
Abstract
In this review, we outline the current trends in the field of machine learning-driven classification studies related to ADME (absorption, distribution, metabolism and excretion) and toxicity endpoints from the past six years (2015–2021). The study focuses only on classification models with large datasets (i.e. more than a thousand compounds). A comprehensive literature search and meta-analysis was carried out for nine different targets: hERG-mediated cardiotoxicity, blood–brain barrier penetration, permeability glycoprotein (P-gp) substrate/inhibitor, cytochrome P450 enzyme family, acute oral toxicity, mutagenicity, carcinogenicity, respiratory toxicity and irritation/corrosion. The comparison of the best classification models was targeted to reveal the differences between machine learning algorithms and modeling types, endpoint-specific performances, dataset sizes and the different validation protocols. Based on the evaluation of the data, we can say that tree-based algorithms are (still) dominating the field, with consensus modeling being an increasing trend in drug safety predictions. Although one can already find classification models with great performances to hERG-mediated cardiotoxicity and the isoenzymes of the cytochrome P450 enzyme family, these targets are still central to ADMET-related research efforts.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the past decade, machine learning (ML) has undergone a definite revival in connection to the emergence of big data and the increase of compute capacities. Some maintain that big data have the potential to challenge the scientific method itself for new discoveries in science, through studying data correlations at a large scale [1]. Either way, present computer facilities allow us to analyze larger and larger data sets. As the computer power (speed and amount of data) increased, machine learning algorithms proliferated and artificial neural networks (ANNs) achieved a renaissance, their key role being further underlined by the appearance of deep learning methods to handle previously unprecedented amounts of data [2]. A consolidated description of big data is given by integrating definitions from practitioners and academics, and mainly deals with analytics related to unstructured data, which constitute 95% of big data [3].
In computer-aided drug design, the application of machine learning methods constitutes the new generation of QSAR modeling, although with the larger amount of training data (wider applicability domain) most authors aim for developing classification, rather than regression models. Recently, Maran et al. have reviewed a large amount of QSAR articles (1 533) on 79 individual endpoints of environmental and medicinal chemistry relevance, from all years up until 2015 [4]. From this plethora of QSAR studies 1235 contained multiple linear regression (MLR) modeling, 226 ANN, 77 support vector machines (SVM), 42 k-nearest neighbors (kNN), 39 decision trees (DT), 35 random forests (RF) and a few others. On one hand, it shows the unchallenged leading role of MLR in the previous decades of QSAR modeling; on the other hand, it reveals the significant effect of the machine learning revolution that is still going on.
Parallel to the unshaken popularity of linear modeling, an unprecedented proliferation of nonlinear machine learning algorithms took place. The present work, as well as other surveys show the most frequently used ML techniques: tree-based methods (e.g., random forest, bagging, boosting and their variants), extensions of artificial neural networks (e.g., deep leaning networks, DNN), support vector machines (SVM), k-nearest neighbors (kNN) and Naïve Bayes (NB). The latter two techniques are involved as standard, classical, well-known techniques mainly for benchmarking; on the other hand, SVM, tree-based algorithms and neural networks are trending now in all aspects of data science. ML algorithms are routinely used in (i) bioactivity [5], as well as property predictions of drug related compounds [6]; (ii) de novo drug design, i.e., generation of new chemical structures of practical interest [7]; (iii) virtual screening [8]; (iv) prediction of reaction pathways [9] and v) compound-protein interactions [10], etc. ML algorithms are mainly aimed at prediction, for which a great selection of descriptors and chemical representations, as well as many ML algorithms can be combined [11]. ML models are trained to recognize structural patterns that differentiate between active and inactive compounds. Understanding the reasons why models are so effective in prediction is a challenging task but of utmost importance to guide drug design [12].
As ML algorithms are easily overfitted, proper validation is of crucial importance. It is an eye-opening conclusion of the review of Maran et al. that reproducible studies (615) are in minority as compared the non-reproducible studies (882) [4]. Although there is no silver bullet that will always produce a reliable estimation of prediction error, a combination of cross-validation techniques achieves consolidated and superb performance in the prediction of unknowns. There are many known and accepted ways for the validation of ML models, such as i) randomization (permutation) tests [13]; ii) the many variants of cross-validation, such as row-wise, pattern-wise, Venetian blinds, contiguous blocks, etc.[14].; iii) repeated double cross-validation [15] iv) internal and external test validation and others. A statistical comparison of cross-validation variants for classification was published recently [16].
ADMET (absorption, distribution, metabolism, excretion and toxicity) properties are crucial for drug design, as they can make or break (usually break) the career of drug candidates. Due to their central role, the present review will concentrate on collecting machine learning classification studies of ADMET-related targets in the last five years, providing a meta-analysis of nine important ADMET endpoints.
Methods
In the past decades, artificial intelligence has escaped the world of science fiction and became a ubiquitous, albeit often hidden, part of our lives. While the self-definition of the field for intelligent agents (autonomous units capable of reacting to environmental changes for a specific goal) is very broad and includes such everyday devices as a simple thermostat, people usually associate artificial intelligence with more complex systems. A prime example for the latter is machine learning, which gradually became a dominating approach in many scientific areas including classification, especially in the case of large datasets. There are several trains of thought to machine learning models (see below), but probably the two most popular, “main” branches are tree-based and neural network-based algorithms. Deep learning methods are mostly neural networks of increased complexity, capable of handling unprecedented amounts of data; a few illustrative examples from the world ADMET endpoints highlight their potential for multitask modeling (predicting multiple endpoints simultaneously) [17, 18].
Tree-based algorithms
Tree-based methods are very popular choices among machine learning techniques, not just in the field of ADME-related in silico modeling. The basic concept of tree-based algorithms is the use of decision trees for classification (and also regression) models. The trees are constructed in the following way: recursive binary splits are performed on the dataset based on the different features, parent and child nodes are created in this way, and the samples are separated into classes based on the majority class of the members in the terminal nodes (without child nodes) [19, 20].
There are new ensemble alternatives of the simple decision trees, such as random forests or gradient boosted trees. In the case of random forests (RT), one can use a voting-based combination of single decision trees for the classification of the objects with a better performance. Gradient boosting is an upgraded version, when the single decision trees are built sequentially with the boosting of the high performance ones and the minimization of the errors [21]. The optimized version of gradient boosted trees is the extreme gradient boosted tree (XGBoost) method, which can handle missing values and with a much smaller chance to overfitting. The tree-based algorithms are useful to handle complex nonlinear problems with imbalanced datasets, although in the case of noisy data they still tend to overfit. The hyperparameters (especially in XGBoost) should be tuned.
Neural networks
Artificial neural networks (ANNs) and their specialized versions such as deep neural networks (DNN) or deep learning (DL) are one of the most common algorithms in the machine learning field, for ADMET-related and other prediction tasks [22, 23]. The basic concept of the algorithm is inspired by the structure of the human brain. Neural networks consist of input layers, hidden layer(s) and output layer(s). The hidden layers include a number of neurons. Every input variable in the input layer has different weights. A nonlinear activation function helps to transform the different linear combination of the input nodes into the output value. The weights are optimized in an iterative process to decrease the error of the prediction, for example with feed-forward or back propagation.
