoa1707914
Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, Lo L, Kling D, Pergola P, Raj D, Libby P, Davidson M, Investigators RESCUE (2021) IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397:2060–2069. https://doi.org/10.1016/S0140-6736(21)00520-1
Lopategi A, Flores-Costa R, Rius B, López-Vicario C, Alcaraz-Quiles J, Titos E, Clària J (2019) Frontline science: specialized proresolving lipid mediators inhibit the priming and activation of the macrophage NLRP3 inflammasome. J Leukoc Biol 105:25–36. https://doi.org/10.1002/JLB.3HI0517-206RR
Gao R, Shi H, Chang S, Gao Y, Li X, Lv C, Yang H, Xiang H, Yang J, Xu L, Tang Y (2019) The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunopharmacol 74:105575. https://doi.org/10.1016/j.intimp.2019.04.022
Wei Z, Fei Y, Wang Q, Hou J, Cai X, Yang Y, Chen T, Xu Q, Wang Y, Li Y-G (2021) Loss of Camk2n1 aggravates cardiac remodeling and malignant ventricular arrhythmia after myocardial infarction in mice via NLRP3 inflammasome activation. Free Radic Biol Med 167:243–257. https://doi.org/10.1016/j.freeradbiomed.2021.03.014
Mottola G, Werlin EC, Wu B, Chen M, Chatterjee A, Schaller MS, Conte MS (2020) Oral Resolvin D1 attenuates early inflammation but not intimal hyperplasia in a rat carotid angioplasty model. Prostaglandins Other Lipid Mediat 146:106401
Valdes AM, Ravipati S, Menni C, Abhishek A, Metrustry S, Harris J, Nessa A, Williams F, Spector TD, Doherty M, Chapman V, Barrett DA (2017) Association of the resolvin precursor 17-HDHA, but not D- or E- series resolvins, with heat pain sensitivity and osteoarthritis pain in humans. Sci Rep 7(1):10748
Krashia P, Cordella A, Nobili A, La Barbera L, Federici M, Leuti A, Campanelli F, Natale G, Marino G, Calabrese V, Vedele F, Ghiglieri V, Picconi B, Di Lazzaro G, Schirinzi T, Sancesario G, Casadei N, Riess O, Bernardini S, Pisani A, Mercuri NB (2019) Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of Parkinson’s disease. Nat Commun 10(1):3945
Acknowledgements
The author is grateful to Mrs. Lucie Lefebvre and Mr. Carlos Lobos-Yevenes for secretarial assistance with the manuscript. Servier Medical Art gallery was used to produce the Graphical Abstract’s illustration. Pictures of SPMs’ chemical structure were obtained from PubChem free database.
Funding
This work is supported by a grant from the Fondation Institut de Cardiologie de Montréal (Canada) allocated to Dr. Roddy Hiram.
Author information
Authors and Affiliations
Contributions
RH performed the review of literature, wrote the manuscript, created the figures and tables, and approved the final version of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The author declare that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hiram, R. Resolution-promoting autacoids demonstrate promising cardioprotective effects against heart diseases. Mol Biol Rep 49, 5179–5197 (2022). https://doi.org/10.1007/s11033-022-07230-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-022-07230-6