
Abstract
The influence of morin hydrate on changes of proliferative, metastatic, and adhesive potential of human ovarian cancer cells concerning the influence of decitabine, and decitabine with trichostatin A, and in comparison to untreated cells, were analyzed. The effect of morin hydrate, decitabine, and trichostatin A were examined in A2780 and SKOV-3 ovarian cancer cell lines using MTS assay, clonogenic assay, adhesion to endothelial HMEC-1 cells, transwell migration assay and cell cycle analysis. The expression level of epithelial to mesenchymal transition (EMT) markers was quantified using PCR Array in relation to the level of global methylation determined with Methylated DNA Quantification Kit. We observed statistically significant inhibition of adhesive and migratory potential of both cell lines and the accumulation of G0/G1 phase A2780 cells after treatment with morin hydrate. Our studies confirmed the influence of morin hydrate on down-regulation of genes considered as up-regulated during EMT, and up-regulation of some genes considered as down-regulated during EMT in A2780 and SKOV-3 cells. Phenotypic changes were associated with molecular changes in cells, eg. decrease of the expression level of genes associated with adhesion, and an increase of genes down-regulated during EMT, after morin hydrate treatment in comparison to untreated control cells in both cell lines, were observed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Epigenetic basics of EMT in cancer
Epigenetic aberrations, including DNA methylation, histone modifications, and microRNA dysregulation play an important role in the development and progression of ovarian cancer. Diet and food ingredients can affect epigenome, both, at the DNA level and histone modification. DNA methylation is a kind of covalent modification at the 5′ position of the cytosine ring (5mC) within CpG dinucleotides and is associated with transcriptionally repressed chromatin. DNA methylation is catalyzed by methyltransferases (DNMT1, DNMT3A, DNMT3B, and DNMT3L) in the presence of the S-adenosyl-l-methionine (AdoMet or SAM). Higher levels of DNMT1, DNMT3A, and DNMT3B have been observed in multiple cancers compared to healthy tissue. Aberrant methylation can lead to cancer by hypermethylation in promoters of tumor suppressor genes, reduced methylation of oncogene promoters, or genome-wide hypomethylation. Among histone modifications (acetylation, methylation, etc.) lysine acetylation catalyzed by several histone acetyltransferases (HATs), neutralize the positive charge of histone, weaken histone-DNA interactions and enhance transcription. The activity of HATs is balanced by histone deacetylases (HDACs—18 enzymes grouped into four classes), which restore the positive charge of the lysine, stabilize the chromatin architecture and act as transcription co-repressors [1]. High levels of HDAC’s cause histone hypoacetylation, being identified in cancer cells [2, 3].
In contrast to various genetic abnormalities, such as somatic mutations or genomic instability, which mainly lead to activation of oncogenes and inactivation of tumor suppressor genes, aberrant epigenetic genes silencing are potentially reversible by a combination of DNA-demethylating agents—DMNTs inhibitors and HDAC inhibitors [4, 5]. The interaction of DNA hypermethylation with histone hypoacetylation silences gene expression, due to the attachment of the 5-methylcytosine binding protein to the target gene promoter followed by HDAC recruitment [6]. The observed synergy between DNMT inhibitors (DNMTi) and HDAC inhibitors (HDACi) seems to be influential in extending the use of DNMTi [2, 7]. The relationship between DNA methylation and histone deacetylation is the basis for using a combination of epigenetic factors that target these mechanisms of gene silencing.
In developed countries, epithelial ovarian cancer (EOC) is one of the most common cancers. Histologically it is divided into five types: high-grade and serous cancer, endometriotic, mucinous, and clear cell carcinoma. Each of the histological subtypes of ovarian epithelial carcinoma shows a clear etiological and molecular pathogenesis and different sensitivity to treatment [8]. Most epithelial ovarian cancers spread through the blood or lymphatic system in the classical metastatic cascade mechanism. In the beginning, a local invasion of primary tumor cells into the surrounding stromal and extracellular matrix (ECM), invasion and spread by blood or lymph vessels, retention in a distant site, extravasation to the parenchyma, and eventually proliferation to form metastases. The basis for invasion and metastasis is the third type of epithelial to mesenchymal transition (EMT) in which non-motile, polarized epithelial cells undergo a series of biochemical changes, becoming mobile non-polar mesenchymal cells with invasive features with characteristic changes at the epigenetic and genetic level [5, 9].
The opposite mesenchymal to epithelial transition (MET) occurs at the metastatic site, allowing for anchored and extravasating cancer cells to regain epithelial features and proliferate into the tumor [9]. Typical biomarkers for EMT are cell-surface proteins, cytoskeletal markers, and extracellular proteins. Downregulation of genes encoding, among others, E-cadherin, occludin, desmoplakin, keratin (type I cytoskeletal 19), caveolin-2, and fibroblast growth factor-binding protein 1 is associated with the 3 type EMT [10,11,12]. On the other hand, up-regulated during EMT in cancer are genes encoding, among others, N-cadherin, α5β1 integrin, αVβ6 integrin, vimentin, β-catenin, caldesmon, moesin, α1(I) collagen, α1(III) collagen, fibronectin, matrix metalloproteinase-9, matrix metalloproteinase-2, stromelysin-1, metalloproteinase inhibitor 1, and versican core protein [11, 13]. EMT involves epigenetic alterations to chromatin modifications at both the DNA and protein level. Transcription factors important for regulation of EMT belong to the three main families such as TWIST (TWIST1 and TWIST2), SNAI (Snail and Slug), and ZEB (ZEB1 and ZEB2) involve various histone-modifying complexes to chromatin, mediating epigenetic silencing of genes [10, 11, 14].