The major difference between traditional neural networks and deep learning is the amount of data and the complexity of the network. DL networks usually consists of several hidden layers, while classical neural networks are using usually just one (or two). In DL, the molecular descriptors are transformed into more abstract levels from layer to layer with the capability to manage complex functions. With such a complex network, overfitting is a possibility thus the network should be tuned [18]. The problem of overfitting is managed in deep neural networks with different improvements such as dropout [24]. Neural networks can be used for both regression and classification problems, and the algorithm can handle missing values and incomplete data. Probably, the biggest disadvantage of the method is the so-called “black-box” modeling; the user has little information on the exact role the provided inputs.
Support vector machine
Support vector machines (SVM) are a classical nonlinear algorithm for classification and regression modeling as well. The basic idea is the nonlinear mapping of the features in a higher dimensional space. A hyperplane is constructed in this space, which can define the class boundaries. Finding the optimal hyperplane needs some training data, and the so-called support vectors [25]. For the optimal separation by the hyperplanes, one should use a kernel function such as a radial basis function, a sigmoidal or a polynomial function [26]. Support vector machines can be applied for binary and multiclass problems as well. SVM works well in high dimensional data and the kernel function is a great strength of the method, although the interpretation of the weights and impact of the variables is difficult.
Naïve Bayes algorithms
Naïve Bayes algorithm is a supervised technique, which is based on the Bayesian theorem and the assumption of the uncorrelated (independent) features in the dataset. It also assumes that no hidden or latent variables influence the predictions (hence the name “naïve”) [27]. It is a simpler and faster algorithm compared to the other ML techniques; however, usually it has a cost in accuracy. Naïve Bayes algorithms are connected to Bayesian networks as well. Individual probability values for each class are calculated to every object separately. The naïve Bayes algorithm is very fast, even in the big data era compared to the other algorithms, but it performs better in the less complex and “ideal” cases.
Nearest neighbor-based algorithms
The k-nearest neighbor algorithm is one of the simplest and most commonly used classification methods [28, 29]. Simple, because this method only needs the calculation of distances between the ligand pairs in the dataset. In the case of k-nearest neighbors, the algorithm considers the group of k nearest compounds (objects) and classifies the compounds/objects according to the majority votes in the class. Lots of variants are existing, such as N-nearest neighbors (N3), which is extended to all the n-1 compounds from k, or binned nearest neighbors (BNN) [30]. Nearest neighbor type algorithms can be used for binary and multiclass classification, and regression as well. These algorithms are easy to understand and intuitive, but they are sensitive to outliers and imbalanced datasets.
ML models on the most prominent ADME targets
Several ADME and toxicity (ADMET) related endpoints have been selected for the comparative analysis of the last five years of research literature. Only classification models and categorical endpoints were selected, thus some important, but mostly regression-based models such as PAMPA or clearance are not covered by this review due to the different trends in these areas. Another focal point of this collection was to limit the considered studies to those with at least one thousand compounds. This way, we could provide a well-defined comparison among the trending algorithms and recent modeling habits.
hERG-mediated cardiotoxicity
The human ether-à-go-go-related gene (hERG) encodes the α subunit of a voltage-gated potassium channel, which is one of the most important antitargets in drug discovery, as the inhibition of this ion channel results in fatal arrhythmia (sudden cardiac death) by prolonging the QT interval of cardiac action potential [31]. As such, significant research efforts are invested into screening compounds against hERG inhibition and developing predictive models to avoid compounds with hERG liabilities in the first place. Conventionally, hERG inhibition is evaluated in patch-clamp electrophysiological assays [32, 33], with thallium-flux assays being a relatively new alternative [34, 35]. The availability of large hERG inhibition datasets in PubChem Bioassay [36] and ChEMBL [37] allows for the development of reliable predictive models for hERG inhibition, with wide applicability domains.
Here, we have collected 15 works from the past five years that employ machine learning-based classification approaches to predict hERG inhibition [38,39,40,41,42,43,44,45,46,47,48,49,50,51]. All of these works apply training datasets of more than 1,000 molecules (and up to tens of thousands in some cases [47, 48]), and an overall majority presents two-class (active vs. inactive) classification (with the notable example of the 2015 study of Braga et al., who have introduced a third class of “weak blockers”) [38]. Categorizing the molecules into the active and inactive classes is usually done by applying common activity thresholds such as 1 µM, 10 µM or their combination, a comprehensive methodological comparison was presented by Siramshetty et al. [44]. Indeed, most of these works use the PubChem and ChEMBL databases as the data source, with a few examples of literature sources or other databases (NCATS, GOSTAR, etc.). Independently of the choice of software and machine learning methods, classification performances are routinely great. The Comparative analysis section contains more details about the performance of the models.
Blood–brain barrier penetration
The blood–brain barrier (BBB) is formed by brain capillary walls and glial cells to prevent harmful substances from entering the brain [52]. The penetration of this natural protective barrier of the central nervous system (CNS) by small molecules can be advantageous (in the case of CNS-directed drug candidates where BBB passage is a requirement of drug action) or disadvantageous. As such, measuring and predicting BBB penetration has been the focus of significant research efforts, particularly in CNS-related drug discovery [53]. As experimental data on BBB penetration is difficult to obtain, there are limited resources available for training machine learning models: there is relatively scarce data on BBB penetration in ChEMBL and PubChem Bioassay. Therefore, most studies rely on a limited number of core literature where experimental logBB (blood–brain distribution coefficient, log(cbrain/cblood)!) values of altogether a few thousand compounds are collected [54, 55].
Here, we have collected seven machine learning classification studies from the past five years[56,57,58,59,60,61,62], with training sets of at least 1000 (and typically around 2000) compounds, employing popular machine learning methods such as random forests of support vector machines. All of these studies apply a two-class (penetrant or BBB + vs. non-penetrant or BBB–) classification scenario, usually with logBB thresholds of + 1, –1 or their combination. In addition to the most popular software choices and dedicated machine learning/deep learning platforms, 2D molecule images also appear as an interesting choice for compound descriptors in the work of Shi et al. [60].
Permeability glycoprotein (P-gp)
Permeability glycoprotein (P-gp) is a membrane protein that plays a pivotal role in the transport of a plethora of substrates through the cell membrane. This means that P-gp (which is expressed in blood–tissue and blood–brain barriers, among many other types of tissues like liver, colon, etc.) is of fundamental importance in pharmacokinetics, by regulating the efflux properties of a drug [63]. Coupled to ATP hydrolysis, P-gp can excrete several substrates out of the cell [64], this is why the over-expression of P-gp is a key factor in multidrug resistance [65]. Additionally, indiscriminate inhibition of P-gp in liver tissue will interfere with the excretion of xenobiotics [17], potentially leading to hepatotoxicity. All this explains why much effort has been devoted to the study of P-gp inhibitors and substrates.
P-gp substrates and inhibitors are usually tested in separate studies and naturally there are more studies with the focus on inhibitors [17, 66,67,68,69,70,71,72] instead of substrates [68, 73]. The use of consensus modeling for this endpoint seems to be a viable option, a good example is the work of Yang et al. [72]. In another specific study of Prachayasittikul and coworkers [70], the authors used SMILES-based descriptors to build a novel classification model using the CORAL software. The pseudo-regression model also shows great promise, with accuracy values over 80%, despite being relatively simple. Finally, among the most recent studies on P-gp inhibition we have the work of Esposito et al. [73], which uses molecular dynamics fingerprints as descriptors. Overall, all methods performed very well, even external validation accuracies were above 0.70. A detailed comparison will be presented in the Comparative analysis section.
