
Abstract
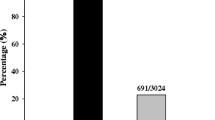
The Pik-h gene in rice confers resistance to several races of rice blast fungus (Magnaporthe oryzae), and has been classified as a member of the Pik cluster, one of the most resistance (R) gene-dense regions in the rice genome. However, the loss of a key mutant isolate has long made it difficult to differentiate Pik-h from other Pik group genes especially from Pik-m. We identified new natural isolates enabling the differentiation between Pik-h and Pik-m genes, and first confirmed the authenticity of the International Rice Research Institute (IRRI) “monogenic” line IRBLkh-K3, and then fine-mapped the Pik-h gene in the Pik cluster. Using 701 susceptible individuals among 3,060 siblings from a cross of IRBLkh-K3×CO39, the Pik-h region was delimited to 270 kb, the narrowest interval among the Pik group genes reported to date, in the cv. Nipponbare genome. Annotation of this genome region first revealed 6 NBS-LRR type R-gene analogs (RGAs), clustered within the central 120 kb, as possible counterparts of Pik-h and 6 other Pik group R genes. Interestingly, the Pik-h region and the cluster of RGAs were shown to be located 130 kb and 230 kb apart from Xa4 and Xa2 bacterial blight resistance genes, respectively, once classified as belonging to the Pik cluster. The closest recombination events were limited to the margins of the Pik-h region, and recombination was suppressed in the core interval with the RGA cluster. This fine-mapping, performed in a short time using an HEGS system, will facilitate utilization of the cluster’s genetic resources and help to elucidate the mechanism of evolution of R-genes. The presence of natural isolates also confirmed that evolution of Pik-h corresponds to pathogen evolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice blast disease, caused by Magnaporthe oryzae, is one of the most serious diseases of rice, an important staple crop for more than half the world’s population. The use of disease resistance (R) genes is the most efficient and environmentally friendly way to control such diseases, and over 50 major rice blast R genes have been mapped to date (Hayashi 2005; Chen et al. 2006; Liu et al. 2005). However, the mechanisms of action of disease resistance genes and their race specificity are still unclear, although several hypotheses have been proposed (Jia et al. 2000; Orbach et al. 2000; Farman et al. 2002; Böhnert et al. 2004). Although isolation of the rice blast R genes has lagged behind efforts in Arabidopsis (Glazebrook et al. 1997; Meyers et al. 2003) and other crops, such as tomato and tobacco (Martin et al. 2003), 7 genes have recently been isolated (Pib; Wang et al. 1999, Pita; Bryan et al. 2000, Pi9; Qu et al. 2006, Pid2; Chen et al. 2006, Pi2; Zhou et al. 2006, Pizt and Pi36; Liu et al. 2007), 6 of which (Pib, Pita, Pi9, Pi2, and Pizt, Pi36) are of the nucleotide binding site-leucine-rich repeat (NBS-LRR) type, while Pi2 encodes a receptor-like kinase. However, our understanding of the mechanism of initial recognition of pathogen signaling by R-genes is still far from complete. In addition, how the plant resistance genes could have evolved to recognize the highly mutable pathogens remains unclear.
More than 600 R-gene analogs of the NBS-LRR type were identified in the rice genome (Bai et al. 2002), and most of these are found in clusters. The rice Pik cluster is located near the telomeric end of the long arm of chromosome 11. This cluster includes the blast resistance genes Pik, Pik-s, Pik-p, Pik-m, Pik-h (Kiyosawa 1972; McCouch et al. 1994), Pi44(t) (Chen et al. 1999), and Pi1 (Yu et al. 1996), and the bacterial blight resistance genes Xa3 (Xiang et al. 2006), Xa4 (Sun et al. 2004), Xa22(t) (Wang et al. 2003), and Xa26 (Sun et al. 2004). This cluster represents one of the most highly concentrated regions of R genes in the rice genome (Inukai et al. 1994; Yu et al. 1996; Chen et al. 1999; Yang et al. 2003). Therefore, analyzing the genomic organization of this Pik cluster should facilitate effective utilization of these genetic resources and a better understanding of the molecular mechanism of R gene evolution. Recently, the Pik, Pik-p, and Pik-m genes were finely mapped (Hayashi et al. 2006). Pik-m was further finely mapped by Li et al. (2007).
