
Abstract
Drug-protein binding plays a key role in determining the pharmacokinetics of a drug. The distribution and protein binding ability of a drug changes over a lifetime, and are important considerations during pregnancy and lactation. Although proteins are a significant fraction in plasma composition, they also exist beyond the bloodstream and bind with drugs in the skin, tissues or organs. Protein binding influences the bioavailability and distribution of active compounds, and is a limiting factor in the passage of drugs across biological membranes and barriers: drugs are often unable to cross membranes mainly due to the high molecular mass of the drug-protein complex, thus resulting in the accumulation of the active compounds and a significant reduction of their pharmacological activity. This review describes the consequences of drug-protein binding on drug transport across physiological barriers, whose role is to allow the passage of essential substances—such as nutrients or oxygen, but not of xenobiotics. The placental barrier regulates passage of xenobiotics into a fetus and protects the unborn organism. The blood–brain barrier is the most important barrier in the entire organism and the skin separates the human body from the environment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Keywords Breast milk, Drug-protein binding, Skin barrier, Protein binding, The blood–brain barrier, The placental barrier.
Drug-protein binding
Following absorption from the gastrointestinal system or direct infusion into bloodstream, a drug can bind with plasma proteins. The main proteins responsible for the binding in plasma are human serum albumin (HSA) and alpha-1-acid glycoprotein (AAG) [1,2,3]. Their concentrations and functions are listed in Table 1 [4,5,6]. While the protein-drug complex is relatively stable, the connection between molecules is reversible: molecules can join and separate, and the equilibrium state is reached a few hours after the administration of a medicine [3].
The structure and properties of the drug determine the extent of both: plasma protein binding (PPB) and protein binding (PB) in the sense of the general process, because these concepts should be distinguished. Lipophilicity (described as logP) and acid–base properties have a significant correlation with binding [7]. Hydrophobic and acidic drugs (e.g. warfarin, ketoprofen, ibuprofen, diazepam) bind preferably to HSA, while AAG connects with the basic ones (e.g. bupivacaine, clindamycine) [6,7,8,9] which should be taken into account while setting the therapy. Binding can also increase the solubility of compounds, especially hydrophobic ones, which would otherwise not be distributed in the aqueous environment of plasma [10]. A connection with the plasma proteins protects compounds from oxidation, lowers their toxicity and increases their half-life; drugs highly bound to the plasma proteins often reveal low first pass-metabolism [10,11,12]. Volume of distribution depends from PB as well and is decreased for drugs highly bound in plasma or increased for those which bind in tissues [13,14,15]. In addition drugs with higher affinity to a binding site on a plasma protein can replace one with lower affinity and such competition can lead to an uncontrolled rise in the concentration of the free, unbound fraction of a drug [16]. This can have serious consequences for narrow therapeutic index (NTID) drugs, where the difference between therapeutic and toxic doses is minimal (e.g. cardenolides, carbamazepine, phenytoin or warfarin [17, 18]) and any changes in the concentration of unbound, active form may be poorly tolerated by the organism. A sudden increase in the unbound fraction of the drug may provide a toxic effect [19]. This can lead to clinical consequences such as high risk of bleeding (warfarin) [19] or cardiac arrest (cardenolides) [20].
Level of protein binding depends on the properties of a drug but also on the surrounding environment for example, the temperature or pH. The latter can change the ionization state of the chemical compound [10, 21, 22]. The degree of plasma protein binding is governed by two variables, these being the unbound fraction of the drug in plasma (fu,p) and the percentage of plasma protein binding (PPB%), as given below in Eqs. 1 and 2 [3]:
An important consideration, often omitted in the literature, is that of drug-protein binding occurring outside the bloodstream. Compounds can bind with macromolecules in skin, breast milk, tissues and organs including the placenta [13, 23, 24], where they become ‘stuck’ and are thus prevented from reaching site of pharmacological action [23, 25]. These drugs may later pass into the plasma but in an uncontrolled way, which disturbs the dosage and the intended result of pharmacotherapy.
Transfer across biological membranes and barriers
The cell membrane is a semipermeable phospholipid bilayer, which separates cell organelles and cytoplasm from the environment. The ability of a molecule to cross the membrane depends on various factors including molecular weight, lipophilicity, ionisation state, the concentration on both sides of the barrier and protein binding [26, 27]. Low-molecular, lipid, unionised and unbound to plasma proteins molecules are reckoned as good penetrators through membranes, although extreme lipophilicity can cause accumulation in lipid environment [28, 29]. The mechanism of passive transport includes: simple diffusion (the undisturbed movement of small, lipophilic and unionized molecules across membrane) and facilitated diffusion, where specialized membrane proteins transport particles across barriers [30]. Active transport acts against the concentration gradient and as such requires energy, which is typically obtained by the hydrolysis of adenosine triphosphate (ATP). One such family of membrane proteins which actively transport drugs and other molecules across membranes is that of the ATP-binding cassette transporters (ABC transporters). They also contribute significantly to the passage of drugs through the blood–brain barrier or placenta [31]. Crossing biological barriers is a far more difficult matter. Their structure is more complex and there are additional mechanisms involved which prevent the passage of xenobiotics. Transfer across each barrier is explained in detail in the appropriate sections of this review. The most important, and the most difficult to pass, is the blood–brain barrier (BBB), which separates crucial organs from the environment.
Binding with HSA and AAG macromolecules affects the pharmacokinetic properties of pharmacologically-active compounds by decreasing their bioavailability and slowing their passage across biological membranes and barriers [32,33,34]; proteins themselves hardly penetrate through the cell membranes [35,36,37]. On the contrary new approaches in target therapy also reveal that drug binding to the protein carrier improves the effectiveness of several pharmacotherapies [38], e.g. a simple but effective mechanism was used in anti-tumour pharmacotherapy. Drug-protein conjugates penetrate into tumour circulation easily, through fenestrated capillaries, and stay trapped inside [39]. Albumin is also used as a protein carrier in commonly used drugs such as levemir, methotrexate, doxorubicin or paclitaxel [40, 41].