Cytochrome P450 enzyme family
The cytochrome P450 enzymes (CYP) have a crucial role in the metabolism of the xenobiotics. The CYP family of enzymes is also involved in drug safety and efficacy, because of the responsibility in drug-drug interactions (DDIs) [74]. In the human body, 57 different CYP isoforms can be found. Out of these, the most important six isoforms (CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4) of the family metabolize more than 95% of the FDA-approved drugs [75].
In recent five years, several machine learning classification models have been developed for the mentioned targets [76,77,78,79,80,81,82,83]. There are several online data sources with experimental results (such as PubChem Bioassay) for the different isoenzymes separately and together as well. The classification models are strongly connected to the PubChem Bioassay database: these datasets were used for almost every model, with one exception [77]. In one specific case, namely the 2C9 isoform, the collected dataset has reached even 35 000 different molecules [74]. It should be emphasized, that the presence of the different CYP isoforms enables the development of multitarget classification models [80, 83]. The performances of the different models are discussed in detail later in the Comparative analysis section.
Acute oral toxicity
Acute toxicity can be defined as oral, dermal or inhalation, but out of the three types, oral toxicity is the most well-known and thoroughly examined. It is an important endpoint from the early stage of drug discovery, since a compound that is hazardous for human health should be filtered out as early as possible [84]. Several machine learning models have been developed for the prediction of the median lethal dose (LD50) values of the compounds in continuous (regression) and categorical (classification) setups as well. Rodents are the most common animals to test the median lethal dose of a compound, thus the usual datasets for machine learning modeling contain this type of data.
In our study, we have summarized the relevant classification models [85,86,87,88]. Different guidelines help in the categorization of the compounds in the different toxicity classes, such as the four-class system of the U.S. Environmental Protection Agency (EPA) [89] or the five-class version of the United Nations Globally Harmonized System of Classification and Labelling (GHS) [90]. Although multiclass classification is more frequent, one can find two-class classifications too, where the datasets are separated into very toxic or non-toxic (positive and negative) classes [87]. For this endpoint, the datasets usually contain more than ten thousand compounds and consensus models are frequently used. More details about these models are discussed later in the Comparative analysis section.
Carcinogenicity
Carcinogens are defined as chemical substances that can cause cancer and therefore, carcinogenicity is one of the most important toxicological endpoints, contributing even to the subsequent withdrawal of several approved drugs [91]. Carcinogenicity is usually tested in animal models [92], which, for ethical (and also economical) reasons, further underpins the importance of developing reliable predictive models to screen out potential carcinogenic liabilities early in the drug discovery process. As such, the prediction of carcinogenicity is the central topic of a vast literature, including early SAR and QSAR studies, and more recently, diverse machine learning approaches based on large training datasets [93,94,95]. It should be noted that structural alert-based systems can also achieve decent accuracies in carcinogenicity prediction [96], further supporting the use of molecular fingerprints in predictive models (as it was dominated in the corresponding literature data from the past five years). All the evaluated models for this target are based on the Carcinogenic Potency Database [97].
Mutagenicity
Genetic toxicity testing is an early alternative of the carcinogenicity tests in the drug discovery processes. Bacterial tests are widespread methods in the pharma industry, and the Salmonella-reverse-mutation assay or Ames test is the in vitro gold standard for the task [98]. The Ames assay was developed by Bruce Ames and his colleagues almost fifty years ago [99], and still this is the most important assay for the determination of the mutagenic potential of compounds. Most of the online mutagenicity databases are based on this in vitro experiment.
In the past five years, several machine learning classification models have been developed for this endpoint [43, 100,101,102,103]. Most of them have applied six to seven thousand compounds for binary classification, mainly based on the Hansen Ames Salmonella mutagenicity benchmark data [104]. The performances were usually a bit lower compared to the other endpoints, especially in binary classification (see more details in the Comparative analysis section).
Respiratory toxicity
Chemical respiratory toxicity can cause serious harm for the human body; moreover, the effects are not always obvious in the early stages [105]. Respiratory toxicity can lead to symptoms such as asthma, bronchitis, pneumonia, rhinitis, etc. Unfortunately, pulmonary drug toxicity is possibly an underdiagnosed cause of lung diseases. Therefore, it is also a major endpoint in ADMET studies. Naturally, in silico models can be useful alternatives to the usually applied animal experiments for the determination of respiratory toxicity.
We can find less publications compared to the other targets in the past few years [106,107,108], but performances are excellent for this endpoint. It is also worth to note that the size of the datasets is much smaller compared to other endpoints. Some commonly used and publicly available databases from the publications are ChemIDplus (TOXNET) (chem.nlm.nih.gov/chemidplus/), PNEUMOTOX (www.pneumotox.com) and ADrecs [109].
Irritation/corrosion
Another important topic is the examination of the skin and eye irritation effects of the different chemicals. REACH requirements should be fulfilled before a compound is entering the market (European legislation, (Regulation EC No 1907/2006)) [110]. This regulation includes the endpoints of skin and eye irritation and serious damage (corrosion). Corrosive compounds can destroy the living tissues in the contact area (irreversible damage), while the irritative substances can cause inflammation (reversible damage) [111].
In this review, we have focused only on eye irritation. Eye irritation and corrosion experiments involve animal testing, preferably rabbits, but in silico approaches could potentially reduce the amount of animal testing in this case as well [110]. We have found three binary classification models from the past five years with more than one thousand compounds in the datasets [112, 113]. Gathering data for these endpoints is harder compared to other targets: usually several databases and literature data were merged into the final datasets for modeling.
Comparative analysis
In this review, 89 different models were evaluated from the relevant literature as a representative set. It is worth mentioning that only those relevant ADME and toxicity targets were used, where the potential use of classification models is supported, i.e., the target variable is categorical, such as inhibitor vs. non-inhibitor, toxic vs. non-toxic, etc. Our aim was to provide a comparison from the relevant publications of the last five years, when the authors used machine learning techniques in a combined or single mode for predicting different ADME-related endpoints in the big data era. The so-called “big data” formalism means different dataset sizes in science; thus, here we considered only those publications for the comparative study, where the datasets contained more than 1000 molecules. The gathering of the publications was closed on February 28, 2021. The final database of the models is shown in the Supplementary material.
Figure 1 shows the distribution among the different targets in the literature dataset. The CYP P450 isoforms (1A2, 2C9, 2C19, 2D6 and 3A4) were treated separately.

Distribution of the targets with percentages (BBB: blood–brain barrier)
In the last five years in machine learning driven in silico classification modeling, the most frequent target was the drug metabolism related cytochrome P450 enzyme family. The distribution is closely uniform for the different isoforms, which can be attributed to the commonly used multi-targets in CYP P450 modeling. Another large proportion (17%) is connected to hERG (cardiotoxicity) modeling, since this target has a crucial role in drug safety as an antitarget and nowadays it is a routine procedure to test compounds for hERG-channel activity in the early stage of drug discovery.
Usually, more than one model is published in the papers, thus it is important to emphasize that only the best model for each target was evaluated from the publications in the following comparison.