Epigenetic modulation of EMT: natural compounds as an alternative epigenetic modulators
Nutritional genomics, dietary supplements, and natural compounds is an emerging field that may lead to reverse the epigenetic marks which are altered during carcinogenesis. Diet has a significant impact on the epigenetic pattern by regulating posttranslational modifications. For this reason, epigenetics has emerged as a topic of interest in the field of nutrigenomics [15]. Nowadays, nutraceuticals, defined as a substance, which has physiological benefit or provides protection against chronic disease, have received considerable interest due to potential nutritional, safety, and therapeutic effects. An interesting example includes anti-cancer compounds known as flavones. Morin (2′,3,4′,5,7-pentahydroxyflavone), a member of the flavanol group [16] exist in high concentrations in many plants such as Morus alba L. (Moraceae) and Otostegia persica (Lamiaceae), and it also exists in figs, apples, onions, tea and cereal grains [17]. The presence of two aromatic rings connected by a c-pyrone ring, in which polar hydroxyl groups are bound at various positions to characterize the chemical structure of morin. All hydroxyl groups are responsible for the free radical-scavenging properties of flavonoids [18]. Morin has a promising anti-cancerous activity by reducing DNA damage and modulating signaling pathways responsible for proliferation and differentiation, which could lead to cell-cycle arrest and apoptosis [19, 20]. Morin downregulated NF-κB which inhibited the inflammatory gene cascade along with the downregulation of several factors such as an inhibitor of apoptosis protein 1 and 2, in cell proliferation, cyclooxygenase-2 and cyclin D1, and invasion such as matrix metalloproteinases [21]. Evidence suggests that polyphenols can modulate chromatin structure through influencing the expression of chromatin remodeling enzymes, HDACs and DNMTs. Some of the flavonols, e.g., quercetin or fisetin, highly structurally similar to morin, were tested for DNMT inhibition, and all inhibited DNMT1-mediated DNA methylation. Additionally, quercetin causes HAT activation and HDAC inhibition [22, 23].
Among chemical compounds that inhibit DNMTs activity, there are distinguish nucleoside analogues and non-nucleoside inhibitors. Nucleoside analogues (i.e. 5-azacytidine, and 5-aza-2′-deoxycytidine—have been approved by the U.S. FDA for clinical use against hematological malignancies) incorporate into DNA and form an irreversible covalent complex with the enzyme triggering its proteasomal degradation [24]. Among HDAC inhibitors there are compounds divided into four classes hydroxamic acids (Trichostatin A—TSA, derivative isolated from Streptomyces), cyclic tetrapeptides, benzamides, and short-chain fatty acids [2].
In this report, we hypothesized that morin exerts the anti-proliferative, anti-metastatic, and anti-adhesive influence on ovarian cancer cell lines in reference to the changes of expression level of genes involved in epithelial to mesenchymal transition, and the methylation level in cells, similar to decitabine, and decitabine in combination with trichostatin A as the example of chemical epigenetic modulators.
Experimental section
Cell culture
Two human ovarian cancer cell lines were used, A2780 and SKOV-3. A2780 was purchased from European Collection of Authenticated Cell Cultures (ECACC®, 93112519) and were cultured in RPMI-1640 medium with 2 mM l-glutamine, 10% (v/v) fetal bovine serum (FBS, Biological Industries) and 0.1% (v/v) gentamicin (50 mg/ml gentamicin sulfate, Biological Industries), in a humidified atmosphere containing 5% CO2, at 37 °C. SKOV-3 were purchased from American Type Culture Collection (ATCC® HTB-77™) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (Biological Industries), and 0.1% (v/v) gentamicin (50 mg/ml gentamicin sulfate, Biological Industries), in a humidified atmosphere containing 5% CO2, at 37 °C. For the adhesion assay, dermal endothelium cells were used. HMEC-1, human microvascular endothelial cells—purchased from American Type Culture Collection (ATCC®, CRL-3243™) were cultured in MCDB131 (Gibco) supplemented with 10 ng/ml Epidermal Growth Factor (EGF, Thermo Fischer Scientific), 1 µg/ml hydrocortisone (Sigma-Aldrich), 10 mM glutamine (Sigma-Aldrich), 10% (v/v) fetal bovine serum (Biological Industries), and 0.1% (v/v) gentamicin (50 mg/ml gentamicin sulfate, Biological Industries), in a humidified atmosphere containing 5% CO2, at 37 °C.
Epigenetic modulators treatment
Morin hydrate (2′,3,4′,5,7-Pentahydroxyflavone, Sigma-Aldrich) 50 mM stock solution in DMSO (dimethyl sulfoxide, BioShop), decitabine (5-aza-2′-deoxycytidine, Sigma-Aldrich) 10 mM stock solution in a DMSO, and trichostatin A ([R-(E,E)]-7-[4-(Dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide, Sigma-Aldrich) 10 µM stock solution in a DMSO were used. Cells were exposed to different concentrations of all modulators (morin hydrate: 150 μM and 200 μM, decitabine: 12.5 μM and 25 μM, trichostatin A: 6.25 nM and 12.5 nM in combination with decitabine 25 μM—for the adhesion assay, migration assay, cell cycle analysis, and global methylation assay, and morin hydrate 150 μM, decitabine 25 μM, decitabine 25 μM with trichostatin A 6.25 nM—for EMT microarray assay) selected after a prior cytotoxicity test evaluation: MTS cell proliferation assay and clonogenic assay of cells in vitro.
Cell viability assay was performed using CellTiter 96®AQueousNon-Radioactive Cell Proliferation Assay (Promega). For the assay SKOV-3 cells were seeded at 6 × 103 cells and A2780 12 × 103/0.32 cm2 growth area in 96-well plates, grown overnight, and treated with all modulators for 24 h (morin hydrate range from 12.5–400 µM, decitabine range from 0.78 to 100 µM, trichostatin A range from 0.78 to 100 nM, additionally the highest used concentration of DMSO was examined). Samples were prepared in triplicates. Following the treatments, MTS assay was provided according to the manufacturer’s protocol. Plates were incubated at 37 °C for 4 h in a humidified CO2 incubator in the dark. The absorbance of each well was measured at 490 nm with ELISA plate reader (Dynex Technologies Triad Multi-Mode Microplate Reader). All treated cells were compared against untreated control cells (considered as 100% viable).
For the clonogenic survival assay, SKOV-3 (5 × 102 cell/well) and A2780 (10 × 102 cell/well) were seeded in 24-well plates. After serum starvation for 16 h cells were treated with all modulators for 24 h (morin hydrate range from 12.5 to 400 µM, decitabine range from 0.78 to 100 µM, trichostatin A range from 0.78 to 100 nM, additionally the highest used concentration of DMSO were examined) in a 37 °C cell culture incubator. Samples were prepared in triplicates. After 24 h, cells were washed, and the culture medium was replaced. After seven days of SKOV-3 incubation and 14 days of A2780 incubation, surviving colonies were washed with PBS and fixed and stained with 0.5% crystal violet (BTL) with 6% glutaraldehyde (Sigma-Aldrich) for 30 min. The cells were photographed and counted using an inverted microscope (Axiovert 40 CFL, Zeiss, magnification × 5). Clonogenicity was expressed as % of colonies in relation to untreated control cells.
For in vitro cell adhesion assay and for in vitro cell migration assays cell culture media supplemented with inhibitors was changed every day until cell harvesting after six days. Untreated control cells were cultured in cell growth media at the same time.
