
Abstract
Background
Hypercholesterolemia is a major determinant of cardiovascular disease-associated morbidity and mortality. Mutations in the LDL-receptor (LDLR) gene are implicated in the majority of the cases with familial hypercholesterolemia (FH). However, the spectrum of mutations in the LDLR gene in Sri Lankan patients has not been investigated. The objective of this study was to report the frequency and spectrum of variants in LDLR in a cohort of Sri Lankan patients with FH.
Methods
A series of consecutive patients with FH, diagnosed according to Modified Simon Broome criteria or Dutch Lipid Clinic Network criteria at the University Medical Unit, Colombo, were recruited. Clinical data was recorded. DNA was extracted from peripheral blood samples. The LDLR gene was screened for genetic variants by Sanger sequencing.
Results
A total of 27 patients [13 (48%) males, 14 (52%) females; age range 24–73 years] were tested. Clinical features found among these 27 patients were: xanthelasma in 5 (18.5%), corneal arcus in 1 (3.7%), coronary artery disease (CAD) in 10 (37%), and a family history of hypercholesterolemia and/or CAD in 24 (88.9%) patients. In the entire cohort, mean total cholesterol was 356.8 mg/dl (±66.4) and mean LDL-cholesterol was 250.3 mg/dl (±67.7). Sanger sequencing of the 27 patients resulted in the identification of known pathogenic missense mutations in 5 (18.5%) patients. Four were heterozygotes for 1 mutation each. They were c.682G > C in 2 patients, c.1720C > A in 1 patient, and c.1855 T > A in 1 patient. One patient with severe FH phenotypes was a compound heterozygote for one known mutation, c.2289G > T, and another missense variant, c.1670C > G (p.Thr557Ser), with unknown functional impact. This latter variant has not been reported in any other population previously.
Conclusions
The frequency of known mutations in the LDLR gene in this cohort of patients was markedly low compared to frequencies reported in other populations. This highlights the likelihood of a complex, polygenic inheritance of FH in Sri Lankan patients, indicating the need for a comprehensive genetic evaluation that includes the screening for mutations in other genes that cause FH, such as APOB, PCSK9, and LDLRAP1.
Similar content being viewed by others

Background
Familial hypercholesterolemia (FH) is an inherited disorder of lipoprotein metabolism characterized by elevated low density lipoprotein (LDL) cholesterol in serum, tendon xanthomas and increased risk of premature coronary artery disease (CAD). FH is primarily an autosomal dominant disorder. The frequency of heterozygous FH is about 1 in 500, while homozygous FH is rare in many populations worldwide [1]. Mutations in the genes that encode proteins involved in LDL uptake and catabolism, the LDL-receptor (LDLR), apolipoprotein-B (APOB), LDL receptor adaptor protein (LDLRAP1) and PCSK9 (PCSK9) are known to cause FH, resulting from defective LDL uptake and degradation. This leads to elevations in plasma LDL-cholesterol levels, resulting in the hypercholesterolemia phenotype, and manifesting as xanthomas, atherosclerosis, CAD and other cardiovascular diseases. The majority (60–80%) of the patients with FH harbor mutations in the LDLR gene, while mutations in the APOB and PCSK9 genes account for a smaller percentage of autosomal dominant FH. Homozygous and compound heterozygous mutations in LDLRAP1 produce a rare autosomal recessive form of FH [1,2,3].