The models were compared based on (i) the applied machine learning algorithm, (ii) the validation protocol, (iii) the used descriptor set, (iv) the modeling type (as consensus/single), (v) the performance of the models and (vi) the dataset size. Naturally, the authors did not always provide these parameters, thus missing values can occur in the dataset.
Consensus modeling means that the model was based on more than one machine learning algorithms and the authors applied various kinds of data fusion options for the development of the consensus model. It was interesting to see that 80% of the models were based on a single algorithm. As consensus modeling is a very common field of in silico machine learning, we have no doubt about the increase of this type of models in the near future, especially for more complex targets.
In QSAR/QSPR modeling, the use of different molecular descriptors, fingerprint variants and other X variables, such as docking score values or molecular dynamics simulation related variables has an important role. Several commercial software and publicly available tools offer the calculation of thousands of descriptors, and the selection of the appropriate ones can have a great effect in the final performance of the models. In Fig. 2, we have collected the used descriptor sets in the best models.

Occurrences of different descriptor types in the classification models
The most frequent combination was the application of classical 1D/2D/3D molecular descriptors with different fingerprints, which was followed by using only molecular descriptors and only fingerprints. Other descriptors, such as SMILES string related descriptors, molecular dynamics (MD) descriptors, 2D molecule images or docking score values are less frequently used, both alone and in combination with the other two favorite types.
Figure 3 shows the occurrences of the different machine learning algorithms. We have classified them into six different groups: tree-based algorithms such as random forests, XGBoost, etc.; neural networks, which includes every algorithm with different network systems; support vector machine-based algorithms; nearest neighbor-based algorithms, such as kNN, 3NN, etc.; Naïve Bayes algorithms; and the rest of them was classified as “Other”. It is important to mention that in the consensus models, all of the used algorithms were classified into the related groups, thus the sum of the occurrences is higher than 89. (If the authors used more than one algorithm from the same type in a consensus model, it was counted only once.)

Occurrences of the different machine learning models in the collected dataset
Tree-based algorithms have clearly dominated in silico classification modeling in the ADME world in the past five years. SVM and neural network-based algorithms are also very common, and only a little amount of models contained algorithms other than the first five group, like logistic regression, LDA, self-organizing maps, SIMCA, etc. [72, 86, 114].
The use of different validation practices for the verification of the models was a divisive factor among the selected publications. We have checked the application of cross-validation (n-fold), internal validation and external validation alone, and in combination. Internal validation meant that the originally used database was split into two parts (training and test), while external validation meant that the authors used another database for the external verification of the model. Moreover, the training-test set splits were also evaluated when internal validation was used. Figure 4 shows the application of the validation types in the publications.

Occurrences of the different types of validations alone and in combination
It is clear that only a relatively small number of publications used all three type of validation. In most cases, cross-validation was used in combination with external test validation. However, it is surprising that in fourteen cases, only internal validation was used, which is at least a questionable practice. Three models were validated only externally, which is also interesting, because without internal or cross-validation, it does not reveal possible overfitting problems. Similar problems can be the use of only cross-validation, because in this case we do not know anything about model performance on “new” test samples.
Those models, where an internal validation set was used in any combination, were further analyzed based on the train–test splits (Fig. 5).

Occurrences of different split ratios in the train/test split of the datasets
Most of the internal test validations used the 80/20 ratio for train/test splitting, which is in good agreement with our recent study about the optimal training-test split ratios [115]. Other common choices are the 75/25 and 70/30 ratios, and relatively few datasets were split in half. It is common sense that the more data we use for training, the better performance we have–up to certain limits.
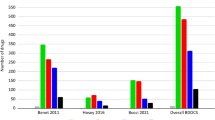
The dataset size was also an interesting factor in the comparison. Even though we had a lower limit of 1000 compounds, we wanted to check the amount of the available data for the examined targets in the past few years. (We did one exception in the case of carcinogenicity, where a publication with 916 compounds was kept in the database, because there was a rather limited number of publications from the last five years in that case.) External test sets were added to the sizes of the datasets. Figure 6 shows the dataset sizes in a Box and Whisker plot with median, maximum and minimum values for each target.

Dataset sizes for each examined target. Figure 6 A is the zoomed version of Fig. 6B, which is visually better for the targets with smaller dataset sizes. The number of molecules are plotted with the use of median, minimum and maximum values
The largest databases belong to the hERG target, while the smallest amount of data is connected to carcinogenicity. We can safely say that the different CYP isoforms, acute oral toxicity, hERG and mutagenicity are the most covered targets. On the other hand, it is an interesting observation that most models operate in the range between 2000 and 10,000 compounds.
In the last section, we have evaluated the performance of the models for each target. Accuracy values were used for the analysis, which were not always given: in a few cases, only AUC, sensitivity or specificity values were determined, these were excluded from the comparisons. While accuracies were selected as the most common performance parameter, we know that model performance is not necessarily captured by only one metric. Figures 7 and 8 show the comparison of the accuracy values for cross-validation, internal validation and external validation separately. CYP P450 isoforms are plotted in Fig. 7, while Fig. 8 shows the rest of the targets.

Comparison of the accuracies for the different classification models for CYP P450 isoforms. Median, minimum and maximum values are plotted for each target

Comparison of the accuracies for the different ADME related targets. Median, minimum and maximum values are plotted
For CYP targets, it is interesting to see that the accuracy of external validation has a larger range compared to internal and cross-validation, especially for the 1A2 isoform. However, dataset sizes were very close to each other in these cases, so it seems that this has no significant effect on model performance. Overall, accuracies are usually above 0.8, which is appropriate for this type of models. In Fig. 8, the variability is much larger. While the accuracies for blood brain barrier (BBB), irritation/corrosion (eye), P-gp inhibitor and hERG targets are very good, sometimes above 0.9, carcinogenicity and hepatotoxicity still need some improvement in the performance of the models. Moreover, hepatotoxicity has the largest range of accuracies for the models compared to the others.
Average accuracies were compared with ANOVA analysis to show the effect of the different machine learning algorithms (only single models with one machine learning algorithm were included). Moreover, average absolute differences of the accuracies were calculated between CV and internal validation, CV and external validation and between external and internal validation (where it was possible). ANOVA analysis was also carried out on these values, which could present the difference in the robustness between the algorithms. Nearest neighbors algorithm was excluded from the comparison, because it was used only in consensus modeling.
Figure 9 shows the results of ANOVA. The machine learning algorithms have no significant effect on the models, but we have to note, that the variances are a bit bigger compared to the target related accuracies, due to the use of average values. On the other hand, in the case of the average absolute differences of the accuracies (b) a significant effect could be detected between the algorithms. We can observe that SVM and Neural networks have somewhat better average accuracies, but their robustness is worse compared to the Tree-based and Naïve Bayes algorithms.

a b ANOVA analysis based on the a average accuracies and b average absolute differences of the accuracies. Machine learning algorithms are plotted in the X axis. The mean values and the 95% confidence intervals are shown in the figures.

