
Abstract
The purpose of this investigation was to characterize a new mutation in the LDL-receptor (LDLR) gene in three families with clinically diagnosed familial hypercholesterolemia (FH) from the South-Eastern part of Poland. Mutational screening with exon by exon sequencing analysis was performed in all probands. The novel mutation c986G>T (Cys308Phe) in the exon 7 of LDLR gene was found in three apparently unrelated probands with FH. Analysis of the receptor activity of peripheral blood lymphocytes by binding and uptake of DiL-LDL showed a significant reduction (by 24% versus healthy control) of the fluorescent label in the lymphocytes of patients heterozygous for this mutation. Concentrations of serum LDL-C in probands before treatment were between 9.5 and 10.5 mmol/l. All patients had corneal arcus and tendon xanthoma. Clinically, families were characterized by premature coronary artery disease. This mutation occurred relatively frequently in our group of patients with FH, but this could be explained by a founder effect since we demonstrated their common ancestors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant disorder, which affects approximately 1/500 individuals worldwide. Familial hypercholesterolemia is clinically characterized by elevations in low density lipoprotein cholesterol (LDL-C), tendon xanthoma, and premature coronary artery disease (CAD). The mortality rate of FH patients is significantly greater than in healthy subjects and several studies have shown, that lipid-lowering treatment with statins reduces coronary heart disease mortality [1–3]. Phenotypic expression of mutations in the LDL-receptor (LDLR) gene varies regarding severity of impairment of LDLR function, depending on class of mutation and also on some not fully elucidated environmental and genetic risk factors. Early identification and treatment of FH patients, especially with familial history of premature atherosclerosis and coronary heart disease, is of importance in prevention of development of premature CAD. Several data indicate that an unequivocal diagnosis of familial hypercholesterolemia, now termed autosomal dominant hypercholesterolemia, can be obtained only by identification of a mutation in LDLR gene, or APOB or PCSK9 genes. In this report we described a new mutation in LDLR gene reoccurring in families with severe risk of premature CAD, recruited from outpatient lipid clinic from South-Eastern part of Poland.
Materials and methods
Material included three probands from apparently unrelated families, in whom a novel LDLR mutation was found. These subjects were recruited from 41 families fulfilling criteria of FH. Enrollment criteria for the patients’ genetic screening based on Simon Broome register [4]. We included patients with definite as well as with possible FH.
Secondary hypercholesterolemia, such as hypothyroidism, diabetes and renal or hepatic disease, were excluded by laboratory tests. Subjects were examined for standard cardiovascular risk factors. Xanthomas were considered to be present if tendons appeared diffusely enlarged or had focal nodularities.
Serum lipids were determined by enzymatic methods using Roche reagents, apolipoproteins (apo) A1 and B were measured by immunoturbidimetric methods.
In all probands exon by exon sequence analysis was performed using direct sequencing of PCR products obtained with primers pairs published by Amsellen et al. [5] (BigDye 3.1 chemistry; ABI 3500 Genetic Analyser, Applied Biosystems—Life Technologies, Carlsbad, CA). In members of the probands’ families we performed a conventional targeted PCR-RFLP analysis to detect the presence of the above described mutation. Amplification primers were: 5′-GCA GGG ACC AAC GAA TGC T-3′ and 5′-CCT TCC TCA CAC TGG CAC TTG TA-3′. The product had 952 bp and was digested with BseMI (Fermentas, USA) restriction endonuclease into fragments of 505, 390 and 57 bp for the wild type 329C allele or 562 and 390 bp for the mutated 329F allele. Thus, transversion of cysteine codon TGC to phenylalanine TTC codon by the end of exon 7 was detected.
In three subjects carrying the novel mutation in LDLR gene we performed analysis of the LDLR activity using peripheral blood lymphocytes measurements of binding and uptake of DiL-LDL [6].