The blood–brain barrier
The blood–brain barrier (BBB) protects the central nervous system (CNS), which controls the whole body. Blood vessels, which are part of the BBB, are lined with tightly-connected endothelial cells. These unique connections between the endothelial cells are called tight junctions (TJs) and adherence junctions (AJs). The BBB is also composed of a basement membrane, glial cells, pericytes and surrounding neurons [42]. The close cell connections, viz. TJs and AJs prevent the passage of molecules through the intercellular space: transport can only take place through the intracellular route, i.e. within the cells [43]. Further defence is provided by unique metabolic activity of the barrier, with enzymes such as γ-glutamyl transpeptidase (γ-GTP) or alkaline phosphatase (AP) enabling chemical decomposition of compounds which can cross the BBB from the bloodstream [42, 44]. The CNS is also protected by the diversity of its routes of xenobiotic transport mechanisms [42, 43, 45]. Of the drug efflux transporters, i.e. those of ABC transporters family P-glycoprotein (P-gp), breast cancer resistance protein (BCRP) and multidrug resistance protein (MRP) demonstrate the highest activity in the BBB [46]. These transporters are responsible for drug distribution into the CNS and they can remove compounds which cross the barrier. Such efflux transporters have various substrates, including anti-cancer drugs, such as doxorubicin or methotrexate, antiepileptics, such as phenytoin and carbamazepine, and antidepressants, such as venlafaxine and paroxetine. While some drugs are not intended to act on the CNS, many others have to penetrate the brain to reach the main site of their activity and achieve successful therapy [46]. Drug transport across the blood–brain barrier has been widely described by Pardridge et al., with a series of articles providing a clear review of various aspects of barrier structure, the transport of drugs across it and the development of drugs for use in the CNS [47,48,49,50,51,52]. New approaches to delivering CNS drugs are also mentioned in other recent articles [53, 54].
The blood–brain barrier protects the CNS from harmful substances but its main role is to provide nutrients and oxygen, essential for the brain structures [42]. Oxygen molecules and drugs with low molecular weight and lipophilic properties can easily cross the BBB by simple diffusion [55]. Nutrition such as glucose, crucial for proper CNS function, or amino acids are carried by specific transporters (e.g. GLUT1 glucose transporter); macromolecules with high molecular mass, such as insulin, are transported in the process of endocytosis [43, 56]. Drugs can pass through the BBB by transmembrane diffusion, especially those that are lightweight or with high lipophilicity, or are carried by transporters, as in the case of glucose [55]. Two parameters (Eqs. 3 and 4) describe the amount of a drug that is passed into the CNS: log BB and log PS [57, 58]. Log BB represents the ratio between drug concentration in the CNS and plasma, while log PS indicates the permeability of certain surface; while the former is easier to obtain and more intuitive to understand, the latter is currently receiving more research attention [57, 58]:
A number of studies have examined protein binding with drugs and their ability to cross the BBB [55, 59,60,61]. Albumin, like other proteins, does not readily pass through the barrier, and its drug-macromolecule complex, cannot cross. Based on this assumption, it appears that drugs which bind more readily to proteins are less able to pass into the CNS (‘free drug theory’ [34, 62]). This may be true for most drugs, but there are some exceptions to the rule. Several drugs which cross the BBB without difficulty, such as benzodiazepines, steroids and a few hormones, demonstrate higher concentrations in the CNS than their unbound plasma fraction would indicate [63,64,65,66]. Similar observations were made by Videbæk et al. (1999) (Table 2) [67]. De Lange and Danhof [68] collected several papers which describe highly bound drugs (oxicams [69], imipramine and desimirpamine [70], isradipine, darodipine [71]) which also penetrate the BBB in surprisingly high extent (Table 2). There are several explanations of this phenomenon. Pardridge et al. claimed that the conformation of the protein changes while interacting with capillary walls and a drug molecule is freed from a complex [64, 65, 72], Tanaka and Mizojiri ended up with similar conclusion [66]. Another idea was protein-mediated transport in which binding with protein (especially AAG) enhances the BBB penetration [62]. Several authors claimed that more permeable structure of capillary endothelium in some regions may be the reason of the increased extraction of a complex into the CNS [67, 68]. There is no doubt that protein binding has a significant role for penetrating BBB; it can either decrease the passage or affect it in the other way with mechanisms still to be discovered. Mentioned studies reveal that in vivo analyses seem to be more applicable in that case. The unique environment in the CNS or interactions between proteins and brain capillaries apparently have a high impact on the matter, therefore there is a substantial difference between in vitro and in vivo results.
The placental barrier
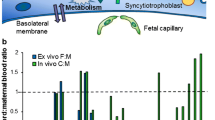
The placenta is a unique connection between mother and fetus. It is formed during the sixth week of pregnancy and exists until the time of birth. Its main function is to deliver nutrients and oxygen to the foetus and to remove waste and metabolites. Throughout the pregnancy, the placenta also adopts other roles: from the tenth week, it also produces hormones such as chorionic gonadotropin (CG), human placental lactogen (HPL), relaxin, progesterone, testosterone, oestrogens etc. and it manifests metabolic activity [73, 74]. Inside the placenta the blood vessels from a mother and a child are tangled together but the blood itself does not mix; despite this, sufficient exchange of substances is maintained between the organisms [73]. Due to its high permeability, the placenta acts more as a filter than an actual barrier [75]. Bacterial cells are retained within the placenta as are macromolecules, such as insulin or heparin, and immunoglobulins, except IgG. Most drugs pass through the placental barrier, including barbiturates, antibiotics, sulphonamides and alcohol [75]. Small molecules cross the barrier by simple diffusion, while drugs also cross by facilitated diffusion or active transport [75] or by endocytosis [73, 76,77,78]. It is assumed that the penetration of drugs through the placenta is limited mainly by protein binding rather than lipophilicity [79]. A significant role in the active transport of drugs is played by the ATP-binding cassette transporters, with the main ones being glycoprotein P and breast cancer resistance protein. They can either transport drug molecules to the fetal side or return them into maternal circulation; of these, the latter function is assumed to be more important, and plays a significant role in forming the placental barrier [77].