Comparison of the applied software packages
Resources
In the past decades, the role of the different programming languages and open-source platforms in QSAR/QSPR modeling rapidly increased. Thus, it is not surprising that in the last five years, the most popular algorithms are connected to Python or R-based packages (see Fig. 10). One can find several machine learning packages for both platforms, however KNIME as a visual JAVA-based platform is also in this competition, because of the useful machine learning-related packages developed especially for classification problems. Several Python-based algorithms have KNIME implementations as well. One good example for this is Weka, which is also a well-known machine learning toolkit [116].
We have compared the software/platform usages in our dataset, where the authors shared this information. LibSVM (https://www.csie.ntu.edu.tw/~cjlin/libsvm/), Weka (https://www.cs.waikato.ac.nz/ml/weka/) and Tensorflow (www.tensorflow.org) software have several implementation options, thus we have decided to present these separately. Sometimes the authors have used more than one platform: these results are added separately to each segment. Almost half of the machine learning model developments are connected to either Python, R studio or KNIME. It is also worth to note, that Orange became a well-known open-source platform in the last couple of years [117]. Naturally, commercial software such as MATLAB or Discovery Studio are covering a smaller portion. Other software includes all the standalone developments (open-source or commercial) such as ADMET predictor (Simulations Plus, Inc., www.simulations-plus.com), PgpRules [68], CORAL [70] or Clementine (SPSS Inc., http://www.spss.com). The latter ones had usually single occurrences in the dataset.
We cannot overlook several useful web-accessible tools for ADMET predictions, such as ADMETlab (http://admet.scbdd.com) [118] or CypReact (https://bitbucket.org/Leon_Ti/cypreact) [119], which are also based on several machine learning models, although this is not the main focus of this review.
Concluding remarks
The prediction of ADMET-related properties plays an important role in drug design as safety endpoints, and it seems that it will stay in this position for a long time. Several of these drug safety targets are connected to harmful or deadly animal experiments, raising ethical concerns, moreover, the cost of most of these measurements is rather high. Thus, the use of in silico QSAR/QSPR models to overcome the problematic aspects of drug safety related experiments is highly supported.
The use of machine learning (artificial intelligence) algorithms is a great opportunity in the QSAR/QSPR world for the reliable prediction of bioactivities on new and complex targets. Naturally, the increasing amount of publicly accessible data is also helping to provide more reliable and extensively applied models. In this review, we have focused on those models, which were based on bigger datasets (above one thousand molecules), to provide a comprehensive evaluation of the recent years’ ADMET-related models in the larger dataset segment. The findings showed the popularity of tree-based algorithms for classification problems. In the aspect of validation, many models still rely on only cross-validation or only internal validation, which signifies a room for improvement in validation practices. The in silico predictions of ADMET parameters have been, and will remain a central question of computational drug discovery and with the increasing databases, fast and efficient open-source platforms for modeling and the development of novel algorithms, we believe that dedicated machine learning models have proven to be indispensable tools.
References
Fillinger S, de la Garza L, Peltzer A et al (2019) Challenges of big data integration in the life sciences. Anal Bioanal Chem 411:6791–6800. https://doi.org/10.1007/s00216-019-02074-9
Panteleev J, Gao H, Jia L (2018) Recent applications of machine learning in medicinal chemistry. Bioorg Med Chem Lett 28:2807–2815. https://doi.org/10.1016/j.bmcl.2018.06.046
Gandomi A, Haider M (2015) Beyond the hype: big data concepts, methods, and analytics. Int J Inf Manage 35:137–144. https://doi.org/10.1016/j.ijinfomgt.2014.10.007
Piir G, Kahn I, García-Sosa AT et al (2018) Best practices for QSAR model reporting: physical and chemical properties, ecotoxicity, environmental fate, human health, and toxicokinetics endpoints. Environ Health Perspect. https://doi.org/10.1289/EHP3264
Lima AN, Philot EA, Trossini GHG et al (2016) Use of machine learning approaches for novel drug discovery. Expert Opin Drug Discov 11:225–239. https://doi.org/10.1517/17460441.2016.1146250
Schneider G Prediction of drug-like properties. In: Madame Curie Biosci. Database [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK6404/
Domenico A, Nicola G, Daniela T et al (2020) De novo drug design of targeted chemical libraries based on artificial intelligence and pair-based multiobjective optimization. J Chem Inf Model 60:4582–4593. https://doi.org/10.1021/acs.jcim.0c00517
Cortés-Ciriano I, Firth NC, Bender A, Watson O (2018) Discovering highly potent molecules from an initial set of inactives using iterative screening. J Chem Inf Model 58:2000–2014. https://doi.org/10.1021/acs.jcim.8b00376
von der Esch B, Dietschreit JCB, Peters LDM, Ochsenfeld C (2019) Finding reactive configurations: a machine learning approach for estimating energy barriers applied to Sirtuin 5. J Chem Theory Comput 15:6660–6667. https://doi.org/10.1021/acs.jctc.9b00876
Lim S, Lu Y, Cho CY et al (2021) A review on compound-protein interaction prediction methods: data, format, representation and model. Comput Struct Biotechnol J 19:1541–1556. https://doi.org/10.1016/j.csbj.2021.03.004
Haghighatlari M, Li J, Heidar-Zadeh F et al (2020) Learning to make chemical predictions: the interplay of feature representation, data, and machine learning methods. Chem 6:1527–1542. https://doi.org/10.1016/j.chempr.2020.05.014
Rodríguez-Pérez R, Bajorath J (2020) Interpretation of compound activity predictions from complex machine learning models using local approximations and shapley values. J Med Chem 63:8761–8777. https://doi.org/10.1021/acs.jmedchem.9b01101
Rücker C, Rücker G, Meringer M (2007) Y-randomization and its variants in QSPR/QSAR. J Chem Inf Model 47:2345–2357. https://doi.org/10.1021/ci700157b
Bro R, Kjeldahl K, Smilde AK, Kiers HAL (2008) Cross-validation of component models: a critical look at current methods. Anal Bioanal Chem 390:1241–1251. https://doi.org/10.1007/s00216-007-1790-1
Filzmoser P, Liebmann B, Varmuza K (2009) Repeated double cross validation. J Chemom 23:160–171. https://doi.org/10.1002/cem.1225
Rácz A, Bajusz D, Héberger K (2018) Modelling methods and cross-validation variants in QSAR: a multi-level analysis $. SAR QSAR Environ Res 29:661–674. https://doi.org/10.1080/1062936X.2018.1505778