In vitro cell adhesion assays
Endothelial HMEC-1 cells were grown to 85–90% confluence in a 24-well plate (seeding density 1 × 105 cell/mL). Ovarian cancer cells, treated with all modulators for six days, were stained with 5 μM CellTrace™ CFSE dye (Invitrogen) in PBS (phosphate-buffered saline, Gibco) for 20 min at 37 °C, protected from light. After incubation, PBS dye solution was removed, and complete culture media were added for 30 min at 37˚C. In the meantime, HMEC-1 were stained with DAPI for 5 min at 37 °C (final conc. 5 μg/ml in PBS, Sigma-Aldrich) and washed with PBS (Gibco). Afterward, the ovarian cancer cells were trypsinized, pelleted and added onto HMEC-1 in the 24-well plate and incubated for 60 min at 37 °C. All experiments were carried out in triplicates. After incubation, each well was gently washed with PBS to remove non-adherent ovarian cancer cells. The adherent ovarian cancer cells were visualized and photographed using an inverted fluorescence microscope Nikon Eclipse Ti (magnification × 100) with a digital camera. The number of adherent ovarian cancer cells was measured with a NIS-Elements Imaging Software at excitation/emission wavelengths of 492 nm/517 nm for CellTrace™ CFSE dye and 354 nm/460 nm for DAPI dye as a ratio to control untreated cells [%].
In vitro cell migration assays
The migration of A2780 and SKOV-3 were analyzed in Transwell chambers ThinCert™ Cell Culture Inserts 24 Well (Greiner bio-one) with 8 µm pore size. Ovarian cancer cells, treated with all modulators for six days, were trypsinized, counted and resuspended in 100 μl serum-free media, and then seeded in the upper chamber (seeding density 0.5 × 105 cell/insert). The lower chamber filled with 600 μl of 10% (v/v) fetal bovine serum media as a chemoattractant. All experiments were carried out in triplicates. After 24 h of incubation, media from the top side of the insert was discarded, and cells were washed with PBS. The non-migrated cells were scraped from the top side of the membrane using a cotton swab, and the migrated cells were fixed in 4% paraformaldehyde (Sigma-Aldrich) for 30 min, then washed with PBS, and stained with 0.1% crystal violet (BTL) for 30 min. The cells were photographed and counted using an inverted microscope (Axiovert 40 CFL, Zeiss, magnification × 200).
Cell cycle analysis
The cell cycle analysis was carried out by flow cytometry after staining with propidium iodide (PI). Ovarian cancer cells were seeded in a 6-well plate (A2780 seeding density 0.6 × 105 cell/cm2, SKOV-3 seeding density 0.3 × 105 cell/cm2), in media supplemented with various concentrations of all modulators, with 24 h media replacement, for 72 h. All experiments were carried out in triplicates. Then adherent cells were harvested, centrifuged 300×g for 5 min, washed with PBS, and centrifuged 300×g for 5 min again. The harvested cells were fixed with 70% ethanol at − 20 °C for 3 h. The 500 μl of fixed cells were washed with PBS, centrifuged, and suspended in 200 μl of Muse® Cell Cycle Assay Kit (Merck) for 30 min in RT (room temperature), protected from light. The cell cycle analysis was performed on Muse™ Cell Analyzer (Merck), and results were analyzed with Muse 1.5 Analysis.
qRT-PCR-based microarray assay
Total RNA was isolated using the ZR-Duet™ DNA/RNA MiniPrep (Zymo Research) according to the manufacturer’s instructions. Total RNA (1 μg) was reverse transcribed to cDNA using the RT2 First Strand Kit (Qiagen), according to the manufacturer’s protocol. The relative quantitation of gene expression was performed by using the Epithelial to Mesenchymal Transition (EMT) PCR Array (Qiagen) in combination with RT2 SYBR® Green qPCR Mastermix (Qiagen), according to the manufacturer’s protocol. Samples were prepared in triplicate. For all reactions recommended thermal profile was used: activation of HotStart DNA Taq Polymerase for 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 1 min at 60 °C, followed by 10 min at 72 °C (Mx 3000p Stratagene). The level of target gene expression was quantified using the relative quantification. The ΔCq value for each sample was determined by calculating the difference between the Cq (quantitation cycle) value of the genes of interest and the Cq value of the reference housekeeping genes (e.g., RPLP0—Ribosomal protein, large, P0 for A2780 cells, HPRT1—Hypoxanthine phosphoribosyltransferase 1 for SKOV-3 cells, and B2M—Beta-2-microglobulin for both) used for normalization, according to the manufacturer’s protocol. That was determined for each unknown sample of cells treated with morin hydrate (150 μM), decitabine (25 μM), decitabine (25 μM) with trichostatin A (6.25 nM) for the control cells. Next, the ΔΔCq value for each sample was determined by subtracting the ΔCq value of the control from the ΔCq value of particular experimental samples. Finally, the normalized level of target gene expression was calculated by using the formula: 2–ΔΔCq.
Global methylation assay
Total DNA was prepared using the ZR-Duet™ DNA/RNA MiniPrep (Zymo Research) according to the manufacturer’s protocol. For the measurement of global DNA methylation status, Methylated DNA Quantification Kit was used (Abcam). To quantify the absolute amount of methylated DNA a standard curve was generated (0.5, 1.0, 2.0, 5.0 and 10.0 ng/µl for 5-mC), and the OD (optical density) values versus the amount of positive control at each concentration point were plotted. The slope (OD/ng) of the standard curve was determined using linear regression. The amount (ng) of methylated DNA (5-mC) was calculated by subtracting the negative control OD from sample OD and divided by the slope (OD/ng) and multiplied by 2, which is a factor to normalize 5-mC in the positive control to 100%, as the positive control contains only 50% of 5-mC, according to the manufacturer’s protocol. The percentage of methylated DNA was calculated by dividing the amount of 5-mC (ng) by the amount of input sample DNA (ng) and multiplying by 100%. Absorbance at 450 nm was measured using with ELISA plate reader (Dynex Technologies Triad Multi-Mode Microplate Reader). Samples were prepared in triplicate.
Statistical analysis
All results were analyzed statistically using the STATISTICA 12.5. Significance was analyzed using ANOVA, followed by post hoc Tukey’s multiple comparison test. The p values for the normalized gene expression were calculated based on a Student’s t-test of the replicate 2–ΔCq values for each gene in the control group and treatment groups. Data are presented as the mean ± standard deviation (SD). A p values less than 0.05 were considered as statistically significant.