Peripheral blood lymphocytes were isolated from heparinized blood by Histopaque gradient centrifugation. The cells were incubated in RPMI medium at the density 105 in 0.15 ml. Binding of LDLR was tested by incubation on ice with 5 to 20 μM labeled LDL (BODIPY FL LDL, Molecular Probes, Eugene, OR) during 1 h. Incorporation of labeled LDL was tested by incubation in a serum free RPMI medium or the medium with 5% fetal calf serum (FCS, Sigma-Aldrich, St. Louis, MO) at 37°C during 2 h. Lymphocytes were analyzed by flow cytometry following additional labeling of the cells using anti-CD3-PerCP antibody (Becton–Dickinson, Franklin Lakes, NJ). Binding of labeled LDL was calculated as an increase of BODIPY fluorescence in CD3-PerCP gated cells over the control ones incubated without the labeled LDL. Similarly, incorporation of LDL was expressed as the ratio of fluorescence over the control incubation without labeled LDL. All experiments were done in triplicates [6–8].
Presence of atherosclerosis was ascertained by measurements of carotid intima media thickness and carotid-femoral pulse wave velocity, using Complior system. Intima-media thickness (IMT) was measured on both proximal and distal wall of the common carotid artery segments, on the left and right sides, 2 cm below the carotid bifurcations. Three measurements at each point were performed using non invasive two dimensional ultrasound with Ultrasonograf GE VIVID 7. We analyzed means and maximal IMT values. The coefficient of variation of IMT measurement was 5%.
Carotid-femoral pulse wave velocity was measured using Complior device. Mean of the 10 measurements was calculated. Coefficient of variation was below 5%.
Results
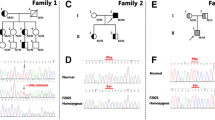
Clinical and biochemical characteristics of probands and examined family members who were heterozygous for the new mutation are presented in Table 1. Pedigrees of the families are presented in Fig. 1. The probands were female born in 1977 (No 62.1), female born in 1969 (No 84.1) and male born in 1974 (No 3.1). They did not smoke, had normal blood pressure and did not practice any kind of regular sport activities. They have a strong family history of premature CAD, all males with high cholesterol died early because of CAD. Patient’s 3.1 father, had two myocardial infarctions (MIs) at the age of 40 and 50 years. Father of patient 62.1, and his two brothers, had MIs also at their forties and underwent coronary by-pass surgery by the age of 50 years. Their sister, however, age 63, also with high cholesterol and confirmed LDLR mutation, had no MI until the time of examination. When extended pedigrees were collected, she turned out to be mother of patient 84.1.

Pedigree of the probands’ families. Family members heterozygous for c986G>T mutation are indicated with filled circles. Detailed pedigree examination revealed that mother of patient 84.1 and father of patient 62.1 were siblings, and daughter (29.1) of another brother of patient 84.4 was also a carrier of this mutation. Patients without numbers were not available for DNA analysis. ? = FH diagnosis based on clinical data, CAD coronary artery disease, MI myocardial infarction, C cholesterol, HDL HDL-cholesterol, LDL LDL-cholesterol, TG triglycerides, # treated with statin
All male relatives, who deceased prematurely from CVD, were heavy smokers, until the time of CVD events. Atherosclerosis, assessed by IMT and PWV measurements, showed higher values in male proband than in persons without mutation, matched by age and sex.
Tendon xanthomas were present in all probands. All patients with mutation had high LDL-C levels, and patient 3.1 had also elevated triglyceride concentration in serum. All FH patients available for lipid examination had normal HDL-C level with exception of father of patient 3, who also had type 2 diabetes.
A novel missense mutation (c986G>T, p C308F or C329F including the leader peptide of the receptor) in exon 7 of the LDLR gene was found in all three probands, of whom two turned out related to each other. Using multiple PCR-based ligation assay (MLPA) copy number abnormalities in LDLR gene were excluded in these subjects. Figure 2 shows results of DNA sequence analysis of exon 7 of LDLR gene in probands heterozygous for the novel mutation. On further targeted screening of their family members we verified that this mutation co-segregated with hypercholesterolemia (Fig. 3). Sister (84.3) and mother (84.4) of patient 84.1 had also high cholesterol levels and were heterozygous carriers of this mutation. The daughter (29.1) of the oldest brother of patient 84.4 also had very high LDL-C concentration and was a carrier of above mutation.