Montanari F, Zdrazil B, Digles D, Ecker GF (2016) Selectivity profiling of BCRP versus P-gp inhibition: from automated collection of polypharmacology data to multi-label learning. J Cheminform 8:7. https://doi.org/10.1186/s13321-016-0121-y
Wenzel J, Matter H, Schmidt F (2019) Predictive multitask deep neural network models for ADME-Tox properties: learning from large data sets. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.8b00785
Zhang MH, Xu QS, Daeyaert F et al (2005) Application of boosting to classification problems in chemometrics. Anal Chim Acta 544:167–176. https://doi.org/10.1016/j.aca.2005.01.075
Breiman L, Friedman JH, Olshen RA, Stone CJ (1984) Classification and regression trees. Wadsworth International Group, Monterey
Chen T, Guestrin C (2016) XGBoost: A scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. ACM, New York, NY, USA, (pp 785–794)
Salt DW, Yildiz N, Livingstone DJ, Tinsley CJ (1992) The use of artificial neural networks in QSAR. Pestic Sci 36(2):161–170. https://doi.org/10.1002/ps.2780360212
Chen H, Engkvist O, Wang Y et al (2018) The rise of deep learning in drug discovery. Drug Discov Today 23(6):1241–1250
Srivastava N, Hinton G, Krizhevsky A et al (2014) Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res 15:1929–1958
Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20(3):273–297
Brereton RG, Lloyd GR (2009) Support vector machines for classification and regression. Analyst 135:230–267
John GH, Langley P (1995) Estimating continuous distributions in Bayesian classifiers. In: UAI’95 Proceedings of the eleventh conference on uncertainty in artificial intelligence (pp 338–345)
Kowalski BR, Bender CF (1972) The K-nearest neighbor classification rule (pattern recognition) applied to nuclear magnetic resonance spectral interpretation. Anal Chem 44:1405–1411. https://doi.org/10.1021/ac60316a008
Kramer O (2013) K-Nearest Neighbors. Dimensionality reduction with unsupervised nearest neighbors. Springer, Berlin Heidelberg, pp 13–23. https://doi.org/10.1007/978-3-642-38652-7_2
Todeschini R, Ballabio D, Cassotti M, Consonni V (2015) N3 and BNN: two new similarity based classification methods in comparison with other classifiers. J Chem Inf Model 55:2365–2374. https://doi.org/10.1021/acs.jcim.5b00326
Vandenberg JI, Perry MD, Perrin MJ et al (2012) hERG K + Channels: structure, function, and clinical significance. Physiol Rev 92:1393–1478. https://doi.org/10.1152/physrev.00036.2011
Polonchuk L (2012) Toward a new gold standard for early safety: automated temperature-controlled hERG test on the PatchLiner®. Front Pharmacol. https://doi.org/10.3389/fphar.2012.00003
Hamill OP, Marty A, Neher E et al (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch-Eur J Physiol 391(2):85–100. https://doi.org/10.1007/BF00656997
Weaver CD, Harden D, Dworetzky SI et al (2004) A Thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen 9:671–677. https://doi.org/10.1177/1087057104268749
Weaver CD (2018) Thallium flux assay for measuring the activity of monovalent cation channels and transporters. In: Shyng SL, Valiyaveetil FI, Whorton M (eds) Potassium channels: methods and protocols. Springer, New York
Bolton EE, Wang Y, Thiessen PA, Bryant SH (2008) PubChem: integrated platform of small molecules and biological activities. In: Dixon DA, Chair RR (eds) Annual reports in computational chemistry. Elsevier, Amsterdam, pp 217–241
Gaulton A, Bellis LJ, Bento AP et al (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40:D1100–D1107. https://doi.org/10.1093/nar/gkr777
Braga RC, Alves VM, Silva MFB et al (2015) Pred-hERG: A Novel web-accessible computational tool for predicting cardiac toxicity. Mol Inform 34:698–701. https://doi.org/10.1002/minf.201500040
Sun H, Huang R, Xia M et al (2017) Prediction of hERG Liability—Using SVM classification Bootstrapping and Jackknifing. Mol Inform 36:1600126. https://doi.org/10.1002/minf.201600126
Konda LSK, KeerthiPraba S, Kristam R (2019) hERG liability classification models using machine learning techniques. Comput Toxicol. https://doi.org/10.1016/j.comtox.2019.100089
Zhang C, Zhou Y, Gu S et al (2016) In silico prediction of hERG potassium channel blockage by chemical category approaches. Toxicol Res (Camb) 5:570–582. https://doi.org/10.1039/c5tx00294j
Li X, Zhang Y, Li H, Zhao Y (2017) Modeling of the hERG K+ Channel blockage using online chemical database and modeling environment (OCHEM). Mol Inform 36:1700074. https://doi.org/10.1002/minf.201700074
Alves VM, Golbraikh A, Capuzzi SJ et al (2018) Multi-Descriptor read across (MuDRA): a simple and transparent approach for developing accurate quantitative structure-activity relationship models. J Chem Inf Model 58:1214–1223. https://doi.org/10.1021/acs.jcim.8b00124
Siramshetty VB, Chen Q, Devarakonda P, Preissner R (2018) The Catch-22 of predicting hERG Blockade using publicly accessible bioactivity data. J Chem Inf Model 58:1224–1233. https://doi.org/10.1021/acs.jcim.8b00150
Siramshetty VB, Nguyen D-T, Martinez NJ et al (2020) Critical assessment of artificial intelligence methods for prediction of hERG channel inhibition in the “Big Data” Era. J Chem Inf Model 60:6007–6019. https://doi.org/10.1021/acs.jcim.0c00884
Liu M, Zhang L, Li S et al (2020) Prediction of hERG potassium channel blockage using ensemble learning methods and molecular fingerprints. Toxicol Lett 332:88–96. https://doi.org/10.1016/j.toxlet.2020.07.003
Kim H, Nam H (2020) hERG-Att: self-attention-based deep neural network for predicting hERG blockers. Comput Biol Chem. https://doi.org/10.1016/j.compbiolchem.2020.107286
Ogura K, Sato T, Yuki H, Honma T (2019) Support vector machine model for hERG inhibitory activities based on the integrated hERG database using descriptor selection by NSGA-II. Sci Rep 9:12220. https://doi.org/10.1038/s41598-019-47536-3
Lee H-M, Yu M-S, Kazmi SR et al (2019) Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinform 20:250. https://doi.org/10.1186/s12859-019-2814-5
Choi K-E, Balupuri A, Kang NS (2020) The study on the hERG blocker prediction using chemical fingerprint analysis. Molecules 25:2615. https://doi.org/10.3390/molecules25112615
Wang Y, Huang L, Jiang S et al (2020) Capsule networks showed excellent performance in the classification of hERG blockers/nonblockers. Front Pharmacol. https://doi.org/10.3389/fphar.2019.01631
Daneman R, Prat A (2015) The blood-brain barrier. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a020412
Kaisar MA, Sajja RK, Prasad S et al (2017) New experimental models of the blood-brain barrier for CNS drug discovery. Expert Opin Drug Discov 12:89–103. https://doi.org/10.1080/17460441.2017.1253676
Abraham MH, Ibrahim A, Zhao Y, Acree WE (2006) A data base for partition of volatile organic compounds and drugs from blood/plasma/serum to brain, and an LFER analysis of the data. J Pharm Sci 95:2091–2100. https://doi.org/10.1002/jps.20595