The rice blast R-gene Pik-h was first reported by Kiyosawa and Murty (1969) from the test line K3 derived from a cross of an Indian cv. HR-22 (indica) and Japanese cv. Sasashigure. The Pik-h gene is expected to be allelic to Pik, because it is effective against a Pik-overcoming mutant isolate, Ken54-20-k +, derived from the Pik-incompatible ken54-20, but other aspects of the resistance spectrum against Japanese and International Rice Research Institute (IRRI) standard races are the same as Pik-m (Hayashi 2005; Tsunematsu et al. 2000), which is also allelic to Pik. Later, Pik-h was shown to be present in a rather wide range of cultivars, including indica cvs. Tetep and Tadukan and the US cv. Dawn (Kiyosawa 1981). However, the key mutant blast isolate, Ken54-20-k +, has been lost, making it difficult to differentiate Pik-h. A “monogenic” line, IRBLkh-K3, was constructed in IRRI by back-crossing from K3 with cv. LTH (Lijiangxintuanheigu), but differentiation of the Pik group monogenic lines with specific races has not been done (Tsunematsu et al. 2000). Although there have been some reports on examining the presence of Pik-h (Fjellstrom et al. 2004; Sharma et al. 2005), these reports included no descriptions of how Pik-h was differentiated from other Pik genes. It is necessary to settle this situation with some clear criteria for the gene. In addition, considering that Pik-h is found in various cvs., as described above, presence of some natural isolates corresponding to mutations in this gene was strongly suggested.
Recently, in the process of screening our Japanese rice blast isolate collection, we found two long sought key isolates that can differentiate Pik-h from the other Pik group genes. This race set will contribute greatly to differentiation between Pik group genes in various cvs. With these isolates, we confirmed the authenticity of the Pik-h gene in IRBLkh-K3. In addition, we confirmed that our fine-mapped gene using the cross CO39×IRBLkh-K3 and race V86010 to be Pik-h. This has localized Pik-h to a significantly narrower segment in the Pik-cluster compared to previous mapping reports, and verified the authenticity of Pik-h as a member of the Pik gene cluster. Mapping was performed efficiently using a high-efficiency genome scanning (HEGS) system involving high lane density polyacrylamide gel electrophoresis (Kawasaki and Murakami 2000). Here, we report the process of verification and fine mapping of the Pik-h gene.
Materials and methods
Plant materials and mapping populations
The original Pik-h line K3 (Kiyosawa and Murty 1969) was obtained from the gene bank of the National Institute of Agrobiological Sciences (NIAS). The Pik-h monogenic line IRBLkh-K3 (indica), derived from K3 and susceptible cv. CO39 (Pik-h: −, Pi-a: +, indica), was provided by IRRI, along with other monogenic lines IRBLk-Ka and IRBLkm-Ts. The 96 F2 individuals of a CO39×IRBLkh-K3 cross were used for preliminary bulked segregant analysis (BSA). After inoculation as described below, 701 susceptible individuals were selected from 3,060 F3 siblings of heterozygous F2 individuals that were selected by segregation of sample progenies. Some of the susceptible (S) and resistant (R) homozygous lines from this cross were used to verify the authenticity of the mapped gene. DNA was extracted by a revised cetyl trimethyl ammonium bromide (CTAB) method for a large population (Xu et al. 2005), and then simple sequence repeat (SSR) and restriction fragment length polymorphism (RFLP)-derived sequence characterized amplified region (SCAR) analysis were performed with the HEGS system (Nihon-Eido, Tokyo, Japan), as described previously (Xu et al. 2005). Samples of approximately 5 μg of DNA were extracted from the leaves (1 g fresh weight) of the selected recombinants using a Kurabo NA2000 DNA isolation machine (Kurabo, Osaka, Japan) for further analysis.
Blast inoculation and disease evaluation
For fine-mapping, phenotypic analysis of F2 and F3 populations, M. oryzae isolate V86010 line, which overcomes minor R-genes against Japanese races in the CO39 background (Imbe et al. 2000), obtained from IRRI, was used for inoculation in an isolated greenhouse. The seedlings at the four- to five-leaf stage (about 10–14 days after sowing) were inoculated with a suspension of M. oryzae conidia (2–3 × 105 spores/ml). The inoculated plants were then placed in darkness in a dew chamber (Koito, Tokyo, Japan) at 100% relative humidity for 24 h at 25°C, and subsequently transferred to a semi-temperature-controlled greenhouse, maintained at around 15–25°C. Disease symptoms of inoculated plants were recorded at 7–10 days after inoculation on a scale of 0–5 in accordance with the Japanese Ministry of Agriculture, Forestry, and Fisheries (MAFF) Microorganism Genetics Resources Manual Vol.18 (Hayashi 2005).
To characterize the authenticity of the Pik-h gene in IRBLkh-K3, in reference to K3, newly collected original Japanese isolates H05-56-1, H05-67-1, H02-20-1 from Ibaraki prefecture, and Kyu89-246 from Miyazaki, were used. For genotyping the Pik-h locus of the F3 plants, F4 progenies were inoculated with the new isolates under the same conditions as used for fine mapping. F4 plants were used for phenotyping to avoid complication of the results by the presence of Pia as much as possible.
Development of SNP markers and physical mapping of the Pik-h locus
Single nucleotide polymorphism (SNP) markers were analyzed by sequencing according to the following method.