During pregnancy, it is difficult to avoid pharmacotherapy, and drug usage has increased in recent years. Drugs are administered in the treatment of chronic diseases such as epilepsy, diabetes, hypertension or they are prescribed temporally to treat infections such as the common cold. Additionally pregnant women often take over-the-counter drugs and dietary supplements, without medical advice [80, 81]. The amount of a drug which crosses the placental barrier is dependent on various factors: its physicochemical properties, pharmacokinetics, the concentration gradient on both sides of the barrier, the differences in pH in between maternal and foetal plasma and the levels of protein binding in both organisms [80, 81]. Protein binding is considered the important property in determining drug transport through the placenta, influencing both the speed and the extent of this process [16, 74].
The distribution of a drug between mother and child is limited also by the concentration of main plasma proteins. These change continually over the course of pregnancy: while the concentration of foetal albumin (alpha-fetoprotein, AFP) is lower than the maternal HSA level during the initial stages of pregnancy, it can be up to 20% higher than maternal HSA at childbirth [16]. The amount of foetal alpha-1-acid glycoprotein also increases with the development of the foetus; however, it never exceeds adult levels, remaining about 30–40% lower [16]. There are also differences in affinities to protein binding sites, with AFP attracting fewer molecules than HSA in adult plasma [82]. Protein binding can also occur in both maternal and foetal tissues; drug molecules can also form a repository in the placenta, from which it can be released in uncontrolled way into the maternal or foetal plasma [16].
In vitro experiments with propofol using a human placenta model by He et al. [79, 83] found propofol clearance to correlate with the concentration of fetal albumin. It appears that the potential to cross into the placenta is significantly dependent on binding with alpha-fetoprotein: an increase of alpha-fetoprotein concentration results in greater drug penetration. It was also found that infiltration across the placental barrier diminishes as the concentration of maternal HSA rises. Elsewhere, [84] it was found that HSA has a great influence on citalopram and fluoxetine placental transport, with its presence in the perfusion solute increased the degree of penetration; this effect was correlated with the affinity of the drugs to HSA: the passage of fluoxetine (PPB% = 94%) was significantly lower than that of citalopram (PPB% = 50%).
The placenta is considered a very weak barrier against xenobiotics and most of the administered drugs can easily cross it. Plasma protein binding appears to affect this process because it significantly limits placental transit, but alpha-fetoprotein concentration increase which can enhance the passage is also an important matter. The accumulation of drugs in the placenta is still underestimated and it needs to be studied in detail to get a clearer picture of the processes that can affect fetal safety during pharmacotherapy.
Skin barrier
The skin is the largest human organ, and one which separates the internal environment from the surroundings and protects it from various pathogens. As a barrier, the skin also prevents the penetration of many chemical compounds. This poses a challenge for the design of dermatological preparations, which are quite common in modern pharmacotherapy, mainly due to their easy and convenient application and lack of side effects typical for the oral administration. Dermatological application can also enhance the systemic activity of a drug [85]. It has previously been assumed that most of the administered drug particles are absorbed into the skin circulation, thus allowing them to pass into the bloodstream, and that the process was regulated by the skin structure and condition, the structure of the drug and the type of pharmaceutical formulation [85, 86]. However, later studies suggest that the most important factors determining skin penetration are the structure and properties of the drug [85]. The permeability of the skin varies across its surface in response to changes in its structure, for example, variation in the numbers of follicles or the thickness of the stratum corneum [87].
Externally administered drugs can bind with the proteins within the skin layers, which can be desirable if only local action is intended: the drug will accumulate at its site of activity and will not cause any adverse systemic effects. However, in the case of transdermal drugs such skin protein binding will disturb their flow into the circulation, slow the passage through the skin and reduce the overall amount of active molecules in the system. Previously, it was found that highly protein binding drugs achieved lower concentration in plasma and the time of skin penetration was longer [88]. A 2008 study [89] examining the different pharmacodynamics of tacrolimus and pimecrolimus with regard to their ability to penetrate the skin found that pimecrolimus is more likely to bind non-specifically with various skin protein than tacrolimus, thus yielding a lower systemic concentration (Table 3). Similarly, benzocaine has also been found to accumulate in the skin through non-specific binding (Table 3) [90].
These results suggest that protein binding in the skin should be carefully studied in case of dermatological formulations, especially for highly protein binding drugs. Albumin is present in the skin [91] so the correlation between binding to HSA in plasma and skin could be a useful tool in pharmaceutical design. Nowadays, only the process of skin sensitisation is widely examined, which is supposed to be the result of non-covalent, reversible binding of various compound with skin proteins, including albumin [92, 93].
Drug penetration into breast milk
During lactation, similarly to pregnancy, it can be difficult to avoid the use of any medicines. Many women abandon breastfeeding when they take drugs, but often unnecessarily. The penetration of most xenobiotics into milk is quite low and only a fraction is typically ingested by an infant [94, 95]. The amount of a drug in breast milk is estimated using the M/P ratio for a particular drug (Eq. 5): this represents the ratio between concentration of the drug in milk and in maternal plasma:
This parameter should be calculated for each drug individually and can be obtained from clinical studies, observations of single, medical cases or derived mathematically, using chemometric methods [96,97,98,99,100,101]. Drugs with M/P value lower than 1 are considered as safe for breast-feeding child.