DNA sequence analysis showing mutation in exon 7 of LDLR gene

PCR-RFLP of family 3, confirming co-segregation of mutation with FH features. Mother (3.3) and son (3.2) of patient 3.1, both with normal blood lipids, were negative for c986 G>T mutation in LDLR gene (healthy controls, HC). PCR of DNA of patient 62.1 also shows presence of mutation. MWM, molecular weight marker (PBR322/AluI). Fragment lengths in base pairs
The results of the functional analysis of the LDLR activity of peripheral blood lymphocytes, evaluating their binding and uptake capacities for DiL-LDL in patients carrying this mutation are presented on Fig. 4. The figure shows mean values of binding and incorporation of the fluorescently labeled LDL in three patients heterozygous for c986G>T mutation in comparison to three matched healthy persons. We observed significantly lower internalization (P < 0.01) in the presence of RPMI medium at the lower concentration of LDL (5 mM/l). In the presence of FCS fortified medium internalization was also lower; however this effect did not reach statistical significance. Analysis of LDLR function in patient 62.1, c986G>T heterozygote, who did not received hypolipidemic treatment because of her pregnancy revealed also reduced binding in addition to lower internalization activity (by 23 and 22% respectively) in comparison to three healthy controls. This finding further confirms that the mutation has a functional consequence on LDL clearance and is the cause of the FH phenotype. However, in patient (3.1), carrying this mutation and treated with statin during last two years, plasma lipid concentrations normalized, and his Dil-LDL binding as well as incorporation were similar to healthy control. In father of patient 3.1 (with type 2 diabetes and thyroid disease; TSH-7,62 μIU/ml, fT4-12,1 ng/l) and with high plasma LDL-cholesterol level despite treatment, binding and incorporation were markedly lower: binding by 10 and 29%, internalization by 36 and 23% at LDL concentration equaled 5 and 20 mM/l, respectively.

Characterization of LDLR binding and internalization functions of peripheral blood monocytes using flow cytometric measurements. Mean fluorescence intensity of peripheral blood lymphocytes in patients with c986G>T mutation and in healthy controls. We explored LDLR expression in three patients: 3.1, 3.4 and 62.1 and 3 healthy controls. ** denotes p < 0.01
A prediction of molecular consequence of the mutation using Polyphen program confirmed that this missense mutation, located in the highly conserved region of LDLR gene, is damaging. Replacement of cysteine by phenylalanine results in a breakdown of disulfide bond at the extracellular domain of LDLR gene, corresponding to the ligand binding site.
Discussion
In this study we described a novel missense mutation c986G>T in exon 7 of LDLR gene. There are several lines of evidence, that mutation of LDLR 308Cys is functionally relevant. First, the mutation showed a clear linkage with hypercholesterolemia trait within families. Sequencing of all exons, promoter and introns did not reveal any other mutations in the LDL-receptor gene. In addition, functional studies of LDLR activity of peripheral blood lymphocytes using binding and uptake of DiL-LDL showed significant reduction of fluorescent label in the cells, in comparison to healthy control. Interestingly, this reduction was observed only with lower concentrations of LDL, but not with higher levels of LDL. However, it is in agreement with data, showing that lower concentrations of LDL in medium might stronger stimulate LDLR activity [7, 9]. Relatively low decrease of binding and internalization could result from the fact, that examined probands were heterozygotes and 50% of receptors retained functional activity. In other studies, products of gene with c986G>A (C308Y) mutation yielded 31% of LDLR activity in transfected COS cells [10], what is in line with our results. Several studies have indicated that human peripheral blood lymphocytes can mirror LDLR activity of hepatocytes and also of other body cells, so measurement of LDLR activity of lymphocytes by fluorescence flow cytometry may be used as a surrogate for hepatic LDLR activity in vivo. The LDLR activity measurement in patients treated with statin shows, that statin therapy increases LDLR activity in carriers of this mutation. This was a possible confounder in the assay in patients from the family number three, especially patient 3.4 who also had type 2 diabetes, similarly his high plasma triglyceride levels could decrease the size of LDL, thus affect binding affinity [11].