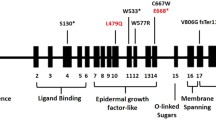
The LDLR spans 45 kb on the short arm of chromosome 19 and comprises 18 exons that are transcribed and translated into five distinct domains which form the cell surface LDL receptor protein. Mutations in the LDLR gene have been classified into 5 groups according to the functional significance of the mutation. Mutations that produce null-alleles with no receptor protein production are classified as ‘class 1’ mutations. The other 4 classes (classes 2–5) include 4 different types of mutations causing a defect in one of the 4 steps in the LDLR uptake and degradation pathway (Class 2: Defects in LDLR transport from the endoplasmic reticulum to the cell surface, Class 3: Defects in the binding of the LDLR to Apolipoprotein B in LDL particles, Class 4: Defects in internalization of the LDL particle-LDLR complex, and Class 5: Defects in recycling of the LDLR protein). Class 1 mutations are known as ‘receptor-negative’ mutations, while the mutations in class 2–5 are commonly described as ‘receptor-defective’ mutations [4, 5]. Association between the different classes of LDLR mutations with the different phenotypic characteristics of FH that include serum LDL-C levels, and clinical manifestations such as tendon xanthomas and risk of CAD have been observed. Patients with receptor negative mutations compared to those with receptor-defective mutations were shown to have higher total cholesterol and LDL-C levels, lower HDL-cholesterol levels and a higher incidence of tendon xanthomas, carotid atherosclerosis and CAD [6,7,8]. Moreover, receptor-negative mutation carriers had a weaker LDL-cholesterol lowering response to statins [8,9,10]. These findings highlight the implications of a definitive genetic diagnosis of FH in clinical surveillance, prognostication and management of FH. It is also likely that the carriers of LDLR mutations, especially those with ‘receptor defective’ mutations would not develop the stigmata of hypercholesterolemia until the later stages of the disease, making it difficult to diagnose the disease clinically at young age. In such cases with suspected FH, molecular genetic evaluation will be beneficial for confirmation of the disease and to initiate the treatment and follow-up at an earlier age, thus preventing cardiovascular complication.
To date nearly 1500 ‘pathogenic’ and ‘likely pathogenic’ variants of the LDLR gene are listed in the ClinVar database. These include a large number of single nucleotide substitutions producing nonsense, missense, frameshift or splice site variants or variants in the promoter or untranslated regions, as well as copy number variants (CNVs) such as duplications, insertions and deletions [11]. It was previously suggested that exons 3 and 4 of the LDLR, which correspond to the ligand-binding domain of the LDLR protein, harbor the majority of the mutations in patients with FH. However, the current understanding is that damaging mutations are distributed throughout the gene without clustering in any specific domains of the LDLR protein. [11, 12]. The presence of many different mutations widely spread throughout the LDLR gene makes a compelling case for complete sequencing of the LDLR coding region in index patients. Identification of a mutation in a patient enables comprehensive genetic counselling. Once an index patient has been genetically diagnosed, cascade screening of the family can identify individuals at risk for increased CAD, and treatment can commence before adulthood. Early treatment, sometimes beginning as early as 9–10 years old, leads to better cardiovascular outcomes in these patients [13].
Despite the extensive heterogeneity of LDLR gene reported among patients with FH, some studies have reported the aggregation of certain types of mutations in different geographic areas and different populations. For example, 3 specific LDLR mutations, FH-North Karelia, FH-Helsinki, and FH-Turku account for almost 80% of FH cases in the Finnish population [14]. As well, a study in the Netherlands revealed that 4 LDLR mutations account for nearly half of the patients with FH [15]. It has been shown that one mutation designated p.Cys681X in the LDLR gene accounts for FH in 81.5% of Lebanese FH patients [16] and an “Iceland-specific” splice-site mutation (c.698 + 2T4C [IVS4 + 2T4C]) in the LDLR gene was identified in 60% of the patients with FH in Iceland [17, 18]. Geographic clustering of these mutations is probably the result of a founder effect. These findings imply that population-specific frequencies and types of LDLR mutations are possible among Sri Lankans. Anecdotal evidence suggests that the incidence of premature CAD is rising in Sri Lanka, and a large number of patients are diagnosed with hypercholesterolemia at a younger age. However, the molecular genetic evaluation of these patients has never been performed in Sri Lanka. Hence our study was designed to assess the frequency and spectrum of LDLR mutations in a cohort of patients who were clinically diagnosed to have FH.