Results
Morin hydrate inhibits the viability and colony formation of A2780 and SKOV-3 cells
The effect of morin hydrate, as well as decitabine and trichostatin A on ovarian cancer cells, were analyzed using MTS assay after treatment for 24 h at various concentrations. Treatment with all modulators caused a decrease in cell viability depending on the dose and cell line as shown in Fig. 1. In A2780 cells IC50 was 43.37 μM ± 3.1 μM for decitabine, 46.86 nM ± 2.48 nM for trichostatin A, and 376.73 μM ± 4.5 μM for morin hydrate. In SKOV-3 cells IC50 was 45.64 μM ± 1.9 μM for decitabine, 29 nM ± 0.78 nM for trichostatin A, and 388.57 μM ± 3.2 μM for morin hydrate.

Effects of decitabine (A), trichostatin A (B), and morin hydrate (C) on the viability of A2780 and SKOV-3 human ovarian cancer cells measured by MTS assay. The cells were treated with indicated concentrations of modulators for 24 h. The data are shown as mean ± SD of triplicate experiments. *ANOVA p < 0.05, **p < 0.01, ***p < 0.001 between the cells treated with the different concentrations of modulators and the untreated control cells
A2780 and SKOV-3 cell survival after treatment with all modulators was established by clonogenic assay. The results indicated that 24 h of all modulators treatment caused the inhibition of colony formation depending on dose in both cell lines, as shown in Fig. 2. A2780 cells appear more sensitive decitabine, slightly less sensitive to trichostatin A and similarly sensitive to morin hydrate as compared to SKOV-3 cells.

Effects of decitabine (A), trichostatin A (B), and morin hydrate (C) on A2780 and SKOV-3 colony formation measured by clonogenic assay. The cells were treated with indicated concentrations of modulators for 24 h and then SKOV-3 cultured for 7 days, and A2780 cultured for 14 days. Data are expressed as colony-forming efficiency calculated as a percentage in relation to untreated control cells. The data are shown as mean ± SD of triplicate experiments. *ANOVA p < 0.05, ***p < 0.001 between the cells treated with the different concentrations of modulators and the untreated control cells
Morin hydrate inhibits the adhesion of A2780 and SKOV-3 cells to HMEC-1 and caused a delay in migratory ability
Treatment with morin hydrate (150 μM) highly reduced the adhesive abilities of A2780 and SKOV-3 cells to human microvascular endothelial cells compared to untreated cells. In A2780, morin hydrate gives even better results than chemical compounds. Interestingly, we noticed a higher effect at smaller concentrations in both cell lines. Representative images of A2780 and SKOV-3 in vitro adhesion to HMEC-1 assay are shown in Fig. 3.

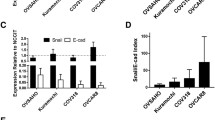
Effects of decitabine, decitabine with trichostatin A and morin hydrate on adhesive abilities of A2780 and SKOV-3 cells (both cell lines stained with CellTrace™ CFSE dye—green) to HMEC-1 human microvascular endothelial cells (stained with DAPI—blue). A Representative images of A2780 cells adhesion assay, Nikon Eclipse Ti, magnification × 100. B Representative images of SKOV-3 adhesion assay, Nikon Eclipse Ti, magnification × 100. C Inhibition of the A2780 and SKOV-3 cells adhesion to HMEC-1 cells, after treatment with morin hydrate compared to control untreated cells and cells treated with decitabine, and decitabine (dec.) with trichostatin A (TSA) [%].The data are shown as mean ± SD of triplicate experiments, ***statistically significant results (p < 0.001). (Color figure online)
We noticed a significant (p < 0.001) delay in migration in both experimental groups of cells treated with morin hydrate (150 μM, 200 μM) compared to untreated control cells. Statistically significant effects were also observed between cells treated with two different concentrations of morin hydrate (p < 0.001). Results obtained in SKOV-3 cells treated with morin hydrate were highly similar to those obtained from cells treated with decitabine, and trichostatin A. Representative images of A2780 and SKOV-3 in vitro transwell migration assay are shown in Fig. 4.

Effects of decitabine, decitabine with trichostatin A and morin hydrate on migratory abilities of A2780 and SKOV-3 cells analyzed in Transwell chambers ThinCert™ Cell Culture Inserts with 8 μm pore sizes. Migrated cells were stained with 0.1% crystal violet after 4% paraformaldehyde fixation. A Representative images of A2780 cells transwell migration assay, Axiovert 40 CFL, Zeiss, magnification × 200. B Representative images of SKOV-3 cells transwell migration assay, Axiovert 40 CFL, Zeiss, magnification × 200. C Inhibition of the A2780 and SKOV-3 cells mobility after treatment with morin hydrate compared to control untreated cells and cells treated with decitabine and decitabine (dec.) with trichostatin A (TSA) [%].The data are shown as mean ± SD of triplicate experiments, ***statistically significant results (p < 0.001). (Color figure online)
Morin hydrate induces G0/G1 phase cell cycle arrest in A2780 cells and G2/M phase cycle arrest in SKOV-3 cells
Morin hydrate induces the A2780 cell cycle arrest at the G0/G1 phase. Both concentration of morin hydrate statistically significant (150 μM—p < 0.001, and 200 μM—p < 0.01) increased the percentage of cells in the G0/G1 phase while the percentage of cells in the S and G2/M phase (p < 0.001) decreased. Analyses of the cell cycle distribution of A2780 cells after exposure to decitabine showed the cells' accumulation in the G2/M phase of the cell cycle compared to untreated cells. Statistically significant (p < 0.001) was also the increase in the number of cells in the S phase. This result was associated with a decrease in the number of cells in the G0/G1 phase. Similar effects were observed in A2780 cells treated with the combination of decitabine and trichostatin A, and statistically significant was the increase in the number of cells in the S phase after treatment with decitabine (25 μM) and trichostatin A (6.25 nM, 12.5 nM) (p < 0.01) (Fig. 3). Analyses of the cell cycle distribution of SKOV-3 cells after exposure to morin hydrate and other epigenetic modulators showed the statistically significant (p < 0.001) cell accumulation in the G2/M phase of the cell cycle compared to untreated cells. Additionally, SKOV-3 cells treated with decitabine (25 μM) and decitabine (25 μM) (p < 0.01) in combination with trichostatin A (6.25 nM, 12.5 nM) (p < 0.001) significantly increased the percentage of cells in the S phase. This percent increase was associated with a decrease in the number of cells in the G0/G1 phase (Fig. 5).