Furthermore, other missense mutation located at the same 308Cys codon of exon 7 (C308G, C308Y) are reported to be associated with FH [5, 12–15].
Eventually, the mutation located at the receptor domain responsible for the ligand binding was predicted to be damaging, when tested by PolyPhen program.
All probands carrying this mutation were characterized by very high levels of LDL-C, however in the male proband from the family three elevated triglyceride levels were also observed. Similar lipid profile was found in his father, who also presented type 2 diabetes. There is increasing number of data, that also patients with phenotype typical for familial combined hyperlipidemia (with high triglyceride levels), especially with markedly increased LDL-C and apo B serum concentration might have mutation in LDLR gene [16].
Families with this mutation had also typical features of severe FH: corneal arcus, tendon xanthoma and premature CAD, affecting males even before the age of 50 years.
The novel mutation c986G>T mutation was located in a highly conserved residues of the epidermal growth factor (EGF)—precursor homology domain of the LDLR gene.
Other mutations of this codon (C308G, C308Y) were described in families of Ashkenazi Jews, and of Canadian, Russian, or Flemish origin. In the study of Amsellem et al., including patients residing in France for at least three generations, mutation C308G caused by T>G transversion at c985 nucleotide of exon 7 was described [5]. Zakharova et al. described C308Y mutation caused by c985G>A transition in two families from population of St.-Petersburg, Russia. In her study, patients with this mutation were characterized by a similar lipid profile to ours, with very high LDL-C cholesterol concentration, corneal arcus, tendon xanthomas and CAD [4]. Mutation at the same nucleotide position as we describe occurred also in Chinese population (C308Y, c986G>A) (12) and in the Netherlands [15]. This was classified as class 2B mutation. Reshef et al. found in one family originated from Poland (Ashkenazi Jews) mutation C308G in LDLR gene [12].
Interestingly, in our population of FH patients this mutation occurred in four out of 14 patients with confirmed LDLR mutation among 41 screened subjects with clinical diagnosis of FH.
In a recent study on molecular pattern of LDLR gene mutation in Poland only one mutation in exon 7 was found [17]. The molecular background of FH in Poland is not clearly defined. Available data include persons living in the central part of Poland and the recruitment scheme of these studies could be biased toward severe manifestation [17]. South-Eastern part of Poland, which Kraków is the main city, has not been screened for familial hypecholesterolemia yet.
In summary we described clinical and molecular characteristic of a novel mutation in exon 7 from Kraków province family.
References
Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, Heeringa J, Witteman JC, Lansberg PJ, Kastelein JJ, Sijbrands EJ (2008) Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. Br Med J 337:a2423
Neil A, Cooper J, Betteridge J, Capps N, Mc Dovell I, Durrington P, Seed M, Humphries SE (2008) Reductions in all-cause, cancer and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolemia: a prospective study. Eur Heart J 29:2625–2633
Avellone G, Di Garbo V, Guarnotta V, Scaglione R, Parrinello G, Purpura L, Torres D, Campisi D (2010) Efficacy and safety of long-term ezetimibe/simvastatin treatment in patients with familial hypercholesterolemia. Int Angiol 29:514–524
Scientific Steering Committee on behalf of the Simon Broome Register Group (1991) Risk of fatal coronary heart disease in familial hypercholesterolemia. Br Med J 303:893–896