Zhang L, Zhu H, Oprea TI et al (2008) QSAR modeling of the blood-brain barrier permeability for diverse organic compounds. Pharm Res 25(8):1902–1914. https://doi.org/10.1007/s11095-008-9609-0
Zhang X, Liu T, Fan X, Ai N (2017) In silico modeling on ADME properties of natural products: classification models for blood-brain barrier permeability, its application to traditional Chinese medicine and in vitro experimental validation. J Mol Graph Model 75:347–354. https://doi.org/10.1016/j.jmgm.2017.05.021
Yuan Y, Zheng F, Zhan C-G (2018) Improved prediction of blood-brain barrier permeability through machine learning with combined use of molecular property-based descriptors and fingerprints. AAPS J 20:54. https://doi.org/10.1208/s12248-018-0215-8
Wang Z, Yang H, Wu Z et al (2018) In silico prediction of blood-brain barrier permeability of compounds by machine learning and resampling methods. Chem Med Chem 13:2189–2201. https://doi.org/10.1002/cmdc.201800533
Roy D, Hinge VK, Kovalenko A (2019) To pass or not to pass: predicting the blood-brain barrier permeability with the 3D-RISM-KH molecular solvation theory. ACS Omega 4(16):16774–16780. https://doi.org/10.1021/acsomega.9b01512
Shi T, Yang Y, Huang S et al (2019) Molecular image-based convolutional neural network for the prediction of ADMET properties. Chemom Intell Lab Syst. https://doi.org/10.1016/j.chemolab.2019.103853
Li X, Fourches D (2020) Inductive transfer learning for molecular activity prediction: next-gen QSAR models with MolPMoFiT. J Cheminform 12:27. https://doi.org/10.1186/s13321-020-00430-x
Shi Z, Chu Y, Zhang Y et al (2021) Prediction of blood-brain barrier permeability of compounds by fusing resampling strategies and eXtreme gradient boosting. IEEE Access 9:9557–9566. https://doi.org/10.1109/ACCESS.2020.3047852
Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW (1998) The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci 95:7024–7029. https://doi.org/10.1073/pnas.95.12.7024
Jones PM, George AM (2004) The ABC transporter structure and mechanism: perspectives on recent research. Cell Mol Life Sci 61:682–699. https://doi.org/10.1007/s00018-003-3336-9
Leslie EM, Deeley RG, Cole SPC (2005) Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol 204:216–237. https://doi.org/10.1016/j.taap.2004.10.012
Prachayasittikul V, Worachartcheewan A, Shoombuatong W et al (2015) Classification of P-glycoprotein-interacting compounds using machine learning methods. EXCLI J 14:958–970
Hinge VK, Roy D, Kovalenko A (2019) Prediction of P-glycoprotein inhibitors with machine learning classification models and 3D-RISM-KH theory based solvation energy descriptors. J Comput Aided Mol Des 33(11):965–971. https://doi.org/10.1007/s10822-019-00253-5
Wang PH, Tu YS, Tseng YJ (2019) PgpRules: a decision tree based prediction server for P-glycoprotein substrates and inhibitors. Bioinformatics 35(20):4193–4195. https://doi.org/10.1093/bioinformatics/btz213
Ngo TD, Tran TD, Le MT, Thai KM (2016) Machine learning-, rule—and pharmacophore-based classification on the inhibition of P-glycoprotein and NorA. SAR QSAR Environ Res 27(9):747–780. https://doi.org/10.1080/1062936X.2016.1233137
Prachayasittikul V, Worachartcheewan A, Toropova AP et al (2017) Large-scale classification of P-glycoprotein inhibitors using SMILES-based descriptors. SAR QSAR Environ Res 28:1–16. https://doi.org/10.1080/1062936X.2016.1264468
CerruelaGarcía G, García-Pedrajas N (2018) Boosted feature selectors: a case study on prediction P-gp inhibitors and substrates. J Comput Aided Mol Des 32(11):1273–1294. https://doi.org/10.1007/s10822-018-0171-5
Yang M, Chen J, Shi X et al (2015) Development of in silico models for predicting p-glycoprotein inhibitors based on a two-step approach for feature selection and its application to Chinese herbal medicine screening. Mol Pharm 12:3691–3713. https://doi.org/10.1021/acs.molpharmaceut.5b00465
Esposito C, Wang S, Lange UEW et al (2020) Combining machine learning and molecular dynamics to predict P-Glycoprotein substrates. J Chem Inf Model 60:4730–4749. https://doi.org/10.1021/acs.jcim.0c00525
Rácz A, Keserű GM (2020) Large-scale evaluation of cytochrome P450 2C9 mediated drug interaction potential with machine learning-based consensus modeling. J Comput Aided Mol Des 34:831–839. https://doi.org/10.1007/s10822-020-00308-y
Kato H (2019) Computational prediction of cytochrome P450 inhibition and induction. Drug Metab Pharmacokinet. https://doi.org/10.1016/J.DMPK.2019.11.006
Pan X, Chao L, Qu S et al (2015) An improved large-scale prediction model of CYP1A2 inhibitors by using combined fragment descriptors. RSC Adv 5:84232–84237. https://doi.org/10.1039/c5ra17196b
Pang X, Zhang B, Mu G et al (2018) Screening of cytochrome P450 3A4 inhibitors via in silico and in vitro approaches. RSC Adv 8:34783–34792. https://doi.org/10.1039/c8ra06311g
Yu L, Shi X, Tian S et al (2017) Classification of cytochrome P450 1A2 Inhibitors and noninhibitors based on deep belief network. Int J Comput Intell Appl 16:1–17. https://doi.org/10.1142/S146902681750002X
Su BH, Tu YS, Lin C et al (2015) Rule-based prediction models of cytochrome P450 inhibition. J Chem Inf Model 55:1426–1434. https://doi.org/10.1021/acs.jcim.5b00130
Lee JH, Basith S, Cui M et al (2017) In silico prediction of multiple-category classification model for cytochrome P450 inhibitors and non-inhibitors using machine-learning method$. SAR QSAR Environ Res 28:863–874. https://doi.org/10.1080/1062936X.2017.1399925
Wu Z, Lei T, Shen C et al (2019) ADMET evaluation in drug discovery. 19. Reliable prediction of human cytochrome P450 inhibition using artificial intelligence approaches. J Chem Inf Model 59:4587–4601. https://doi.org/10.1021/acs.jcim.9b00801
Nembri S, Grisoni F, Consonni V, Todeschini R (2016) In silico prediction of cytochrome P450-Drug interaction : QSARs for CYP3A4 and CYP2C9. Int J Mol Sci 17:914. https://doi.org/10.3390/ijms17060914
Li X, Xu Y, Lai L, Pei J (2018) Prediction of human cytochrome P450 inhibition using a multitask deep autoencoder neural network. Mol Pharm 15:4336–4345. https://doi.org/10.1021/acs.molpharmaceut.8b00110
Yang H, Sun L, Li W et al (2018) In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front Chem 6:1–12. https://doi.org/10.3389/fchem.2018.00030
Xu Y, Pei J, Lai L (2017) Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model 57:2672–2685. https://doi.org/10.1021/acs.jcim.7b00244
Gadaleta D, Vuković K, Toma C et al (2019) SAR and QSAR modeling of a large collection of LD 50 rat acute oral toxicity data. J Cheminform. https://doi.org/10.1186/s13321-019-0383-2
Ballabio D, Grisoni F, Consonni V, Todeschini R (2019) Integrated QSAR models to predict acute oral systemic toxicity. Mol Inform 38:1800124. https://doi.org/10.1002/minf.201800124
Li X, Kleinstreuer NC, Fourches D (2020) Hierarchical quantitative structure—activity relationship modeling approach for integrating binary, multiclass, and regression models of acute oral systemic toxicity. Chem Res Toxicol. https://doi.org/10.1021/acs.chemrestox.9b00259