Breast milk is regarded as the best nourishment for a newborn infant, and for first six months of life, it can be its only food. Milk is produced in the mammary glands by specialised cells called lactocytes [102]. It is composed of a mixture of water and carbohydrates, proteins, lipids, vitamins and various other nutrients [103], with the composition changing over the course of lactation [104, 105]. During the first stage of lactation, breast milk, colostrum, is composed mainly of structural proteins and proteins which support the immune system e.g. lactoferrin or immunoglobulins [106, 107]. This is later replaced by transitional milk, which has higher levels of carbohydrates and lipids and it is more nutritious than colostrum. Mature milk is produced around the third week after birth, and it consists of around 7% carbohydrates, 4% lipids and less than 1% proteins [102, 106, 108]. This is later replaced by transitional milk, which has higher levels of carbohydrates and lipids and it is more nutritious than colostrum. Mature milk is produced around the third week after birth, and it consists of around 7% carbohydrates, 4% lipids and less than 1% proteins [109].
Drugs mostly penetrate into breast milk by simple diffusion along a concentration gradient. This process is also limited by various factors connected with the compound structure: molecular weight, lipophilicity, protein binding or pKa [94, 110]. The pKa of a drug plays an important role on its accumulation in milk: the mean pH of breast milk ranges from 7.1–7.2 while that of plasma is around 7.4 [102, 105]. Weak bases become ionized in breast milk, trapping them inside the mammary gland and preventing their return to maternal plasma [111]. In addition, drugs with high lipophilicity can also accumulate in the lipid phase of breast milk, and while protein binding can prevent the passage of molecules into milk, drugs also bind with the breast milk proteins themselves [112]. The composition of the protein phase consists of alpha-S1, alpha-S2, beta- and kappa- caseins, alpha-lactoalbumin, beta-lactoglobulin, plasma albumin and lactoferrin, as well as immunoglobulins A, M, G and lysozyme and alpha-1-acid glycoprotein [104, 113]. However, drug binding is typically weaker in breast milk than in plasma [111].
Drug transfer into breast milk is still a difficult subject for in vivo study. Although clinical studies have been performed, they are usually based on very small groups of subjects or describe individual cases. Short-term use of drugs, during infection for example, seems to be less problematic than in the case of long-term pharmacotherapy. Women suffering from chronic conditions such as multiple sclerosis, epilepsy or psychiatric disorders, or those undergoing anticancer therapy, often want to maintain breast-feeding. A review by Constantinescu et al. [114] examined the usage of various immunosuppressive drugs, including azathioprine, belatacept, corticosteroids, cyclosporine A, everolimus, sirolimus and tacrolimus, during lactation. A study of methylprednisolone levels in the breast milk of two lactating women, one of them after renal transplantation and the other with multiple sclerosis [115, 116] found that methylprednisolone passes poorly into milk, which could be related with its high PPB%, estimated to be around 79% (Table 4) [116].
A 2013 study of antiepileptic drugs by Davanzo et al. [117] reviewed a body of pharmacokinetic and clinical data, including relevant infant dose (RID), and toxicity guidelines taken from LactMed [118] and Hale [119]. Older-generation drugs such as carbamazepine, phenobarbital, phenytoin and valproic acid were found to be relatively safe, even phenobarbital, which weakly binds with plasma proteins in maternal plasma (20–45% [120]). The overall conclusion was that neither pharmacokinetic or literature toxicity parameters are good predictors of the drug penetration into breast milk. The penetration of cisplatin across the placenta [121] and into breast milk (Table 4) [121, 122] was also studied. The drug was found to demonstrate poor penetration into milk as its platinum ion binds strongly with plasma proteins [120, 122]. However, cisplatin is contraindicated during lactation, probably due to the fact that that it accumulates during repeated dosage.
Postpartum depression or anxiety also requires a long-term treatment. SSRIs (selective serotonin reuptake inhibitors) are believed to be the safest drugs for lactating women because their high PPB% values, among other factors, prevent them from crossing readily into milk (Table 4) [123]. One exception is paroxetine, as it has been linked with an increased risk of heart dysfunction [124]. A detailed reviews about CNS drugs usage during lactation by Eberhard-Gran et al. and by Weissman et al. [125, 126]. They provide data regarding drug secretion into breast milk and recommendations for use. The latter study also points out the negative correlation between PB and M/P values [125]. Further information about the use of antidepressants is also given in a review by Lanza di Scalea and Wisner [127].
In most of the cases described, where the excretion of the drug into milk is very low, one of the main reasons mentioned is high plasma protein binding. This may indicate that milk penetration may be the most PPB dependent of all the barriers described in this review. Another issue to consider is milk protein binding, which further reduces the amount of medicine an infant ingests. Experts claim that breastfeeding should not be interrupted during pharmacotherapy unless it is necessary, which seems to be a reasonable solution. However, there should be sufficient evidence that the medicine is safe for breastfed infants or wouldn’t interrupt the lactation.
Summary
Drug-protein binding has a significant influence on the pharmacokinetic properties of most compounds. It can limit the bioavailability of active compounds by controlling their passage through biological membranes; however, binding to plasma proteins allows hydrophobic drugs to be transported in the aqueous environment of the human organism. The drug-protein complex is less likely to cross the placental barrier or to enter breast milk, which decreases the negative effect of medicines on breastfeeding infants; however, some drugs can accumulate in placental tissues or in milk by binding with proteins in these regions, and upon their later release, enter the foetus or infant in uncontrolled way. Passage through the blood–brain barrier is more complicated by mechanisms which protect the central nervous system, such as active efflux and the use of strong protein binding mechanisms. Additional unknown mechanisms that lead to the penetration of several protein-bounded drugs make this matter even more complex. Skin penetration is an important issue for transdermal drugs because they have a strong impact on their bioavailability and protein-binding interacts with this process.
Protein binding is relatively simple to study in vitro, but its effect on crossing biological barriers in a living organism could be difficult to grasp with these methods. This is probably due to the complexity of the entire barrier-crossing process and additional side-effects that simply cannot be obtained in the laboratory.