To find unique sequences, 6 bacterial artificial chromosome (BAC) clones of Oryza sativa L. cv. Nipponbare sequences in the International Rice Genome Sequencing Project (IRGSP) database (http://rgp.dna.affrc.go.jp/IRGSP/), within the region narrowed by the above fine mapping, were used for BLAST analysis (http://www.ncbi.nlm.nih.gov) against the complete rice genome sequence. Sequences found to be unique in the genome were used to design primers with the Primer3 software (http://fokker.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The uniqueness of each primer in the genome was confirmed again by BLAST analysis. The PCR products, showing a single band on 1% agarose gels, were directly sequenced using an ABI BigDye® Terminator v3.1 Cycle Sequencing Kit and ABI 3100 DNA sequencer (Applied Biosystems, Foster City, CA). In case in which normal PCR amplification with Takara Ex Taq polymerase (Takara Bio, Otsu, Shiga, Japan) was difficult, Phusion™ High-Fidelity DNA Polymerase (Finnzymes, Espoo, Finland) was used in accordance with the manufacturer’s protocol.
The physical locations of RFLP-SCAR and SSR markers were checked in the Gramene marker database (http://www.gramene.org), in addition to BLAST searches as described above. These and SNP marker loci in Nipponbare were determined from the genome sequences and checked by graphical genotyping with close recombinants. The location of the Pik-h gene was determined by phenotyping of the same recombinants, and a physical map around this region was constructed based on the Nipponbare genome database.
Results
Confirmation of Pik-h gene authenticity
In the Pik cluster blast resistance genes, the Pik, Pik-s, Pik-p, and Pi1 have already been differentiated by Japanese and Philippine races from Pik-h, and only the differentiation of Pik-h and Pik-m has been difficult (Hayashi 2005; Tsunematsu et al. 2000).
With the newly collected blast isolates H05-56-1, H05-67-1, and H02-20-1 from Ibaraki Prefecture, and Kyu89-246 from Miyazaki, the original line K3 and the IRRI Pik-h, Pik-m, and Pik monogenic lines IRBLKh-K3, IRBLkm-Ts, and IRBLk-Ka were tested for their resistance reactions (Table 1). The last Pik line was added as a reference. In addition, the background of these IRRI monogenic lines, LTH, was also tested. As shown in Table 1, the new isolates, H05-56-1 and H02-20-1, differentiated Pik-h in K3 from Pik and Pik-m, and confirmed the authenticity of Pik-h in IRBLKh-K3. It is notable that the resistance reaction of Pik-h to the new races is not strong (grade 2 in Table 1, with numerous small stopping lesions with brown fringes, without sporulation; Fig. 1) compared to other known races seen as grade 1 in Table 1. Interestingly, such mild reaction of Pik-h was also reported previously against the mutant isolate Ken54-20-k + (Kiyosawa and Murty 1969).

Reaction of the Pik cluster blast resistance genes Pik-h, Pik-m, and Pik against the new differential blast race H05-56-1. For inoculation, a conidia suspension of 2–3 × 105/ml was sprayed onto all test plants at the same time, and after standing in a dew chamber for 24 h, the plants were moved to a greenhouse, and diagnosed on the 10th day after inoculation. (a) R-homozygous line RC-R Kh-102, from the cross of CO39×IRBLkh-K3 with only the Pik-h gene (Table 1). The resistance reaction of Pik-h varied from non-lesion type to this extent of small stopping-type lesions with browned fringes, but there were no progressive lesions or sporulation. (b) IRBLkm-Ts with Pik-m. (c) IRBLk-Ka with Pik. In (b) and (c), plants show susceptible reactions with sporulating progressive lesions, although sometime lesion density is lower than in (a). Pik seems more susceptible to this race than Pik-m
To confirm the authenticity of the F2 and F3 populations from IRBLkh-K3×CO39, used for subsequent fine mapping, some R and S homozygous lines of F3 against V86010 from siblings were also checked for their reaction at their F4 generation, to avoid complication with Pia, known to be present in CO39 (Tsunematsu et al. 2000), as much as possible. As shown in Table 1, the reactions of S homozygous siblings, RC-S Kh-2, 3, and 4, can be interpreted consistently as the absence of Pik-h and presence or segregation of Pi-a, and R homozygous lines RC-R Kh-101, 102, and 103 confirmed that the R gene of interest was Pik-h. The presence of Pia in RC-R Kh-101 and RC-S Kh-2, -3, -4 is by chance, and differentiated by stronger resistance reaction of grade 1 in Table 1, because the mapping isolate V86010 was selected to overcome Pia in CO39 (see Tsunematsu et al. 2000).