Effect of decitabine, decitabine with trichostatin A and morin hydrate on A2780 and SKOV-3 cell cycle analyzed by flow cytometry after staining with propidium iodide. A Representative profiles of the A2780 cell cycle. Comparison of cells treated with 150 μM and 200 μM morin hydrate; control, untreated cells; cells treated with 12.5 μM and 25 μM decitabine; cells treated with 25 μM decitabine and trichostatin A 6.25 nM and 12.5 nM. B Representative profiles of the SKOV-3 cell cycle. Comparison of cells treated with 150 μM and 200 μM morin hydrate; control, untreated cells; cells treated with 12.5 μM and 25 μM decitabine; cells treated with 25 μM decitabine and trichostatin A 6.25 nM and 12.5 nM. C Average percent of A2780 and SKOV-3 cells in various phases. The data are shown as mean ± SD of triplicate experiments. Statistically significant results **(p < 0.01), ***(p < 0.001) cells treated with morin hydrate, decitabine and decitabine (dec.) with trichostatin A (TSA) compared to control untreated cells. (Color figure online)
Measurement of transcriptomic responses to morin hydrate in A2780 and SKOV-3 cells
Fold change 2–ΔΔCq is the normalized gene expression 2–ΔCq in all the test samples divided the normalized gene expression 2–ΔCq in the control group. Fold regulation represents fold-change results in a biologically relevant way. Fold change values greater than one indicates an up-regulation, and the fold regulation is equal to the fold change; on the other hand, fold-change values less than one indicate a down-regulation, and the fold regulation is the negative inverse of the fold change. Fold changes in analyzed genes in cells treated with morin hydrate, decitabine, and trichostatin A, and additionally, comparison of fold changes of genes involved in EMT in A2780 and SKOV-3 cells are shown in Fig. 6.

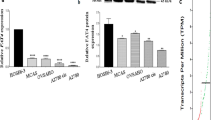
Effect of decitabine, decitabine with trichostatin A and morin hydrate on the expression of EMT related markers in A2780 and SKOV-3 cells. A Comparison of the average fold change of genes considered as up-regulated during EMT in A2780 and SKOV-3. With an indication of a significant change in the level of expression after using the compounds tested. The data are shown as mean ± SD of triplicate experiments, statistically significant results *(p < 0.05), **(p < 0.01), ***(p < 0.001). B Comparison of the average fold change of genes considered as down-regulated during EMT in A2780 and SKOV-3. With an indication of a significant change in the level of expression after using the compounds tested. The data are shown as mean ± SD of triplicate experiments, statistically significant results *(p < 0.05), **(p < 0.01), ***(p < 0.001). C Average fold change of all analyzed genes (in alphabetical order) in A2780 and SKOV-3 cells treated with morin hydrate (150 μM), decitabine (25 μM), decitabine (25 μM) with trichostatin A (6.25 nM) and comparing to control cells (p-values less than 0.05 are indicated in red), genes considered as up-regulated during EMT are marked in yellow, and genes considered to be reduced in EMT are marked in green—with an indication of a significant change in the level of expression after using the decitabine, decitabine with trichostatin A and morin hydrate. (Color figure online)
Analysis of expression of genes involved in EMT in A2780 cells have shown a statistically significant decrease of expression of genes involved in cell migration and motility AHNAK, JAG1, TGFB1 after morin hydrate, decitabine and decitabine with trichostatin A, ERBB3 after decitabine, CAV2, NODAL after decitabine, and decitabine with trichostatin A. As well as ECM molecules and those involved in cell adhesion: SERPINE after morin hydrate and decitabine with trichostatin A, COL1A2, ITGA5, MMP3 after morin hydrate and all used compounds, BMP1, BMP7, COL5A2, ITGB1, FN1, NODAL, STAT3, and VCAN after decitabine, and decitabine with trichostatin A. We observed a statistically significant decrease of ERBB1 (EGFR) gene involved in cells’ differentiation and proliferation after morin hydrate and all compounds treatment. Additionally, our results have shown a statistically significant decrease of TMEF1 and TMEM132A genes considered as up-regulated during EMT after all used compounds [8, 10, 23]. Analysis of expression of genes involved in EMT in SKOV-3 cells have shown statistically significant decrease of genes involved in cell migration and motility: AHNAK, FN1, and STAT3 after morin hydrate and all used compounds, and MST1R after decitabine and decitabine with trichostatin A. We observed statistically significant decrease of expression of genes involved in cell adhesion: VCAN after morin hydrate and all used compounds, and PTK2 after decitabine treatment. Additionally, our results showed a statistically significant decrease of CAMK2N1 and GNG11 genes considered as up-regulated during EMT after all used compounds.
Morin hydrate reduces global methylation in SKOV-3 cells and only partly in A2780 cells
Global DNA methylation in ovarian cancer cells treated with morin hydrate, decitabine, and trichostatin A was analyzed with Methylated DNA Quantification Kit in comparison to control untreated cells. The highest reduction in the level of A2780 methylation was observed in cells treated with morin hydrate (150 μM) (p < 0.05), decitabine (25 μM) with trichostatin A (6.25 nM) (p < 0.05), and decitabine (25 μM, 12.5 μM). Interestingly, an increase in the level of methylation was observed in A2780 cells treated with morin hydrate (200 μM) and decitabine (25 μM) with trichostatin A (12.5 nM). The highest reduction in the level of SKOV-3 methylation was observed in cells treated with morin hydrate (150 μM, 200 μM) (p < 0.01) and decitabine (25 μM) with trichostatin A (6.25 nM) (p < 0.001). All obtained results were statistically significant (Fig. 7).

The influence of decitabine, decitabine with trichostatin A and morin hydrate on changes in the level of global DNA methylation in A2780 and SKOV-3 cells compared to control untreated cells [%]. The data are shown as mean ± SD of triplicate experiments, statistically significant results *(p < 0.05), **(p < 0.01), ***(p < 0.001)
Discussion
Epithelial ovarian cancer is the most lethal gynecological malignancy, and tumor heterogeneity has been blamed for treatment failure. Epigenetic treatment regimens being tested in clinical or preclinical studies could provide promising novel treatment approaches and hope for improving patient survival [25]. In addition to standard cancer chemotherapy, an essential and new approach is the use of nutrigenomics, which possesses an enormous potential that can apply for the prevention and management of certain carcinomas and diseases. There is some evidence that dietary polyphenols substantially take part in the prevention of, among others, breast, skin, colorectal, prostate, pancreatic and lung cancers [26, 27]. This evidence was the reason for investigating the effect of morin hydrate on ovarian cancer cells. Morin hydrate is a naturally occurring polyphenolic flavonol compound which is an isomeric form of quercetin. It has been recently investigated for its many biological activities, such as anti-inflammatory, antioxidant, and as well as anticancer [28, 29]. In our study, we investigated the influence of morin hydrate on viability and clonogenicity of A2780 and SKOV-3 cells. Then, the migration and adhesion capacity of the tested cells, and the cell cycle analysis after morin hydrate treatment were performed. Next, we measured the transcriptomic responses to morin hydrate in A2780 and SKOV-3 cells compared to decitabine and decitabine with trichostatin A. Finally, the global methylation level in cells was examined. All tests were performed in relation to the untreated control of cancer cells. A2780 and SKOV-3 cells treated with decitabine, and decitabine in combination with trichostatin A, were positive control of the morin hydrate influence on cells in our studies.