Chemical hazard classification and labeling - US EPA. www.epa.gov/sites/production/files/2015-09/documents/ghscriteria-summary.pdf
Globally harmonized system of classification and labelling of chemicals (GHS)https://pubchem.ncbi.nlm.nih.gov/ghs/
Onakpoya IJ, Heneghan CJ, Aronson JK (2016) Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med 14:10. https://doi.org/10.1186/s12916-016-0553-2
Jacobs AC, Brown PC (2015) Regulatory forum opinion piece*. Toxicol Pathol 43:605–610. https://doi.org/10.1177/0192623314566241
Li X, Du Z, Wang J et al (2015) In silico estimation of chemical carcinogenicity with binary and ternary classification methods. Mol Inform 34:228–235. https://doi.org/10.1002/minf.201400127
Zhang H, Cao ZX, Li M et al (2016) Novel naïve Bayes classification models for predicting the carcinogenicity of chemicals. Food Chem Toxicol 97:141–149. https://doi.org/10.1016/j.fct.2016.09.005
Zhang L, Ai H, Chen W et al (2017) CarcinoPred-EL: novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci Rep 7:2118. https://doi.org/10.1038/s41598-017-02365-0
Benigni R, Bossa C, Tcheremenskaia O, Giuliani A (2010) Alternatives to the carcinogenicity bioassay: in silico methods, and the in vitro and in vivo mutagenicity assays. Expert Opin Drug Metab Toxicol 6:809–819. https://doi.org/10.1517/17425255.2010.486400
Fitzpatrick RB (2008) CPDB: carcinogenic potency database. Med Ref Serv Q 27:303–311. https://doi.org/10.1080/02763860802198895
Escobar PA, Kemper RA, Tarca J et al (2013) Bacterial mutagenicity screening in the pharmaceutical industry. Mutat Res-Rev Mutat Res 752:99–118. https://doi.org/10.1016/j.mrrev.2012.12.002
Ames BN, Durston WE, Yamasaki E, Lee FD (1973) Carcinogens are mutagens: a simple test system. Mutat Res 21:209–210
Zhang H, Kang YL, Zhu YY et al (2017) Novel naïve Bayes classification models for predicting the chemical Ames mutagenicity. Toxicol Vitr 41:56–63. https://doi.org/10.1016/j.tiv.2017.02.016
Li S, Zhang L, Feng H et al (2021) MutagenPred-GCNNs: a graph convolutional neural network-based classification model for mutagenicity prediction with data-driven molecular fingerprints. Interdiscip Sci Comput Life Sci 13:25–33. https://doi.org/10.1007/s12539-020-00407-2
CerruelaGarcía G, García-Pedrajas N, Luque Ruiz I, Gómez-Nieto MÁ (2018) An ensemble approach for in silico prediction of Ames mutagenicity. J Math Chem 56:2085–2098. https://doi.org/10.1007/s10910-018-0855-z
Zhang J, Mucs D, Norinder U, Svensson F (2019) LightGBM: an effective and scalable algorithm for prediction of chemical toxicity-application to the Tox21 and mutagenicity data sets. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.9b00633
Hansen K, Mika S, Schroeter T et al (2009) Benchmark data set for in silico prediction of Ames mutagenicity. J Chem Inf Model 49:2077–2081. https://doi.org/10.1021/ci900161g
Kubo K, Azuma A, Kanazawa M et al (2013) Consensus statement for the diagnosis and treatment of drug-induced lung injuries. Respir Investig 51:260–277. https://doi.org/10.1016/j.resinv.2013.09.001
Lei T, Chen F, Liu H et al (2017) ADMET evaluation in drug discovery. Part 17: development of quantitative and qualitative prediction models for chemical-induced respiratory toxicity. Mol Pharm 14:2407–2421. https://doi.org/10.1021/acs.molpharmaceut.7b00317
Zhang H, Ma JX, Liu CT et al (2018) Development and evaluation of in silico prediction model for drug-induced respiratory toxicity by using naïve Bayes classifier method. Food Chem Toxicol 121:593–603. https://doi.org/10.1016/j.fct.2018.09.051
Wang Z, Zhao P, Zhang X et al (2021) In silico prediction of chemical respiratory toxicity via machine learning. Comput Toxicol. https://doi.org/10.1016/j.comtox.2021.100155
Cai MC, Xu Q, Pan YJ et al (2015) ADReCS: an ontology database for aiding standardization and hierarchical classification of adverse drug reaction terms. Nucleic Acids Res 43:D907–D913. https://doi.org/10.1093/nar/gku1066
Verheyen GR, Braeken E, Van Deun K, Van Miert S (2017) Evaluation of existing (Q)SAR models for skin and eye irritation and corrosion to use for REACH registration. Toxicol Lett 265:47–52. https://doi.org/10.1016/j.toxlet.2016.11.007
(ECHA) European chemicals agency (2015) Chapter R.7a: Endpoint specific guidance in: guidance on information requirements and chemical safety assessment. https://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf
Verma RP, Matthews EJ (2015) Estimation of the chemical-induced eye injury using a weight-of-evidence (WoE) battery of 21 artificial neural network (ANN) c-QSAR models (QSAR-21): Part I: Irritation potential. Regul Toxicol Pharmacol 71:318–330. https://doi.org/10.1016/j.yrtph.2014.11.011
Wang Q, Li X, Yang H et al (2017) In silico prediction of serious eye irritation or corrosion potential of chemicals. RSC Adv 7:6697–6703. https://doi.org/10.1039/c6ra25267b
Shoombuatong W, Prathipati P, Prachayasittikul V, Schaduangrat N (2017) Towards predicting the cytochrome P450 modulation : from QSAR to proteochemometric modeling. Current Drug Metab. https://doi.org/10.2174/1389200218666170320121932
Rácz A, Bajusz D, Héberger K (2021) Effect of dataset size and train/test split ratios in QSAR/QSPR multiclass classification. Molecules 26(4):1111
Hall M, Frank E, Holmes G et al (2009) The WEKA data mining software. ACM SIGKDD Explor Newsl 11:10–18. https://doi.org/10.1145/1656274.1656278
Demsar J, Curk T, Erjavec A et al (2013) Orange: data mining toolbox in Python. J Mach Learn Res 14:2349–2353. https://doi.org/10.5555/2567709.2567736
Dong J, Wang N-N, Yao Z-J et al (2018) ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform 10:29. https://doi.org/10.1186/s13321-018-0283-x
Tian S, Djoumbou-Feunang Y, Greiner R, Wishart DS (2018) CypReact: a software tool for in silico reactant prediction for human cytochrome P450 enzymes. J Chem Inf Model 58:1282–1291. https://doi.org/10.1021/acs.jcim.8b00035
Acknowledgements
The work of D.B. is supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences and the ÚNKP-20-5 New National Excellence Program of the Ministry for Innovation and Technology.
Funding
Open access funding provided by ELKH Research Centre for Natural Sciences. National Research, Development and Innovation Office of Hungary (OTKA, contract Nos K 119269 and K 134260 and PD134416). University of Florida: startup grant: RAMQ. Hungarian Academy of Sciences: János Bolyai Research Scholarship: DB. Ministry for Innovation and Technology of Hungary: ÚNKP-20-5 New National Excellence Program: DB
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rácz, A., Bajusz, D., Miranda-Quintana, R.A. et al. Machine learning models for classification tasks related to drug safety. Mol Divers 25, 1409–1424 (2021). https://doi.org/10.1007/s11030-021-10239-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-021-10239-x