The typical resistance reaction of Pik-h on RC-R Kh-102 inoculated with the isolate H05-56-1 is shown in Fig. 1a. Although several small non-sporulating lesions appeared on the leaves, these were all of the stopping type with browned fringes, and did not extend as in the case of progressive lesions in the susceptible reaction, as shown in Fig. 1b and c. Other Pik group monogenic lines with Pik-m (IRBLkm-Ts: Fig. 1b) and Pik (IRBLk-Ka: Fig. 1c) developed severe progressive lesions with gray fringes and sporulation with the same isolate. In Pik-m, the lesion density was lower than in the case of Pik-h. This lower density of susceptible lesions than that of the small lesions in resistant reactions is often observed in rice blast spray inoculation systems.
Segregation of Pik-h in the F2 population
One hundred fifty-five F2 plants derived from crosses of IRBLkh-K3×CO39 were examined for the presence or absence of Pik-h with the M. oryzae isolate V86010, which can overcome Pia and other minor R genes against Japanese isolates in CO39. The segregation of resistant and susceptible progeny was 117:38 in the F2 populations, fitting a 3:1 ratio (χ2 = 0.32, P > 0.05) indicating that IRBLkh-K3 carries a single dominant R gene against the isolate V86010.
Fine mapping of the Pik-h locus
Forty-two DNA markers were selected from the RGP (http://rgp.dna.affrc.go.jp/) data (Kaji and Ogawa 1996; Hayasaka et al. 1996) in the region within 10 cM from Pik (110.1–120 cM) on Chromosome 11, and modified for PCR analysis. Twelve polymorphic markers were selected by preliminary BSA analysis using F2 individuals (10 resistant and 10 susceptible plants were selected to make pools). An example of BSA is presented in the side four lanes of Fig. 2. By screening 96 of the 155 F2 individuals, the Pik-h region was narrowed to ∼2 cM flanked by the markers RM224 and Y6855RA (Figs. 2, 3). Then, 701 susceptible individuals were selected from among 3,060 F3 hetero-siblings, and recombinants within the markers were selected with HEGS (Fig. 2). Among these, the 13 closest recombinants were selected between the flanking markers RM224 and Y6855RA, both linked to the Pik-h locus at 0.9 cM (Fig. 3).

An example of HEGS/SSR (RM224) screening of recombinants around the blast resistance gene Pik-h, from susceptible F3 individuals of CO39×IRBLkh-K3. Right side four lanes show an example of bulked segregant analysis with the F2 population. M: ΦX174 DNA-HaeIII marker; C: CO39; K: IRBLkh-K3; BS: susceptible bulk; BR: resistant bulk of F2, including heterozygotes. The arrows R and S indicate resistant and susceptible type bands, respectively

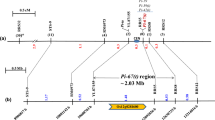
Genetic and physical maps around the Pik-h region at the end of the long arm of chromosome 11, based on Nipponbare BACs (a), and comparison with the previous reports of fine-mapping of Pik cluster genes (b). (a) Marker locations on the chromosome and BAC clones, with recombination data in parentheses. The cumulative numbers of recombinants of markers to the R-gene per tested individual are shown at the top in parentheses. Genetic distances between the markers and the Pik-h gene region are indicated under the chromosome in Kosambi units. The details of the Pik-h region in the Nipponbare physical map are shown on the bottom with 54 predicted ORFs. There is a still unsequenced gap of 40 kb near the right end of the region, although a bridge clone covering the gap was recently found. There are six resistance gene analogs (RGAs); RGA1 to 6, as candidate counterparts of the real Pik-h, and possibly also of the other Pik-group R-genes. There are also 11 transposon-related ORFs indicated by gray boxes. (b) Previously reported Pik-group genes regions. A: Pik-m (Kaji and Ogawa 1996); B: Pik (Hayasaka et al. 1996); C: Pik-s and Pik-h (Fjellstrom et al. 2004). D: Pik-m, Pik-p, and Pik (Hayashi et al. 2006); E: Pik-m (Li et al. 2007). Comparison was performed using the Nipponbare genome database with cM addresses. All the blast resistance gene regions overlap as Pik clusters, while recently cloned bacterial blight resistance genes Xa4 and Xa26, (hatched rectangle on the right) were outside of the Pik-h region
Within this region, all the Nipponbare sequences obtained from IRSGP were segmented to sub-fragments of about 10 kb, and these were checked for their uniqueness in the genome by BLAST. From the unique regions, 90 PCR primers were designed using the Primer3 software and named such as Kh-(no.)F/R, depending on the optimal sequencing primers directions (Table 2). PCR was performed with these primers for IRBLkh-K3, and 19 primer pairs were found to amplify single bands. Among them, 8 bands properly positioned in the map were sequenced, and the SNPs between the parent cvs. were used as markers to determine the sites of close recombination events by sequencing with an ABI 3100 DNA sequencer.