We showed morin hydrate-induced inhibition of cell viability and inhibition of A2780 and SKOV-3 cell colony formation. Statistically significant in higher concentration. We observed no colonies after 400 μM of morin hydrate treatment in SKOV-3 cells, and we observed statistically significant colony-forming efficiency at the level of 68 ± 1% in A2780 and SKOV-3 cells after 200 μM morin hydrate.
We observed statistically significant inhibition of adhesion of A2780 cells to human microvascular endothelial cells (HMEC-1), wherein the highest inhibitory (65%) effect showed morin (150 μM), almost at the same level as decitabine (25 μM) which reduced adhesion by an average of 56.64%, and decitabine (25 μM) in combination with trichostatin A (6.25 nM) which reduced adhesion by an average of 53.85% in comparison to untreated cells. On the other hand, we observed statistically significant inhibition of adhesion of SKOV-3 cells to human microvascular endothelial cells (HMEC-1) after treatment with all compounds except morin (200 μM). Notably, other used concentrations we received similar results, wherein high inhibitory effect showed morin (150 μM) reducing adhesion by an average of 57.72%, decitabine (25 μM) in combination with trichostatin A (12.5 nM) reducing adhesion by an average of 66.7%, and decitabine (25 μM) reducing adhesion by an average of 50.41% [30, 31]. Similar results Lee JH et al. observed and indicated that morin might have anti-cancer activity by inhibiting MDA-MB-231 adhesion to HUVEC [30].
When analyzing the results of changes in the rate of migration, we observed the differential migratory ability of A2780 cells with statistically significant inhibition of mobility after treatment with all compounds in comparison to untreated cells. We also observed statistically significant inhibition of migratory ability of SKOV-3 cells after treatment with all compounds, wherein high influence on slowing down the migration rate showed morin (150 μM) reducing the mobility by an average of 67.35%, morin (200 μM) reducing the mobility by an average of 67%, decitabine in combination with trichostatin A (25 μM and 6.25 nM—73.15%, 25 μM and 12.5 nM—68.52%), and decitabine (25 μM) reducing the migratory ability by an average of 57.1%, all in comparison to untreated cells [32].
Our research revealed an influence on the cell cycle of SKOV-3 cells treated with epigenetic modulators. Analyses of the cell cycle distribution of SKOV-3 cells after exposure to all used compounds showed statistically significant cell accumulation in the G2/M phase of the cell cycle in a dose-dependent manner compared to the control groups of untreated cells. A similar effect on A549 and HCT116 cancer cells were observed at the G2/M phase of the cell cycle after decitabine treatment observed Shin et al. [32]. SKOV-3 cells treated with decitabine (25 μM) in combination with trichostatin A (6.25 nM, 12.5 nM) statistically significantly increased the percentage of cells in the S phase. This observation was associated with a decrease in the number of cells in the G0/G1 phase. A similar effect on the HeLa cancer cell cycle arrest at the G2/M phase after morin treatment observed Zhang et al. [19].
The potential influence of epigenetic inactivation on genes expression, which is down-regulated during epithelial to mesenchymal transition, may be confirmed. In this study, we observed an increase in their expression after treatment with morin hydrate and all other compounds. The most important changes relate to statistically significant up-regulation of CDH1 after all compounds which are significant since the loss of function of this gene is thought to contribute to cancer progression by increasing proliferation and metastasis. We observed statistically significant up-regulation of DSC2 (protein encoded by this gene constitutes the adhesive proteins of the desmosome cell–cell junction in epithelial cells), NUDT13, DESI1 (PPPDE2), RGS2, OCLN which encodes an integral membrane protein that is required for cytokine-induced regulation of the tight junction paracellular permeability barrier, and TSPAN13 which plays a role in the regulation of cell development, after morin hydrate (150 µM) treatment and KRT19, whose protein is responsible for the structural integrity of epithelial cells, after decitabine (25 μM). All the mentioned changes observed in A2780 cells. In SKOV-3 cells we observed statistically significant up-regulation of IL1RN after morin hydrate and after all other used compounds, CAV2 (the protein encoded by this gene may function as a tumor suppressor), CDH1, DSP (encoded the component of the protein which links adjacent cells), FGFBP1, KRT19, MST1R, NUDT13, OCLN, and SPP1 after morin hydrate treatment, TFPI2, TIMP1 (the natural inhibitor of the matrix metalloproteinases) and TSPAN13 after morin hydrate, and decitabine with trichostatin A, COL5A2 and ERBB3 after decitabine and decitabine with trichostatin A. RGS2 after decitabine with trichostatin A. Due to this increased expression, cells are less susceptible to metastasis. Importantly, our studies suggested down-regulation of SMAD2, which mediates the signal of the TGF-β, and thus regulates multiple cellular processes, such as cell proliferation, apoptosis, and differentiation, in A2780 and SKOV-3 after decitabine. We observed downregulation of SNAI2, a transcriptional repressor that binds to E-box motifs and is also likely to repress E-cadherin transcription, and involved in epithelial-mesenchymal transitions and has anti-apoptotic activity, in A2780 cells after decitabine, and decitabine with trichostatin A (both statistically significant), and in SKOV-3 after decitabine and decitabine with trichostatin A, and downregulation of SNAI3 in SKOV-3 cells after decitabine and decitabine with trichostatin A. Additionally, we observed statistically significant downregulation of STAT3, the signal transducer and activator of transcription 3 which mediates the expression of a variety of genes in response to cell stimuli, and thus plays a crucial role in many cellular processes such as cell growth and apoptosis, in A2780 after decitabine and decitabine with trichostatin A, and SKOV-3 after morin hydrate and all other compounds. STAT3 is the significant intrinsic transcription factor in the induction of the EMT and in the pathogenesis of cancer. IL‐6/JAK2/STAT3 activation enhances metastasis by induction of EMT by the upregulation of EMT‐inducing transcription factors (Snail, ZEB1, JUNB, and TWIST‐1) and increases cell motility by focal adhesion kinase (FAK) activation [33]. In A2780 and SKOV-3 (statistically significant) cells after decitabine and decitabine with trichostatin A TWIST1 downregulation was observed, which is overexpressed in multiple human cancers, and the encoded protein promotes tumor cell invasion and metastasis. A similar effect Lo et al. observed and indicated that TWIST1 expression is suppressed by EGFR and JAK/STAT3 inhibitors [34]. Noteworthy, Yue et al. observed and indicated that the inhibition of EGFR or STAT3 activity repressed Survivin, VEGF and VIM expression and the colony-forming potential, viability, and migration of ovarian cancer cells [35]. It is important to remember that EMT is not only the cellular dynamic process but also the inherent nature of the cell, which forms a spectrum ranging from epithelium to mesenchyme as a function of the expression level of EMT markers, what is observed in ovarian cancer [36].