Amsellem S, Briffaut D, Carrié A, Rabès JP, Girardet JP, Fredenrich A, Moulin P, Krempf M, Reznik Y, Vialettes B, de Gennes JL, Brukert E, Benlian P (2002) Intronic mutations outside of Alu-repeat-rich domains of the LDL receptor gene are a cause of familial hypercholesterolemia. Hum Genet 111:501–510
Raungaard B, Jensen HK, Brorholt-Petersen JU, Heath F, Faergeman O (2000) Functional characterization of two low density lipoprotein receptor gene mutations by fluorescence flow cytometric assessment of receptor activity in stimulated human T-lymphocytes. Clin Genet 57:110–115
Raungaard B, Heath F, Brorholt-Petersen JU, Jensen HK, Faergeman O (1999) Flow cytometric assessment of LDL receptor activity in peripheral blood mononuclear cells compared to gene mutation detection in diagnosis of heterozygous familial hypercholesterolemia. Cytometry 36:52–59
Cuthbert JA, East CA, Bilheimer DW, Lipsky PE (1986) Detection of familial hypercholesterolemia by assaying functional low-density lipoprotein receptors on lymphocytes. N Engl J Med 314:879–883
Lahne K, Urdal P, Leren TP, Tonstad S, Ose I (1995) Standardization of a flow cytometric method for measurement of low-density lipoprotein receptor activity on blood mononuclear cells. Cytometry 20:290–295
Chang JH, Pan JP, Tai DY, Huang AC, Li PH, Ho HL, Hsieh HL, Chou SC, Lin WL, Lo E, Chang CY, Tseng J, Su MT, Lee-Chen GJ (2003) Identification and characterization of LDL receptor gene mutations in hyperlipidemic Chinese. J Lipid Res 44:1850–1858
Galeano NF, Milne R, Marcel YL, Walsh MT, Levy E, Nguyen TD, Gleeson A, Arad Y, Witte L, Al-Haideri M, Rumsey SC, Deckelbaum RJ et al (1994) Apoprotein B structure and receptor recognition of triglyceride-rich low density lipoprotein (LDL) is modified in small LDL but not in triglyceride-rich LDL of normal size. J Biol Chem 269:511–519
Reshef A, Nissen H, Triger L, Hensen TS, Eliav O, Schurr D, Safadi R, Gare M, Leitersdorf E (1996) Molecular genetics of familial hypercholesterolemia in Israel. Hum Genet 98:581–586
Mak YT, Pang CP, Tomlinson B, Zhang J, Chan YS, Mak TW, Masarei JR (1998) Mutations in the low density lipoprotein receptor gene in Chinese familial hypercholesterolemia patients. Arterioscler Thromb Vasc Biol 18:1600–1605
Zakharova FM, Damgaard D, Mandelshtam MY, Golubkov VI, Nissen PH, Nilsen GG, Stenderup A, Lipovetsky BM, Konstantinov VO, Denisenko AD, Vasilyev VB, Faergeman O (2005) Familial hypercholesterolemia in St-Petersburg: the known and novel mutations found in the low density lipoprotein receptor gene in Russia. BMC Med Genet. 8(6):6
Fouchier SW, Defesche JC, Umans-Eckenhaudsen MW, Kastelein JP (2001) The molecular basis of familial hypercholesterolemia in The Netherlands. Hum Genet 109:602–615
Civeira F, Jarauta E, Cenarro A, Garcia-Otin Al, Tejedor D, Zambon D, Mallen M, Ros E, Pocovi M (2008) Frequency of low-density lipoprotein receptor gene mutations in patients with a clinical diagnosis of familial combined hyperlipidemia in a clinical setting. J Am Coll Cardiol 52:1554–1556
Chmara M, Wasag B, Zuk M, Kubalska J, Wegrzyn A, Bednarska-Makaruk M, Pronicka E, Wehr H, Defesche JC, Rynkiewicz A, Limon J (2010) Molecular characterization of Polish patients with familial hypercholesterolemia: novel and recurrent LDLR mutations. J Appl Genet 51:95–106
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s11033-015-3879-5.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Waluś-Miarka, M., Sanak, M., Idzior-Waluś, B. et al. A novel mutation (Cys308Phe) of the LDL receptor gene in families from the South-Eastern part of Poland. Mol Biol Rep 39, 5181–5186 (2012). https://doi.org/10.1007/s11033-011-1314-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-1314-0