Methods
Patient recruitment
Patients were selected from consecutive patients seen at the University Medical Unit, University of Colombo, Sri Lanka between October 2011 and March 2012. Patients who were clinically diagnosed with FH were recruited for the study. Diagnosis of FH was performed according to either the Modified Simon Bloom criteria [19] or the Dutch Lipid Clinic Network criteria [20, 21]. Personal and family history of CAD, peripheral vascular disease (PVD), cerebrovascular disease (CVD) and hypercholesterolemia were evaluated and the family data was entered in to a three-generation pedigree chart. Relevant details including the serum lipid levels (i.e. Total cholesterol, LDL-cholesterol and HDL-cholesterol) at the time of initial presentation were obtained from patient’s previous clinic records. All the patients selected for this study were clinically evaluated for peripheral stigmata of hypercholesterolemia (i.e. tendon xanthomas, xanthelasma and corneal arcus). Detailed examination of the cardiovascular and central nervous systems was performed.
LDLR sequencing
Genomic DNA was extracted from peripheral whole blood samples and screened for LDLR mutations by standard bi-directional Sanger sequencing. All 18 exons of the LDLR gene from all the different transcripts, and approximately 50 bp of flanking intronic sequences at each exon-intron boundary were amplified by polymerase chain reaction. Primer sequences are available upon request. Sequences were analyzed using Sequencher (Genecodes) and compared to the human LDLR reference sequence (Genome Reference Consortium Human Build 38 patch release 7 (GRCh38.p7), NM_000527.4). All the identified variants were confirmed by re-sequencing. Written informed consent for the participation in this study and for the genetic testing was obtained from all the participants. The study was approved by the Ethics Review Committee, Faculty of Medicine, University of Colombo, Sri Lanka.
Results
A total of 27 patients, 13 (48%) males and 14 (52%) females were recruited for the study. The age of the patients ranged between 24 and 73 years (mean 50.1±12.2). Most of the patients (22; 81.5%) did not have any peripheral stigmata of hypercholesterolemia. Xanthelasma was observed in 5 (18.5%) patients and corneal arcus in one (3.7%). None of the patients had tendon xanthoma. The most common phenotype of FH in the study cohort was CAD, which was observed in 10 (37%) patients, 5 of whom had triple vessel disease, and 4 had double vessel disease as determined by coronary angiography, all requiring surgical interventions. One (3.7%) patient had a past history of CVD. None of the patients had peripheral vascular disease (PVD). The majority (24; 88.9%) of the patients had a family history of hypercholesterolemia and/or CAD. Seventeen (63.0%) patients had at least one first degree relative with hypercholesterolemia, and 16 (59.2%) had at least one first degree relative with CAD. In the entire cohort, mean total cholesterol was 356.8 mg/dl (±66.4) and mean LDL-cholesterol was 250.3 mg/dl (±67.7).
When the coding region of the LDLR gene was sequenced, pathogenic mutations were detected in 5 (18.5%) patients. Phenotypic characteristics of these mutation-positive patients are summarized in Table 1.
Four of the 5 patients were heterozygotes for 1 previously identified mutation each. Two of them (mother and son) had the same missense mutation designated c.682G > C (p. Glu228Gln) in exon 4 of the LDLR gene. Missense mutations c.1720C > A (p.Arg574Ser) in exon 12 and c.1855 T > A (p.Phe619Leu) in exon 13 were detected in another 2 patients. One patient was a compound heterozygote for a missense mutation c.2289G > T (p.Glu763Asp) in exon 15 and another missense variant, c.1670C > G (p.Thr557Ser) in exon 11 (Fig. 1a). The c.1670C > G is a novel variant that has not been reported in any other population before. This compound heterozygous 52 year old female patient was diagnosed with triple vessel disease by coronary angiography. There is a strong family history of CAD and hypercholesterolemia among several first degree relatives of this patient (Fig. 1b). She had the highest levels of total cholesterol and LDL-cholesterol among the five mutation-positive patients.