In silico mapping of the Pik-h region and annotation of ORFs and RGAs
The above 8 SNP markers around the Pik-h region were located on a contig of 6 BAC clones of Nipponbare flanked by RM224 and Kh-38F (Fig. 3a) in reference to INE (http://rgp.dna.affrc.go.jp/E/giot/INE.html). Finally, the Pik-h region was delineated by the two closest flanking SNP markers, Kh-45F and Kh-A-3R on the north and south sides, respectively, with 5 and 2 recombination events on each side, spanning three BAC clones [OSJNBb0049B20 (131 kb), OSJNBa0047M04 (109 kb) and OSJNBa0036K13 (136 kb)]. According the complete sequence of Nipponbare chromosome 11 from IRGSP Release Build 4.0 Pseudomolecules of the Rice Genome, the physical distance between these two markers was estimated to be about 238 kb in length excluding a gap between OSJNBa0047M04 and OSJNBa0036K13. However, a gap-bridging BAC clone (110 kb; CHEF electrophoresis) was recently found, and by mapping of its end sequences and those of adjoining clones, the gap size was determined to be about 40 kb (RGP-team; Dr. T. Matsumoto, personal communication). Therefore, the Pik-h region size was delimited to about 270 kb, including the yet to be sequenced gap region. In comparison with other reports of Pik group genes, the Pik-h region was the narrowest and in the center of the previously reported regions (Fig. 3b).
The annotation of the expected ORFs in the region were determined by RiceGAAS (Rice Genome Automated Annotation System, http://ricegaas.dna.affrc.go.jp/: data not shown), and 54 ORFs were predicted; among these clones, 6 RGAs were found clustered near the center of the Pik-h region (Fig. 3a). All the RGAs were of the nucleotide binding site leucine rich repeat (NBS-LRR) type, ranging in size from 1.6–3.4 kb. The cluster of 6 RGAs was 230 kb apart from the recently cloned Xa4 and Xa26 (Fig. 3b). The presence of 6 RGAs as a cluster in the center of not only Pik-h region but also of all Pik group gene regions is of special interest, because this suggests that some of the RGAs may be counterparts of not only Pik-h, but also of other Pik group genes.
Although there were 7 recombination events in this region, all occurred in the marginal regions near the flanking markers (Fig. 3a). The four markers, Kh-67, Kh-44F, Kh-83F, and Kh-85F, within about 200 kb of core region showed no recombination with the Pik-h gene in these F3 siblings (Fig. 3a).
Discussion
There have been several genetic studies of the rice blast resistance genes in the Pik region. This region is on the near-telomeric end of the long arm of chromosome 11, between RFLP markers G181 and R543, closely linked with RM224 and R1506 (Kaji and Ogawa, 1996; Hayasaka et al. 1996; Yu et al. 1996; Fjellstrom et al. 2004; Hayashi et al. 2006; Li et al. 2007). The Pik-h gene was identified in the experimental line K3, which was derived from a cross of Indian cv. HR-22 and Japanese cv. Sasashigure (Kiyosawa and Murty 1969), by suppressing the isolate Ken54-20-k +, which has a mutation in Avr-Pik and was separated from a lesion developed in the incompatible Ken54-20 inoculation against lines with the Pik resistance gene. Therefore, Pik-h is expected to be allelic to Pik, and indeed there was no recombination between the resistances against the wild-type isolate Ken54-20 and mutant isolate Ken54-20-k +, i.e., between Pik and Pik-h (Kiyosawa and Murty 1969). Further, the resistance spectra of the Pik group genes are identical for the 12 IRRI isolates used for IRBL Pi monogenic line development (Tsunematsu et al. 2000) except for a slight difference in Pi1. The resistance spectra of Pik, Pik-m, and Pik-h group genes are also very similar to the Japanese standard differentiating set of 31 blast races (Hayashi 2005).
Later, Pik-h was found to be widely distributed in several cvs. as in Indian Charnak, Vietnamese Te-tep, Philippine Tadukan, Russian Roshia 33, American Dawn (Kiyosawa 1981), and Japanese cvs., such as Fuji120, Mutsunishiki, and Chugoku 31 (Kiyosawa 1978). Thus, Pik-h was found more frequently than Pik-p or Pik-m with each single cv. known at that time. However, the detailed analysis of Pik-h has been severely hindered due to the loss of the key differentiating mutant isolate Ken54-20-k +. For example, although Fjellstrom et al. (2004) reported that Pik-h is rather widely distributed in American rice cvs., his paper lacks a critical description of the race system differentiating Pik cluster genes, except the easily discernable Pik-s. With the conventional USA (Dr. Y. Jia, personal communication) and Philippine (Tsunematsu et al. 2000) race set, it is not possible to discriminate Pik-h from some other closely related Pik group genes, such as Pik-m. Therefore, our new identification of natural Pik-h-differentiating isolates will facilitate utilization of Pik group genes in breeding, and analysis of evolution of the R genes in the Pik cluster, and also that of the AvrPik group genes in blast fungus, and of their recognition mechanism.