References
Sun H, Zhao H (2016) Physiologic drug distribution and protein binding. in: applied biopharmaceutics & pharmacokinetics. McGraw-Hill Education, 259
Kratz F, Elsadek B (2012) Clinical impact of serum proteins on drug delivery. J Control Release 161:429–445. https://doi.org/10.1016/j.jconrel.2011.11.028
Schmidt S, Gonzalez D, Derendorf H (2010) Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci 99:1107–1122. https://doi.org/10.1002/jps.21916
Spinella R, Sawhney R, Jalan R (2016) Albumin in chronic liver disease: structure, functions and therapeutic implications. Hepatol Int 10:124–132. https://doi.org/10.1007/s12072-015-9665-6
Caraceni P, Domenicali M, Tovoli A et al (2013) Clinical indications for the albumin use: still a controversial issue. Eur J Intern Med 24:721–728. https://doi.org/10.1016/j.ejim.2013.05.015
Israili ZH, Dayton PG, Frederick J, Di C (2001) Human alpha-1-glycoprotein and its interactions with drugs. Drug Metab Rev 33:161–235. https://doi.org/10.1081/DMR-100104402
Ghuman J, Zunszain PA, Petitpas I et al (2005) Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol 353:38–52. https://doi.org/10.1016/j.jmb.2005.07.075
Yamasaki K, Chuang VTG, Maruyama T, Otagiri M (2013) Albumin-drug interaction and its clinical implication. Biochim Biophys Acta - Gen Subj 1830:5435–5443. https://doi.org/10.1016/j.bbagen.2013.05.005
Otagiri M (2005) A molecular functional study on the interactions of drugs with plasma proteins. Drug Metab Pharmacokinet 20:309–323. https://doi.org/10.2133/dmpk.20.309
Chechłacz M, Korytowska N (2017) Związki wiążące się z białkami osocza u ludzi: Znaczenie w terapii oraz metody oznaczania wolnej frakcji. Biul Wydz Farm WUM 6:50–59
Svennebring A (2016) The connection between plasma protein binding and acute toxicity as determined by the LD50 value. Drug Dev Res 77:3–11. https://doi.org/10.1002/ddr.21286
Zhang F, Xue J, Shao J, Jia L (2012) Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today 17:475–485. https://doi.org/10.1016/j.drudis.2011.12.018
Heuberger J, Schmidt S, Derendorf H (2013) When is protein binding important? J Pharm Sci 102:3458–3467. https://doi.org/10.1002/jps.23559
Hong L, Rosenbaum S (2014) Developmental pharmacokinetics in pediatric populations. J Pediatr Pharmacol Ther 19:262–276
Korzekwa K, Nagar S (2017) Drug distribution part 2. predicting volume of distribution from plasma protein binding and membrane partitioning. Pharm Res 34:544–551. https://doi.org/10.1007/s11095-016-2086-y
Polin RA, Abman SH, Rowitch D, Benitz WE (2017) Developmental pharmacology and pharmacokinetics. In: fetal and neonatal physiology. Elsevier B.V., pp 187–249
Greenberg RG, Melloni C, Wu H et al (2016) Therapeutic index estimation of antiepileptic drugs: a systematic literature review approach. Clin Neuropharmacol 39:232. https://doi.org/10.1097/WNF.0000000000000172
Tamargo J, Le Heuzey JY, Mabo P (2015) Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide. Eur J Clin Pharmacol 71:549–567. https://doi.org/10.1007/s00228-015-1832-0
Mullokandov E, Ahn J, Szalkiewicz A (2014) Protein binding drug-drug interaction between warfarin and tizoxanide in human plasma. Austin J Pharmacol Ther
El-Mallakh RS, Brar KS, Yeruva RR (2019) Cardiac glycosides in human physiology and disease: update for entomologists. Insects 10:102
Cohen L (2004) Plasma protein-binding methods in drug discovery. in: optimization in drug discovery: In vitro methods. Humana Press, 111–222
Howard M, Hill J, Galluppi G, McLean M (2010) Plasma protein binding in drug discovery and development. Comb Chem High Throughput Screen 13:170–187. https://doi.org/10.2174/138620710790596745
Deb PK, Al-Attraqchi O, Prasad MR, Tekade RK (2018) Protein and tissue binding: implication on pharmacokinetic parameters. In: advances in pharmaceutical product development and research. Academic Press, 371–399
Faed EM (1981) Protein binding of drugs in plasma, interstitial fluid and tissues: effect on pharmacokinetics. Eur J Clin Pharmacol 21:77–81
Sun H, Zhao H (2012) Physiologic drug distribution and protein binding. In: applied biopharmaceutics & pharmacokinetics. McGraw-Hill Education, 259–308
Peck T, Hill S, Williams M (2008) Drug passage across the cell membrane. In: pharmacology for anaesthesia and intensive care, Third Edition. Cambridge University Press, 1–10
Benet LZ, Hosey CM, Ursu O, Oprea TI (2016) BDDCS, the Rule of 5 and drugability. Adv Drug Deliv Rev 101:89–98. https://doi.org/10.1016/j.addr.2016.05.007
Kok-Yong S, Lawrence L (2015) Drug distribution and drug elimination. In: basic pharmacokinetic concepts and some clinical applications
Waring MJ (2010) Lipophilicity in drug discovery. Expert Opin Drug Discov 5:235–248. https://doi.org/10.1517/17460441003605098
Tymoczko JL, Berg JM, Stryer L (2009) Biochemia. Wydawnictwo Naukowe PWN, Warszawa
Bamburowicz-Klimkowska M, Bogucka U, Szutowski M (2011) Funkcje transporterów typu ABC. Biul Wydz Farm WUM 3:34–40
Zhivkova ZD (2017) Quantitative structure—pharmacokinetics relationships for plasma protein binding of basic drugs. J Pharm Pharm Sci 20: 349–359. https://doi.org/10.18433/J33633
Di L, Kerns EH (2016) In vivo environments affect drug exposure. In: Drug-Like Properties. pp 15–28