a- sequencing results, b- Pedigree of the patient with two variants in the LDLR gene. Arrow – proband
The details of the 5 missense mutations detected in the present study are shown in Table 2. We have also identified 14 synonymous variants, and 16 (59.2%) patients were homozygous for the synonymous variant c.1413A > G (p.Arg417Arg).
Discussion
This study is the first to report the mutations in the LDLR gene in the Sri Lankan population. Detailed descriptions of the phenotypic characteristics of the Sri Lankan patients with FH are also lacking in scientific literature. Phenotypically, the most notable observation in the present study was the rarity of the peripheral stigmata of hypercholesterolemia in our study cohort. Tendon xanthoma, which is generally considered specific for the FH was reported to be present in 30–60% of patients in Western populations, more commonly among patients with receptor negative mutations [6, 22]. In contrast, tendon xanthomas were not observed in any of the patients in the present study. Statistically significant association between FH and CAD has been demonstrated in studies done in both European and Asian populations [5]. A similar trend was observed in our cohort with a high percentage of patients with CAD requiring surgical interventions. Our results also highlight the value of obtaining detailed family histories of the patients clinically diagnosed with FH.
Four of the five mutations detected in the present study have been previously reported. The missense mutation FH Iraq (c.682G > C), which was detected in 2 related patients in the present study was first reported in 1992 in African Americans [4]. This mutation is in the exon 4 which is within the ligand binding domain (i.e. exon 2–6). The majority of LDLR mutations reported to-date are in the exon 4. However, this might have arisen from a selection bias, since many early studies assessed the clinical and functional relevance of mutations in exon 4, and targeted screening of exon 4 in many studies on FH may have contributed to this observation [5]. It has been reported that even a single amino acid substitution in the ligand binding domain of the LDLR protein causes defective binding of the LDLR to apoB-100 apolipoproteins in the LDL particles, thereby impairing the cellular uptake of LDL, and leading to elevations in serum LDL-cholesterol levels [4]. Since the first report of this mutation in African Americans 1992, it has been described in several other populations in Europe [15, 23, 24], Israel [25] and recently, in Japan [26]. Neither of the two patients with this mutation in the present study had a significant medical history other than hypercholesterolemia. However marked differences were observed in the total cholesterol and LDL-cholesterol levels at the initial presentation between these 2 patients with the same familial mutation (S3 and S26 in Table 1). Similarly, intra-familial variations in the clinical expression of FH among patients with the same LDLR mutation has been observed in other family-based studies [27, 28]. A case-control study conducted in the French-Canadian population has also suggested gender-specific pleiotropic effects of LDLR mutations [29]. Together, this highlights that the phenotypic characteristics of the patients with FH are determined by their genetic as well as other risk factors. This is an important fact that has to be taken into account in counselling mutation-positive patients and in planning their management and follow-up.
The heterozygous missense mutation, c.1855 T > C (p.Phe619Leu) in exon 13 of the LDLR gene was detected in one patient in our study, and has been previously reported in a study from the United Kingdom [30]. The missense mutation c.1720C > A (p.R574S) in exon 12 detected in another patient was described recently in a 13-year old Chinese boy with FH. This mutation was predicted to be pathogenic using function prediction bioinformatics tools [31]. However, this mutation is not listed in any of the human genome variant databases.