In the present study, for genetic analysis of Pik-h, we used the monogenic line IRBLkh-K3 with the LTH (Lijiangxintuanheigu) genetic background (Tsunematsu et al. 2000). The original report on IRBL monogenic lines (Tsunematsu et al. 2000) did not differentiate the Pik group lines with race reactions, probably because of the lack of appropriate differentiating races, and in confidence regarding the donor lines. However, it became necessary to confirm the authenticity of the Pik-h gene in IRBLkh-K3 and siblings of its cross with CO39 to refute the claim of “Pik-h cloning” by Sharma et al. (2005). As shown in Table 1, we have verified that the gene conferring resistance against V86010 in the cross was actually Pik-h.
We have also verified that the Pik-h gene maps both genetically and physically to the same region as the Pik gene cluster with higher resolution than in any previous report (Fig. 3a, b), confirming the association of Pik-h with the cluster. Use of the HEGS system greatly facilitated the fine mapping, also in the SSR marker system, while hitherto HEGS has mainly been used with AFLP marker systems (Hori et al. 2003). One marker analysis of 768 susceptible siblings was performed by overnight electrophoresis of 800 lanes with 8 set gel plates each with 100 lanes by a single researcher.
Pik-h gene was mapped within an interval of about 270 kb, including a gap of 40 kb that has yet to be sequenced, which was bridged by a recently found clone, based on the sequences of cv. Nipponbare. Within 54 predicted ORFs (RGP Rice Genome Annotation Database: http://ricegaas.dna.affrc.go.jp/) in this region, six were RGAs with the NBS-LRR structure. We suppose that some of these may be counterparts of the real Pik group genes, including Pik-h, although the genome structure around this region will be very different between japonica and indica, as described in the following section. Therefore, this report may facilitate cloning of other Pik cluster genes. Especially, Pik-s is found in several domestic and modern japonica lines although it is absent in Nipponbare. This indicates that the sequence differences between the Nipponbare RGA and the Pik-s gene are rather small, and it is possible that one of the RGAs identified here may be the counterpart. Therefore, we are currently also attempting to amplify the Pik-s gene using the sequences of the 6 RGAs shown in Fig. 3a.
Although we have 4 co-segregation markers within this region, it was still not easy to localize the R gene candidate further by simply enlarging the size of the mapping population, because recombination in 200 kb around the target region is highly suppressed, and seven recombination events occurred only in the region near the flanking markers (Fig. 3a). Therefore, construction of a new BAC library from a Pik-h-containing resistant cv. will be needed for cloning of Pik-h. Indeed, no data are available on the regions in the indica Kasalath (http://rgp.dna.affrc.go.jp/E/publicdata/kasalathendmap/index.html) or 9311 (Rice Genome Database, http://rise.genomics.org.cn/rice/index2.jsp) genome, corresponding to the Nipponbare BAC OSJNBb0049B20 to OSJNBa0036K13 in Fig. 3a. Although Chinese indica cv. Minghui 63, which contains Xa26(t), has a BAC contig that includes Y6855RA and R1506, slightly overlapping with our Pik-h region (Sun et al. 2004) with R1506, their rather short contig of Minghui 63 (around 100 kb) corresponds to a very long (more than 0.5 Mb) region in the Nipponbare map (Fig. 3). This absence of genome data in indica cvs. is likely due to the hypervariability of the genome in this region, as often seen around several resistance genes. Clustering of resistance genes other than Pik genes, such as the Xa gene group, has been reported in this region. However, we speculate that these Xa resistance genes, such as Xa3, Xa4, and Xa26, are not completely allelic to the Pik-group rice blast genes. As the resistance spectra of the Pik-group genes are so close that the epitopes recognized by them, i.e., Avr gene products of the blast races, are thought to be very similar, and will be different from the bacterial epitopes recognized by Xa gene products. Indeed, the Pik-h region and the cluster of RGAs were shown to be located 130 kb and 230 kb apart from Xa4 and Xa26, respectively (Fig. 3b). However, the markers developed in the present study will be useful for research and breeding to introduce other resistance genes in this region.