Bohnert T, Gan LS (2013) Plasma protein binding: from discovery to development. J Pharm Sci 102:2953–2994. https://doi.org/10.1002/jps.23614
Yang NJ, Hinner MJ (2015) Getting across the cell membrane: an overview for small molecules, peptides, and proteins. Methods Mol Biol 1266:29–53. https://doi.org/10.1007/978-1-4939-2272-7_3
Zhang R, Qin X, Kong F et al (2019) Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv 26:328–342. https://doi.org/10.1080/10717544.2019.1582730
Pellegatti M, Pagliarusco S, Solazzo L, Colato D (2011) Plasma protein binding and blood-free concentrations: which studies are needed to develop a drug? Expert Opin Drug Metab Toxicol 7:1009–1020. https://doi.org/10.1517/17425255.2011.586336
Vhora I, Patil S, Bhatt P, Misra A (2015) Protein- and Peptide-drug conjugates: an emerging drug delivery technology. In: advances in protein chemistry and structural biology. 98:1–44
Maeda H, Wu J, Sawa T et al (2000) Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 65:271–284. https://doi.org/10.1016/S0168-3659(99)00248-5
Larsen MT, Kuhlmann M, Hvam ML, Howard KA (2016) Albumin-based drug delivery: harnessing nature to cure disease. Mol Cell Ther 4:3. https://doi.org/10.1186/s40591-016-0048-8
Feng J, Zhao C, Wang L et al (2018) Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics 8:2094–2106. https://doi.org/10.7150/thno.22069
Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD (2006) Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 1:223–236. https://doi.org/10.1007/s11481-006-9025-3
Brzezińska K, Ziaja M (2012) Struktura i funkcje bariery krew-mózg: the structure and role of blood-brain barrier. Postępy Biol Komórki 39:84–99
Meyer J, Mischeck U, Veyhl M et al (1990) Blood-brain barrier characteristic enzymatic properties in cultured brain capillary endothelial cells. Brain Res 514:305–309. https://doi.org/10.1016/0006-8993(90)91425-G
Taylor EM (2005) Efflux transporters and the blood-brain barrier. Nova Science Publishers, New York
Löscher W, Potschka H (2005) Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2:86–98. https://doi.org/10.1602/neurorx.2.1.86
Pardridge WM (2002) CNS Drug design based on principles of blood-brain barrier transport. J Neurochem. https://doi.org/10.1046/j.1471-4159.1998.70051781.x
Pardridge WM (2005) The blood-brain barrier: Bottleneck in brain drug development. NeuroRx. https://doi.org/10.1602/neurorx.2.1.3
Pardridge WM (2011) Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS 8:7
Pardridge WM (2012) Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 32:1959–1972
Pardridge WM (2015) Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin Pharmacol Ther 97:347–361
Pardridge WM (2019) Blood-brain barrier and delivery of protein and gene therapeutics to brain. Front Aging Neurosci 11:373. https://doi.org/10.3389/fnagi.2019.00373
Dong X (2018) Current strategies for brain drug delivery. Theranostics 8:1481–1493. https://doi.org/10.7150/thno.21254
Bors LA, Erdö F (2019) Overcoming the blood-brain barrier: challenges and tricks for CNS drug delivery. Sci Pharm 87:6. https://doi.org/10.3390/scipharm87010006
Banks WA (2009) Characteristics of compounds that cross the blood-brain barrier. BMC Neurol 9:S3. https://doi.org/10.1186/1471-2377-9-S1-S3
Ballabh P, Braun A, Nedergaard M (2004) The blood–brain barrier: an overview. Neurobiol Dis 16:1–13. https://doi.org/10.1016/j.nbd.2003.12.016
Pardridge WM (2004) Log(BB), PS products and in silico models of drug brain penetration. Drug Discov Today 9:392–393
Misra A, Ganesh S, Shahiwala A, Shah SP (2003) Drug delivery to the central nervous system: a review. J Pharm Pharm Sci 6:252–273
Kakee A, Terasaki T, Sugiyama Y (1996) Brain efflux index as a novel method of analyzing efflux transport at the blood-brain barrier. J Pharmacol Exp Ther 277:1550–1559
Kalvass JC, Maurer TS (2002) Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23:327–338. https://doi.org/10.1002/bdd.325
Kalvass JC, Maurer TS, Pollack GM (2007) Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo P-glycoprotein efflux ratios. Drug Metab Dispos 35:660–666. https://doi.org/10.1124/dmd.106.012294
Pardridge WM, Sakiyama R, Fierer G (1983) Transport of propranolol and lidocaine through the rat blood-brain barrier: primary role of globulin-bound drug. J Clin Invest 71:900–908. https://doi.org/10.1172/JCI110844
Jones DR, Hall SD, Jackson EK et al (1988) Brain uptake of benzodiazepines: effects of lipophilicity and plasma protein binding. J Pharmacol Exp Ther 245:816–822
Kobiler D, Lustig S, Shapira S (2001) The role of plasma protein binding in drug delivery to brain. In: blood-brain barrier: drug delivery and brain pathology. Kluwer Academic/Plenum Publishers, 311–321
Pardridge WM, Fierer G (1990) Transport of tryptophan into brain from the circulating, albumin-bound pool in rats and in rabbits. J Neurochem 54:971–976. https://doi.org/10.1111/j.1471-4159.1990.tb02345.x
Tanaka H, Mizojiri K (1999) Drug-protein binding and blood-brain barrier permeability. J Pharmacol Exp Ther 288:912–918
Videbæk C, Ott P, Paulson OB, Knudsen GM (1999) Blood-brain barrier transport and protein binding of flumazenil and iomazenil in the rat: implications for neuroreceptor studies. J Cereb Blood Flow Metab 19:948–955. https://doi.org/10.1097/00004647-199909000-00002