The global methylation level in both cell lines treated with all epigenetic modulators was analyzed. In A2780 cells morin hydrate (150 μM) (p < 0.01), decitabine (25 μM) with trichostatin A (6.25 nM) (p < 0.01), decitabine (25 μM, 12.5 μM) decrease the methylation level in comparison to untreated cells. On the other hand, morin hydrate (200 μM) and decitabine (25 μM) with trichostatin A (12.5 nM) increase the methylation level in A2780 cells. It is important to remember that in cancer cells, methylation and demethylation regulating mechanisms are much less regulated and lead to DNA methylation instability when gains or losses of methylation. Importantly, our research suggests that morin hydrate (150 μM, 200 μM) had a significant (p < 0.01) inhibitory effect on the global DNA methylation level in SKOV-3cells. We obtained a similar effect after decitabine (25 μM) with trichostatin A (6.25 nM) (p < 0.001) cell treatment. Similar results observed Xie Q et al. in MCF-7 and MDA-MB-231 human breast cancer cells treated with genistein (100 μM) in vitro. Noteworthy, morin hydrate as a catechol polyphenol, may indirectly inhibit DNMTs and DNA methylation, through changing intracellular concentrations of S-adenosyl methionine (SAM) [23, 37,38,39]. Hypomethylation and hypermethylation are found in the same tumor although in the different sequences, it had been noticed that there is a dependence between demethylation and de novo methylation mechanisms during tumorigenesis [38]. Ovarian cancer is generally characterized by the instability of the epigenome at the DNA and chromatin levels. It is necessary to examine the observed quantitative changes by qualitative methods targeted at the demethylation of tumor suppressor gene promoters to distinguish with changes in methylation in regulatory areas of oncogenes where reduced level affects transcription, often increasing it.
In conclusion, instability of the DNA methylation could contribute to oncogenesis by affecting classical oncogenes, but evidence suggests a greater involvement in the activation of genes associated with tumor invasion and metastasis. We observed the correlation between the decrease of methylation level and the changes of phenotypic cell features, e.g., reduction of cell mobility and reduction of their adhesive abilities after morin hydrate in a concentration of 150 µM treatment in A2780 and SKOV-3 cell lines. Furthermore, these observations correlate with a decrease of expression of genes characteristic for the mesenchymal type of cells. We also observed increase of genes that expression level is down-regulated while EMT during carcinogenesis which is more significant to confirm our assumptions about the potential regulative mechanisms of DNA methylation and histone deacetylation [31] on the level of gene expression and the validity of the use of compounds with potential inhibitory activity on DNA methyltransferases. Our research confirmed that the most significant was the usage of morin hydrate (150 μM) and decitabine (25 μM) with trichostatin A (6.25 nM) as a control compound, in comparison to untreated cells, what we observed in both ovarian cancer cell lines. We can assume, morin, as an example of anti-cancer flavones with a potential epigenetic modulation activity, may become a vital nutraceutical substance helpful in the standard anti-cancer therapy as well as in the prevention of carcinogenesis, in the near future.
Abbreviations
- ATCC:
-
American type culture collection
- DNMT:
-
DNA methyltransferase
- ECACC:
-
European collection of authenticated cell cultures
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial to mesenchymal transition
- EOC:
-
Epithelial ovarian cancer
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- MET:
-
Mesenchymal to epithelial transition
- SAM:
-
S-sdenosyl-l-methionine
- TSA:
-
Trichostatin A
References
Sun L, Fang J (2016) Epigenetic regulation of epithelia-mesenchymal transition. Cell Mol Life Sci 73(23):4493–4515
Smith HJ, Straughn JM, Buchsbaum DJ, Arend RC (2017) Epigenetic therapy for the treatment of epithelial ovarian cancer: a clinical review. Gynecol Oncol Rep 20:81–86
Earp MA, Cunningham JM (2015) DNA methylation changes in epithelial ovarian cancer histotypes. Genomics 106(6):311–321
Subramaniam D, Thombre R, Dhar A, Anant S (2014) DNA methyltransferases: a novel target for prevention and therapy. Front Oncol 4:1–13
Serrano-Gomez SJ, Maziveyi M, Alahari SK (2016) Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer 15(1):1–14
Momparler RL, Côté S, Momparler LF, Idaghdour Y (2014) Epigenetic therapy of acute myeloid leukemia using 5-aza-2’-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin Epigenet 6(1):1–12
Pathania R et al (2016) Combined inhibition of DNMT and HDAC blocks the tumorigenicity of cancer stem-like cells and attenuates mammary tumor growth. Cancer Res 76(11):3224–3235
Pratheeshkumar P et al (2018) FoxM1 and β-catenin predicts aggressiveness in Middle Eastern ovarian cancer and their co-targeting impairs the growth of ovarian cancer cells. Oncotarget 9(3):3590–3604
Klymenko Y, Kim O, Stack MS (2017) Complex determinants of epithelial: mesenchymal phenotypic plasticity in ovarian cancer. Cancers (Basel) 9(8):1–32
Bedi U, Mishra VK, Wasilewski D, Scheel C, Johnsen S (2014) Epigenetic plasticity: a central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 5(8):2016–2029
Loret N, Denys H, Tummers P, Berx G (2019) The role of epithelial-to-mesenchymal plasticity in ovarian cancer progression and therapy resistance. Cancers 11(838):1–22
Cummins PM (2012) Occludin: one protein, many forms. Mol Cell Biol 32(2):242–250