Considering the character of the Pik-h gene and the region discussed above, the claim of Sharma et al. (2005) about “cloning of the Pik-h gene from Tetep” is very doubtful. First, they presented no race characterization verifying their gene’s authenticity. Second, their “locus” is located more than 10 cM (JRGP RFLP 2000 Map) apart from the Pik cluster. Third, our PCR comparison of their “Pik-h ORF” between Tetep and IRBLkh-K3 revealed 5.5% deviation at the amino acid level (data not shown), a difference that is too large to consider their “gene” to also be functional in IRBLkh-K3. Fourth, the possibility that their “gene” is another R-gene is also small because their annotated “LRR and NBS sequences” are only 20 to 30 amino acids in length, which is one order shorter than the normal R-genes. Thirty amino acids can scarcely constitute a single unit of an LR “repeat.” Such structures will not be able to function as a ligand recognizing domain or a nucleotide binding site. Finally, no concrete evidence of complementation was presented at the 4th International Rice Blast Conference in Oct. 2007, in Changsha, China, to which they submitted an abstract.
A BAC library of a cultivar containing the Pik-h is being constructed to develop a real contig around the gene, and to finally clone the gene and elucidate the genomic character of this highly R-gene clustered region.
References
Bai J, Pennill LA, Ning J, Lee SW, Ramalingam J, Webb CR, Zhao B, Sun Q, Nelson JC, Leach JE, Hulbert SH (2002) Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res 12:1871–1884
Böhnert HU, Fudal I, Dioh W, Tharreau D, Notteghem JL, Lebrun MH (2004) A putative polyketide synthase/peptide synthetase from Magnaporthe grisea signals pathogen attack to resistant rice. Plant Cell 16:2499–2513
Bryan GT, Wu KS, Farrall L, Jia Y, Hershey HP, McAdams SA, Faulk KN, Donaldson GK, Tarchini R, Valent B (2000) A single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene Pi-ta. Plant Cell 12:2033–2045
Chen DH, Dela Vina M, Inukai T, Mackill DJ, Ronald PC, Nelson RJ (1999) Molecular mapping of the blast resistance genes, Pi44(t), derived from a durably resistant rice cultivar. Theor Appl Genet 98:1046–1053
Chen XW, Shang JJ, Chen DX, Lei CL, Zou Y, Zhai WX, Liu GZ, Xu JH, Ling ZZ, Cao G, Ma BT, Wang YP, Zhao XF, Li SG, Zhu LH (2006) A B-lectin receptor kinase gene conferring rice blast resistance. Plant J 46:794–804
Farman ML, Eto Y, Nakao T, Tosa Y, Nakayashiki H, Mayama S, Leong SA (2002) Analysis of the structure of the AVR1-CO39 avirulence locus in virulent rice-infecting isolates of Magnaporthe grisea. Mol Plant-Microbe Interact 15:6–16
Fjellstrom R, Conaway-Bormans CA, McClung AM, Marchetti MA, Shank AR, Park WD (2004) Development of DNA markers suitable for marker assisted selection of three Pi genes conferring resistance to multiple Pyricularia grisea pathotypes. Crop Sci 44:1790–1798
Glazebrook J, Rogers EE, Ausubel FM (1997) Use of Arabidopsis for genetic dissection of plant defense responses. Annu Rev Genet 31:547–569
Hayasaka H, Miyao A, Yano M, Matsunaga K, Sasaki T (1996) RFLP mapping of a rice blast resistance gene Pik. Breed Sci 46(Suppl 2):68
Hayashi N (2005) MAFF Microorganism genetic resources manual. Vol 18. Rice blast fungus. Natl Inst of Agrobiological Sciences
Hayashi K, Yoshida H, Ashikawa I (2006) Development of PCR-based allele-specific and InDel marker sets for nine rice blast resistance genes. Theor Appl Genet 113(2):251–260
Hori K, Kobayashi T, Shimizu A, Sato K, Takeda K, Kawasaki S (2003) Efficient construction of high-density linkage map and its application to QTL analysis in barley. Theor Appl Genet 107(5):806–813
Imbe T, Tsunematsu H, Kato H, Khush GS (2000) Genetic analysis of blast resistance in IR varieties and resistant breeding strategy. In: Tharreau D, Lebrun MH, Talbot NJ, Notteghem JL (eds) Advances in rice blast research. Kluwer Academic Publishers, pp 34–44
Inukai T, Nelson RJ, Zeigler RS, Sarkarung S, Mackill DJ, Bonnan JM, Takamure I, Kinoshita T (1994) Allelism of blast resistance genes in near isogenic lines of rice. Phytopathology 84:1278–1283
Jia Y, McAdams SA, Bryan GT, Hershey HP, Valent B (2000) Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. EMBO J 19:4004–4014
Kaji R, Ogawa T (1996) RFLP mapping of a blast resistance gene, Pik-m, in rice. Breed Sci 46(Suppl 1):70
Kawasaki S, Murakami Y (2000) Genome Analysis of Lotus japonicus. J Plant Res 113:497–506