One of the two variants [c.2289G > T (p.Glu763Asp)] detected in the patient with compound heterozygosity has been previously reported in a study conducted in Western Australia. This missense variant has been classified as a ‘mutation of unknown pathogenicity’ because of the different interpretations of pathogenicity in different function prediction tools [32]. The other variant identified in this compound heterozygote patient, c.1670C > G (p.Thr557Ser), is not listed in any of the human genome/exome variant databases and also has not been described. This variant leads to a conservative change in amino acid from Threonine to Serine at position 557. It is predicted as ‘disease causing variant’ and ‘probably damaging variant’ in MutationTaster [33] and PolyPhen-2 [34] databases respectively, and as ‘neutral/tolerated’ in the PROVEAN/SIFT [35, 36] database. Further in-vitro studies are required to assess the functional impact of this variant. Considering the uncertain pathogenicity of the first mutation, c.2289G > T, in this patient, it is very likely that this second novel mutation has an additive damaging effect, giving rise to the severe phenotypic expression in this patient. This is in concurrence with previous studies that described a more severe clinical and biochemical phenotype associated with biallelic (homozygous or compound heterozygous) FH mutations [1, 5, 32, 37]. Moreover, the diagnosis of compound heterozygosity for LDLR mutations has important therapeutic implications. These patients are likely to have poor response to statins, because they are lacking functional LDL receptors in the liver that can be up-regulated by statins [37]. The pathogenicity of the novel variant identified in this study could have been tested by carrying out co-segregation studies looking for the co-inheritance of the variant with elevated LDL-C levels in family members. However, the interpretation of family data may be complicated by the overlay of environmental factors that influence lipid levels and by the presence of other genetic variants that raise or lower LDL-C in the family. Still, we could not perform the segregation studies on this family due to the unapproachability of these family members for testing.
The frequency of LDLR mutations in our cohort of patients is very low (18.5%) compared to the population frequency of 60–80% that has been described [1,2,3]. Possible reasons for this low frequency of LDLR mutations include the presence of mutations in other genes that cause FH (i.e. APOB, PCSK9, and LDLRAP1) that were not tested in the present study, and the likelihood of polygenic inheritance of FH in Sri Lankan patients. There may also be other genes associated with FH that have yet to be identified. Mutations in the intronic regions of the LDLR gene outside the amplified exonic and flanking regions were not detected by the testing methodology of the present study. Major structural rearrangements of the LDLR gene would also have been missed by this technique contributing to the low mutation detection rate. This highlights the need of a comprehensive gene panel that includes the sequencing of the whole LDLR gene and other genes with a recognized or a potential role in the pathogenesis of hypercholesterolemia, for genetic diagnosis. Though expensive at present, this method will allow the definite molecular genetic diagnosis of the FH in index patients. The identification of a mutation in index patients enables the cascade screening of family members [38]. It has been shown that cascade screening increases the percentage of patients receiving treatment for FH [39,40,41] resulting in a significant improvement in the serum lipid parameters following the genetic diagnosis [41, 42]. These studies highlighted the value of cascade screening in early diagnosis and treatment of FH, which will lead to a reduction in mortality and morbidity associated with premature coronary artery disease in these patients.
Conclusions
This is the first report of the spectrum of LDLR variants in a cohort of Sri Lankan patients with FH. We report the discovery of a novel variant and the low contribution of pathogenic variants in the LDLR gene to FH in this cohort. This highlights the need for a compressive study involving all the genes implicated in FH to understand the genetic aetiology of the condition in our population.
Abbreviations
- ApoB:
-
Apolipoprotein-B
- CAD:
-
Coronary artery disease
- CVD:
-
Cerebrovascular disease
- FH:
-
Familial hypercholesterolemia
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- LDLR:
-
LDL-receptor
- PVD:
-
Peripheral vascular disease
References
Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4:214–25.
Varret M, Abifadel M, Rabès JP, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet. 2008;73:1–3.
Youngblom E, Pariani M, Knowles JW. Familial Hypercholesterolemia. 2014 Jan 2 [Updated 2016 Dec 8]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2017. https://www.ncbi.nlm.nih.gov/books/NBK174884/. Accessed 10 Aug 2017.
Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–66.
Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160:407–20.
Bertolini S, Cantafora A, Averna M, Cortese C, Motti C, Martini S, et al. Clinical expression of familial hypercholesterolemia in clusters of mutations of the LDL receptor gene that cause a receptor-defective or receptor-negative phenotype. Arterioscler Thromb Vasc Biol. 2000;20:e41–52.