de Lange ECM, Danhof M (2002) Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin Pharmacokinet 41:691–703. https://doi.org/10.2165/00003088-200241100-00001
Jolliet P, Simon N, Brée F et al (1997) Blood-to-brain transfer of various oxicams: effects of plasma binding on their brain delivery. Pharm Res 14:650–656. https://doi.org/10.1023/A:1012165414610
Lin TH, Sawada Y, Sugiyama Y et al (1987) Effects of albumin and α1-acid glycoprotein on the transport of imipramine and desipramine through the blood-brain barrier in rats. Chem Pharm Bull 35:294–301. https://doi.org/10.1248/cpb.35.294
Urien S, Pinquier JL, Paquette B et al (1987) Effect of the binding of isradipine and darodipine to different plasma proteins on their transfer through the rat blood-brain barrier: drug binding to lipoproteins does not limit the transfer of drug. J Pharmacol Exp Ther 242:349–353
Pardridge WM (1988) Recent advances in blood-brain barrier transport. Annu Rev Pharmacol Toxicol 28:25–39. https://doi.org/10.1146/annurev.pharmtox.28.1.25
Van Der Aa EM, Peereboom-Stegeman JH, Noordhoek J et al (1998) Mechanisms of drug transfer across the human placenta. Pharm World Sci 20:139–148. https://doi.org/10.1023/A:1008656928861
Griffiths SK, Campbell JP (2015) Placental structure, function and drug transfer. Contin Educ Anaesthesia, Crit Care Pain 15:84–89. https://doi.org/10.1093/bjaceaccp/mku013
Traczyk WZ (2013) Fizjologia człowieka w zarysie. PZWL, Warszawa
Jamroziak K, Kowalczyk M, Robak T (2002) Białko oporności raka piersi ABCG2 (BCRP/MXR/ABCP) nowy transporter z nadrodziny ABC związany z opornością wielolekową. Acta Haematol Pol 33:403–416
Iqbal M, Audette MC, Petropoulos S et al (2012) Placental drug transporters and their role in fetal protection. Placenta 33:137–142. https://doi.org/10.1016/j.placenta.2012.01.008
Rubinchik-Stern M, Eyal S (2012) Drug interactions at the human placenta: What is the evidence? Front Pharmacol 3:126. https://doi.org/10.3389/fphar.2012.00126
He YL, Tsujimoto S, Tanimoto M et al (2000) Effects of protein binding on the placental transfer of propofol in the human dually perfused cotyledon in vitro. Br J Anaesth 85:281–286. https://doi.org/10.1093/bja/85.2.281
Evseenko D, Paxton JW, Keelan JA (2006) Active transport across the human placenta: impact on drug efficacy and toxicity. Expert Opin Drug Metab Toxicol 2:51–69. https://doi.org/10.1517/17425255.2.1.51
Gedeon C, Koren G (2006) Designing pregnancy centered medications: drugs which do not cross the human placenta. Placenta 27:861–868. https://doi.org/10.1016/j.placenta.2005.09.001
Unadkat J, Dahlin A, Vijay S (2005) Placental drug transporters. Curr Drug Metab 5:125–131. https://doi.org/10.2174/1389200043489171
He YL, Seno H, Sasaki K, Tashiro C (2002) The influences of maternal albumin concentrations on the placental transfer of propofol in human dually perfused cotyledon in vitro. Anesth Analg 94:1312–1314. https://doi.org/10.1097/00000539-200205000-00048
Heikkinen T, Ekblad U, Laine K (2002) Transplacental transfer of citalopram, fluoxetine and their primary demethylated metabolites in isolated perfused human placenta. BJOG An Int J Obstet Gynaecol 109:1003–1008. https://doi.org/10.1016/S1470-0328(02)01467-2
Cross SE, Roberts MS (1999) Targeting local tissues by transdermal application: understanding drug physicochemical properties that best exploit protein binding and blood flow effects. Drug Dev Res 46:309–315. https://doi.org/10.1002/(SICI)1098-2299(199903/04)46:3/4%3c309:AID-DDR17%3e3.0.CO;2-H
Benfeldt E, Serup J, Menné T (1999) Effect of barrier perturbation on cutaneous salicylic acid penetration in human skin: in vivo pharmacokinetics using microdialysis and non-invasive quantification of barrier function. Br J Dermatol 140:739–748. https://doi.org/10.1046/j.1365-2133.1999.02859.x
Nielsen JB, Benfeldt E, Holmgaard R (2016) Penetration through the skin barrier. Curr Probl Dermatology 49:103–111. https://doi.org/10.1159/000441549
Hwang S, Bayne W (1984) Influence of protein binding on the accumulation and depletion of drug from the skin. J Pharm Sci 73:710–712
Weiss HM, Fresneau M, Moenius T et al (2008) Binding of pimecrolimus and tacrolimus to skin and plasma proteins: implications for systemic exposure after topical application. Drug Metab Dispos 36:1812–1818. https://doi.org/10.1124/dmd.108.021915
Dalvi UG, Zatz JL (1982) Effect of skin binding on percutaneous transport of benzocaine from aqueous suspensions and solutions. J Pharm Sci 71:824–826. https://doi.org/10.1002/jps.2600710728
Divkovic M, Pease CK, Gerberick GF, Basketter DA (2005) Hapten-protein binding: From theory to practical application in the in vitro prediction of skin sensitization. Contact Dermat 53:189–200. https://doi.org/10.1111/j.0105-1873.2005.00683.x
Aleksic M, Thain E, Gutsell SJ et al (2007) The role of non-covalent protein binding in skin sensitisation potency of chemicals. Cutan Ocul Toxicol 26:161–169. https://doi.org/10.1080/15569520701212282
Settivari RS, Rowlands JC, Wilson DM, et al (2017) Application of evolving computational and biological platforms for chemical safety assessment. In: A Comprehensive Guide to Toxicology in Nonclinical Drug Development. 843–867
Hotham N, Hotham E (2015) Drugs in breastfeeding. Aust Prescr 38:156–159. https://doi.org/10.18773/austprescr.2015.056