Kiyosawa S (1972) Genetics of blast resistance. Rice breeding IRRI. Manila, Philippines. pp 203–225
Kiyosawa S (1978) Identification of blast-resistance genes in some rice varieties. Jpn J Breed 28:287–296
Kiyosawa S (1981) Gene analysis for blast resistance. Oryza 18:196–203
Kiyosawa S, Murty VVS (1969) The inheritance of blast-resistance in india rice variety, HR-22. Jpn J Breed 19:269–276
Li L, Wang L, Jing J, Li Z, Lin F, Pan QH (2007) The Pik m gene, conferring stable resistance to isolates of Magnaporthe oryzae, was finely mapped in a crossover-cold region on rice chromosome 11. Mol Breed 20:179–188
Liu X, Wang L, Chen S, Lin F, Pan Q (2005) Genetic and physical mapping of Pi36 (t), a novel rice blast resistance gene located on rice chromosome 8. Mol Genet Genomics 274:394–401
Liu X, Lin F, Wang L, Pan Q (2007) The in silico map-based cloning of Pi36, a rice coiled-coil nucleotide-binding site leucine-rich repeat gene that confers race-specific resistance to the blast fungus. Genetics 176:2541–2549
Martin GB, Bogdanove AJ, Sessa G (2003) Understanding the functions of plant disease resistance proteins. Annu Rev Plant Biol 54:23–61
McCouch SR, Nelson RJ, Tohme J, Zeigler RS (1994) Mapping of blast resistance genes in rice. In: Rice blast disease. In: Zeigler RS, Leong SA, Teng PS (eds) CABI and IRRI, Wallingford, Oxon, UK, pp 167–186
Meyers BC, Kozik A, Griego A, Kuang HH, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15:809–834
Orbach MJ, Farrall L, Sweigard JA, Chumley FG, Valent B (2000) A telomeric avirulence gene determines efficacy for rice blast resistance gene Pi-ta. Plant Cell 12:2019–2032
Qu S, Liu G, Zhou B, Bellizzi M, Zeng L, Dai L, Han B, Wang GL (2006) The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site-leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172:1901–1914
Sharma TR, Madhav MS, Singh BK, Shanker P, Jana TK, Dalal V, Pandit A, Singh A, Gaikwad K, Upreti HC, Singh NK (2005) High-resolution mapping, cloning and molecular characterization of the Pik-h gene of rice, which confers resistance to Magnaporthe grisea. Mol Genet Genomics 274:569–578
Sun XL, Cao YL, Yang ZF, Xu CG, Li XH, Wang SP, Zhang QF (2004) Xa26, a gene conferring resistance to Xanthomonas oryzae pv oryzae in rice, encodes an LRR receptor kinase-like protein. Plant J 37(4):517–527
Tsunematsu H, Yanoria MJT, Ebron LA, Hayashi N, Ando I, Kato H, Imbe T, Khush GS (2000) Development of monogenic lines of rice for rice blast resistance. Breed Sci 50:229–234
Wang ZX, Yano M, Yamanouchi U, Iwamoto M, Monna L, Hayasaka H, Katayose Y, Sasaki T (1999) The Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J 19:55–64
Wang C, Tan M, Xu X, Wen G, Zhang D, Lin X (2003) Localizing the bacterial blight resistance gene, Xa22(t), to a 100-kilobase bacterial artificial chromosom. Phytopathology 93:1258–1262
Xiang Y, Cao YL, Xu CG, Li XH, Wang SP (2006) Xa3, conferring resistance for rice bacterial blight and encoding a receptor kinase-like protein, is the same as Xa26. Theor Appl Genet 113:1347–1135
Xu X, Kawasaki S, Fujimura T, Wang CT (2005) A protocol for high-throughput extraction of DNA from rice leaves. Plant Mol Biol Rep 27:291–295
Yang Z, Sun X, Wang S, Zhang Q (2003) Genetic and physical mapping of a new gene for bacterial blight resistance in rice. Theor Appl Genet 106:1467–1472
Yu Z, Mackill DJ, Bonman JM, McCouch S, Guiderdoni E, Notteghem JL, Tanksley SD (1996) Molecular mapping of genes for resistance to rice blast (Pyricularia grisea Sacc). Theor Appl Genet 93:859–863
Zhou B, Qu SH, Liu GF, Dolan M, Sakai H, Lu GD, Bellizzi M, Wang GL (2006) The eight amino acid differences within three leucine-rich repeats between Pi2 and Piz-t resistance proteins determine the resistance specificity to Magnaporthe grisea. Mol Plant-Microbe Interact 19:1216–1228
Acknowledgments
We are grateful to Dr. XQ. Liu (South-Central University for Nationalities of China) and Dr. R. Babu for their technical assistance and helpful advice in the data analysis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Xu, X., Hayashi, N., Wang, C.T. et al. Efficient authentic fine mapping of the rice blast resistance gene Pik-h in the Pik cluster, using new Pik-h-differentiating isolates. Mol Breeding 22, 289–299 (2008). https://doi.org/10.1007/s11032-008-9175-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-008-9175-5