Meola J et al (2013) Caldesmon: new insights for diagnosing endometriosis. Biol Reprod 88(5):1–8
Nantajit D, Lin D, Li JJ (2015) The network of epithelial-mesenchymal transition: potential new targets for tumor resistance. J Cancer Res Clin Oncol 141(10):1697–1713
Carlos-Reyes A et al (2019) Dietary compounds as epigenetic modulating agents in cancer. Frontiers 10:1–14
Panche AN, Diwan AD, Chandra SR (2016) Flavonoids: an overview. J Nutr Sci 5:1–15
Yue M, Zeng N, Xia Y, Wei Z, Dai Y (2018) Morin exerts anti-arthritic effects by attenuating synovial angiogenesis via activation of peroxisome proliferator activated receptor-γ. Mol Nutr Food Res 62:1
Singh MP, Cho H, Kim JT, Baek KE, Lee H, Kang S (2019) Morin hydrate reverses cisplatin resistance by impairing PARP1/HMGB1-dependent autophagy in hepatocellular carcinoma. Cancers 11(7):986
Zhang Q et al (2018) Molecular mechanism of anti-cancerous potential of Morin extracted from mulberry in Hela cells. Food Chem Toxicol 112:466–475
Choudhury A, Chakraborty I, Banerjee TS, Vana DR, Adapa D (2017) Efficacy of morin as a potential therapeutic phytocomponent: insights into the mechanism of action. Int J Med Res Heal Sci 6(11):175–194
Sinha K, Chowdhury S, Sil P (2018) Phytochemicals and Human Health. In: Rani V, Yadav U (eds) Functional food and human health. Springer, Singapore, pp 201–226
Busch C, Burkard M, Leischner C, Lauer UM, Frank J, Venturelli S (2015) Epigenetic activities of flavonoids in the prevention and treatment of cancer. Clin Epigenet 7(1):1–18
Stepanić V, Novak Kujundžić R, Gall Trošelj K (2014) Epigenome, cancer prevention and flavonoids and curcumin in epigenetics and epigenomics. IntechOpen, London
Pechalrieu D, Etievant C, Arimondo PB (2017) DNA methyltransferase inhibitors in cancer: from pharmacology to translational studies. Biochem Pharmacol 129:1–13
Jia D et al (2017) Distinguishing mechanisms underlying EMT tristability. Cancer Converg 1(1):2
Jin H et al (2014) Morin, a flavonoid from Moraceae, suppresses growth and invasion of the highly metastatic breast cancer cell line MDA-MB-231 partly through suppression of the Akt pathway. Int J Oncol 45(4):1629–1637
Nasir A, Bullo NMH, Ahmed Z, Imtiaz A, Yaqoob E et al (2019) Nutrigenomics: Epigenetics and cancer prevention: a comprehensive review. Crit Rev Food Sci Nutr 7:1
Jangida AK et al (2019) Designing of fatty acid-surfactant conjugate based nanomicelles of morin hydrate for simultaneously enhancing anticancer activity and oral bioavailability. Colloids Surf B 175:202
Sinha K, Ghosh J, Sil PC (2018) Morin and its role in chronic diseases. In: Crusio WE, Lambris JD, Radeke HH, Rezaei N (eds) Advances in experimental medicine and biology. Springer, New York, pp 453–471
Lee JH et al (2016) Morin, a flavonoid from moraceae, inhibits cancer cell adhesion to endothelial cells and EMT by down-regulating VCAM-1 and N-cadherin. Asian Pac J Cancer Prev 17(7):3071–3075
Meng F, Sun G, Zhong M, Yu Y, Brewer MA (2013) Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2′-deoxycytidine on ovarian cancer. Br J Cancer 108(3):579–586
Shin DY, Sung Kang H, Kim GY, Kim WJ, Yoo YH, Choi YH (2013) Decitabine, a DNA methyltransferases inhibitor, induces cell cycle arrest at G2/M phase through p53-independent pathway in human cancer cells. Biomed Pharmacother 67(4):305–311
Jin W (2020) Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial–mesenchymal transition. Cells 9(217):1–24
Lo HW, Hsu SC, Xia WY, Cao XY, Shih JY, Wei YK, Abbruzzese JL, Hortobagyi GN, Hung MC (2007) Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res 67:9066–9076
Yue P, Zhang X, Paladino D, Sengupta B, Ahmad S, Holloway RW, Ingersoll SB, Turkson J (2012) Hyperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene 31:2309–2322
Miow QH, Tan TZ, Ye J, Lau JA, Yokomizo T, Thiery J-P, Mori S (2015) Epithelial–mesenchymal status renders differentialresponses to cisplatin in ovarian cancer. Oncogene 34:1899–1907
Fouad MA et al (2018) Impact of global DNA methylation in treatment outcome of colorectal cancer patients. Front Pharmacol 9:1173
Link A, Balaguer F, Shen Y, Lozano JJ, Leung H-CE et al (2013) Curcumin modulates DNA methylation in colorectal cancer cells. PLoS ONE 8(2):e57709
Xie Q et al (2014) Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosom Cancer 53:422–431
Acknowledgements
We thank Dr Dominik Bieg for help with statistical data analyses, and M.Sc. Jakub Rech for help with collecting the results of clonogenic assay.
Funding
The research was supported by Medical University of Silesia in Katowice, Poland: KNW-2-B24/N/8/K, KNW-2-B13/N/9/K and KNW-1-182/N/8/B, KNW-1-095/N/9/B.
Author information
Authors and Affiliations
Contributions
Conceptualization: Ewa Nowak, Ilona Bednarek, Methodology: Ewa Nowak, Daniel Sypniewski, Ilona Bednarek, Software: Ewa Nowak, Validation: Ewa Nowak, Formal Analysis: Ewa Nowak, Ilona Bednarek, Investigation: Ewa Nowak, Resources: Ewa Nowak, Ilona Bednarek, Data Curation: Ewa Nowak, Writing—Original Draft Preparation: Ewa Nowak, Writing—Review & Editing: Ilona Bednarek, Visualization: Ewa Nowak, Ilona Bednarek, Supervision: Ilona Bednarek, Project Administration: Ewa Nowak, Ilona Bednarek, Funding Acquisition: Ewa Nowak, Ilona Bednarek.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nowak, E., Sypniewski, D. & Bednarek, I. Morin exerts anti-metastatic, anti-proliferative and anti-adhesive effect in ovarian cancer cells: an in vitro studies. Mol Biol Rep 47, 1965–1978 (2020). https://doi.org/10.1007/s11033-020-05293-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05293-x