Bertolini S, Pisciotta L, Rabacchi C, Cefalù AB, Noto D, Fasano T, et al. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis. 2013;227:342–8.
Santos PC, Morgan AC, Jannes CE, Turolla L, Krieger JE, Santos RD, et al. Presence and type of low density lipoprotein receptor (LDLR) mutation influences the lipid profile and response to lipid-lowering therapy in Brazilian patients with heterozygous familial hypercholesterolemia. Atherosclerosis. 2014;233:206–10.
Miltiadous G, Xenophontos S, Bairaktari E, Ganotakis M, Cariolou M, Elisaf M. Genetic and environmental factors affecting the response to statin therapy in patients with molecularly defined familial hypercholesterolaemia. Pharmacogenet Genomics. 2005;15:219–25.
Koeijvoets KC, Rodenburg J, Hutten BA, Wiegman A, Kastelein JJ, Sijbrands EJ. Low-density lipoprotein receptor genotype and response to pravastatin in children with familial hypercholesterolemia. Circulation. 2005;112(20):3168–73.
ClinVar (TM). National Center for Biotechnology Information, National Library of Medicine, Bethesda. Available from http://www.ncbi.nlm.nih.gov/clinvar/. Accessed 12 Aug 2017.
Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet. 2012;76(5):387–401.
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European atherosclerosis society. Eur Heart J. 2013;34:3478–90.
Vuorio AF, Aalto-Setälä K, Koivisto UM, Turtola H, Nissen H, Kovanen PT, et al. Familial hypercholesterolaemia in Finland: common, rare and mild mutations of the LDL receptor and their clinical consequences. Ann Med. 2001;33:410–21.
Fouchier SW, Defesche JC, Umans-Eckenhausen MA, Kastelein JJ. The molecular basis of familial hypercholesterolemia in the Netherlands. Hum Genet. 2001;109:602–15.
Abifadel M, Rabès JP, Jambart S, Halaby G, Gannagé-Yared MH, Sarkis A, et al. The molecular basis of familial hypercholesterolemia in Lebanon: spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum Mutat. 2009;30:E682–91.
Gudnason V, Sigurdsson G, Nissen H, Humphries SE. Common founder mutation in the LDL receptor gene causing familial hypercholesterolaemia in the Icelandic population. Hum Mutat. 1997;10:36.
Dedoussis GV, Schmidt H, Genschel J. LDL-receptor mutations in Europe. Hum Mutat. 2004;24:443–59.
Minhas R, Humphries SE, Qureshi N, Neil HA. Controversies in familial hypercholesterolaemia: recommendations of the NICE guideline development group for the identification and management of familial hypercholesterolaemia. Heart. 2009;95:584–7.
Walma EP, Visseren FL, Jukema JW, Kastelein JJ, Hoes AW, Stalenhoef AF. The practice guideline ‘diagnosis and treatment of familial hypercholesterolaemia’ of the Dutch health care insurance board. Ned Tijdschr Geneeskd. 2006;150(1):18–23.
Civeira F, International Panel on Management of Familial Hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55–68.
Civeira F, Castillo S, Alonso R, Merino-Ibarra E, Cenarro A, Artied M, et al. Tendon xanthomas in familial hypercholesterolemia are associated with cardiovascular risk independently of the low-density lipoprotein receptor gene mutation. Arterioscler Thromb Vasc Biol. 2005;25:1960–5.
Leren TP, Manshaus T, Skovholt U, Skodje T, Nossen IE, Teie C, et al. Application of molecular genetics for diagnosing familial hypercholesterolemia in Norway: results from a family-based screening program. Semin Vasc Med. 2004;l:75–85.
Humphries SE, Cranston T, Allen M, Middleton-Price H, Fernandez MC, Senior V, et al. Mutational analysis in UK patients with a clinical diagnosis of familial hypercholesterolaemia: relationship with plasma lipid traits, heart disease risk and utility in relative tracing. J Mol Med. 2006;84:203–14.