Lauterbach R (2011) Stosowanie leków a karmienie piersią. Pediatr po Dyplomie 15:77–85
Atkinson HC, Begg EJ (1988) Relationship between human milk lipid-ultrafiltrate and octanol-water partition coefficients. J Pharm Sci 77:796–798. https://doi.org/10.1002/jps.2600770916
Atkinson H, Begg E (1988) Prediction of drug concentrations in human skim milk from plasma protein binding and acid-base characteristics. Br J Clin Pharmacol 25:495–503. https://doi.org/10.1111/j.1365-2125.1988.tb03334.x
Atkinson HC, Begg EJ, Darlow BA (1988) Drugs in human milk: clinical pharmacokinetic considerations. Clin Pharmacokinet 14:217–240. https://doi.org/10.2165/00003088-198814040-00003
Atkinson UC, Begg EJ (1990) Prediction of drug distribution into human milk from physicochemical characteristics. Clin Pharmacokinet 18:151–167. https://doi.org/10.2165/00003088-199018020-00005
Agatonovic-Kustrin S, Tucker IG, Zecevic M, Zivanovic LJ (2000) Prediction of drug transfer into human milk from theoretically derived descriptors. Anal Chim Acta 418:181–195. https://doi.org/10.1016/S0003-2670(00)00963-6
Abraham MH, Gil-Lostes J, Fatemi M (2009) Prediction of milk/plasma concentration ratios of drugs and environmental pollutants. Eur J Med Chem 44:2452–2458
Florea M (2014) Laktacja i karmienie piersią: Przegląd piśmiennictwa. Perinatol Neonatol i Ginekol 7:165–170
Newton E (2004) Breastmilk: the Gold Standard. Clin Obstet Gynecol 47:632–642
Lis J, Orczyk-Pawiłowicz M, Ka̧tnik-Prastowska I (2013) Białka mleka ludzkiego zaangażowane w procesy immunologiczne. Postepy Hig Med Dosw 67:529–547. https://doi.org/10.5604/17322693.1051648
Fleishaker JC, Desai N, McNamara PJ (1987) Factors affecting the milk to plasma drug concentration ratio in lactating women: physical interactions with protein and fat. J Pharm Sci 76:189–193. https://doi.org/10.1002/jps.2600760302
Kowalska D, Gruczyńska E, Bryś J (2015) Mleko matki-pierwsza żywność w życiu człowieka. Probl Hig Epidemiol 96:387–398
Rak K, Bronkowska M (2014) Immunologiczne znaczenie siary Hygeia Public Heal 49:249–254
Jenness R (1979) The composition of human milk. Semin Perinatol 3:225–239. https://doi.org/10.1007/BF02748197
Riodan, Jan; Wambach K (2009) Breastfeeding and human Lactation. Jones & Bartlett Learning; 4th edition
Spencer JP, Gonzalez LS, Barnhart DJ (2001) Medications in the breast-feeding mother. Am Fam Physician 64:119
Breitzka RL, Sandritter TL, Hatzopoulos FK (1997) Principles of drug transfer into breast milk and drug disposition in the nursing infant. J Hum Lact 13:155–158. https://doi.org/10.1177/089033449701300219
Gentile S, Rossi A, Bellantuono C (2007) SSRIs during breastfeeding: Spotlight on milk-to-plasma ratio. Arch Womens Ment Health 10:39–51
Ballard O, Morrow AL (2013) Human milk composition. nutrients and bioactive factors. Pediatr Clin North Am 60:46–74. https://doi.org/10.1016/j.pcl.2012.10.002
Constantinescu S, Pai A, Coscia LA et al (2014) Breast-feeding after transplantation. Best Pract Res Clin Obstet Gynaecol 28:1163–1173. https://doi.org/10.1016/j.bpobgyn.2014.09.001
Coulam C, Moyer T, Jiang N, Zincke H (1982) Breast-feeding after renal transplantation Transpl Proc 14:605–609
Cooper SD, Felkins K, Baker TE, Hale TW (2015) Transfer of methylprednisolone into breast milk in a mother with multiple sclerosis. J Hum Lact 31:237–239. https://doi.org/10.1177/0890334415570970
Davanzo R, Dal Bo S, Bua J et al (2013) Antiepileptic drugs and breastfeeding. Ital J Pediatr 39:50. https://doi.org/10.1186/1824-7288-39-50
Drugs and lactation database (LactMed). https://toxnet.nlm.nih.gov/pda/lactmed.htm. Accessed 11 Nov 2019
Hale TW (2012) Medications and mother’s milk, 15th ed. Pharmasoft Medical Publishing
Drugbank. https://www.drugbank.ca/drugs/DB01174. Accessed 20 Nov 2019
Hale TW (2012) Medications and mother’s milk, 15th ed. Pharmasoft Medical Publishing. https://doi.org/10.1515/JPM.2011.015
Hays KE, Ryu RJ, Swisher EM et al (2013) Duration of cisplatin excretion in breast milk. J Hum Lact 29:469–472. https://doi.org/10.1177/0890334413479671
Williams AS (2007) Antidepressants in pregnancy and breastfeeding. Aust, Prescr
Norris MM (2013) Use of antidepressants during pregnancy and lactation. Ment Heal Clin. https://doi.org/10.9740/mhc.n163520
Weissman AM, Levy BT, Hartz AJ et al (2004) Pooled analysis of antidepressant levels in lactating mothers, breast milk, and nursing infants. Am J Psychiatry 161:1066–1078. https://doi.org/10.1176/appi.ajp.161.6.1066
Eberhard-Gran M, Eskild A, Opjordsmoen S (2006) Use of psychotropic medications in treating mood disorders during lactation: Practical recommendations. CNS Drugs 20:187–198
Lanza Di Scalea T, Wisner KL (2009) Antidepressant medication use during breastfeeding. Clin Obstet Gynecol 52:483. https://doi.org/10.1097/GRF.0b013e3181b52bd6
Acknowledgements
This work was supported by an internal grant of the Medical University of Lodz no. 502–34-106.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest, financial or otherwise.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wanat, K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol Biol Rep 47, 3221–3231 (2020). https://doi.org/10.1007/s11033-020-05361-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05361-2