Reshef A, Nissen H, Triger L, Hensen TS, Eliav O, Schurr D, et al. Molecular genetics of familial hypercholesterolemia in Israel. Hum Genet. 1996;98:581–6.
Mabuchi H. Half a century Tales of familial hypercholesterolemia (FH) in Japan. J Atheroscler Thromb. 2017;24:189–207.
Kotze MJ, Davis HJ, Bissbort S, Langenhoven E, Brusnicky J, Oosthuizen CJ. Intrafamilial variability in the clinical expression of familial hypercholesterolemia: importance of risk factor determination for genetic counselling. Clin Genet. 1993;43:295–9.
Levy E, Minnich A, Cacan SL, Thibault L, Giroux LM, Davignon J, et al. Association of an exon 3 mutation (Trp66→ Gly) of the LDL receptor with variable expression of familial hypercholesterolemia in a French Canadian family. Biochem Mol Med. 1997;60:59–69.
Roy M, Sing CF, Betard C, Davignon J. Impact of a common mutation of the LDL receptor gene, in French-Canadian patients with familial hypercholesterolemia, on means, variances and correlations among traits of lipid metabolism. Clin Genet. 1995;47:59–67.
Sun XM, Patel DD, Knight BL, Soutar AK. Comparison of the genetic defect with LDL-receptor activity in cultured cells from patients with a clinical diagnosis of heterozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1997;17:3092–101.
Du R, Fan LL, Lin MJ, He ZJ, Huang H, Chen YQ, et al. Mutation detection in Chinese patients with familial hypercholesterolemia. Spring. 2016;5:2095.
Hooper AJ, Nguyen LT, Burnett JR, Bates TR, Bell DA, Redgrave TG, et al. Genetic analysis of familial hypercholesterolaemia in Western Australia. Atherosclerosis. 2012;224:430–4.
Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–8.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013;76:7.20.1-7.20.41.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Ned RM, Sijbrands E. Cascade screening for familial hypercholesterolemia (FH). Plos Curr. 2011;3:RRN1238. https://doi.org/10.1371/currents.RRN1238.
Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet. 2001;357:165–8.
Leren TP. Cascade genetic screening for familial hypercholesterolemia. Clin Genet. 2004;66:483–7.
Huijgen R, Kindt I, Verhoeven SB, Sijbrands EJ, Vissers MN, Kastelein JJ, et al. Two years after molecular diagnosis of familial hypercholesterolemia: majority on cholesterol-lowering treatment but a minority reaches treatment goal. PLoS One. 2010;5:e9220.
Leren TP, Finborud TH, Manshaus TE, Ose L, Berge KE. Diagnosis of familial hypercholesterolemia in general practice using clinical diagnostic criteria or genetic testing as part of cascade genetic screening. Public Health Genomics. 2008;11:26–35.
Acknowledgements
We are grateful to the patients who generously participated in this study.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
CSP collected the phenotypic and genotypic data, carried out the literature survey and wrote the manuscript. JRDKR collected the clinical data and the patients’ samples for the analysis and drafted the manuscript. GRC performed the clinical assessment of the patients and supervised the study. KPS performed the gene sequencing and analysis. RRS performed the gene sequencing and analysis and revised the manuscript. RWJ participated in the design of the study and supervised the study. VHWD supervised all aspects of the study and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol and the form of consent were approved by the Ethics Review Committee, Faculty of Medicine, University of Colombo, Sri Lanka. Written informed consent for the participation in this study and for the genetic testing was obtained from all the participants.
Consent for publication
All the participants provided written informed consent for the publication of the results of this study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Paththinige, C.S., Rajapakse, J.R.D.K., Constantine, G.R. et al. Spectrum of low-density lipoprotein receptor (LDLR) mutations in a cohort of Sri Lankan patients with familial hypercholesterolemia – a preliminary report. Lipids Health Dis 17, 100 (2018). https://doi.org/10.1186/s12944-018-0763-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-018-0763-z