
Abstract
The severe acute respiratory coronavirus 2 (SARS-CoV-2) infection demonstrates a highly variable and unpredictable course. Several reports have claimed a smoker’s paradox in coronavirus disease 2019 (COVID-19), in line with previous suggestions that smoking is associated with better survival after acute myocardial infarction and appears protective in preeclampsia. Several plausible physiological explanations exist accounting for the paradoxical observation of smoking engendering protection against SARS-CoV-2 infection. In this review, we delineate novel mechanisms whereby smoking habits and smokers’ genetic polymorphism status affecting various nitric oxide (NO) pathways (endothelial NO synthase, cytochrome P450 (CYP450), erythropoietin receptor (EPOR); β-common receptor (βcR)), along with tobacco smoke modulation of microRNA-155 and aryl-hydrocarbon receptor (AHR) effects, may be important determinators of SARS-CoV-2 infection and COVID-19 course. While transient NO bioavailability increase and beneficial immunoregulatory modulations through the above-mentioned pathways using exogenous, endogenous, genetic and/or therapeutic modalities may have direct and specific, viricidal SARS-CoV-2 effects, employing tobacco smoke inhalation to achieve protection equals self-harm. Tobacco smoking remains the leading cause of death, illness, and impoverishment.
Similar content being viewed by others
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, the cause of the coronavirus disease 2019 (COVID-19) pandemic, is a continuing global threat to human health and economies that despite increasing vaccinations has, to date, infected almost 700 million people, while its death toll is approaching seven million [1]. Tobacco smoking is the cause of another unending and hugely devastating pandemic and the leading cause of worldwide death, illness, and impoverishment [2]. Tobacco smoke is a known human carcinogen [3] and an established cause for a multitude of cardiovascular, pulmonary, metabolic, and neoplastic conditions, all vastly detrimental to human health [4]. These facts cannot be overemphasized. Mainstream tobacco smoke contains over 100,000 chemicals and more than 400 individual gaseous components, with nitrogen (58%), carbon dioxide (13%), oxygen (12%), carbon monoxide (3.5%), and hydrogen (0.5%) predominating [3]. With its content of over 4000 identified harmful chemical substances, among which over 70 carcinogens [5], fueled by the pharmacological and toxicological effects of the highly addictive alkaloid, nicotine [6], tobacco smoking will kill over half of its users and over 8 million annually worldwide [2, 7]. One-fifth of global deaths in males are attributable to smoking, and more than 16 million Americans are living with a disease caused by smoking [7]. Second-hand smoke, the smoke that fills enclosed spaces when people burn tobacco products, is also a known human carcinogen, causing serious cardiovascular and respiratory disease and more than 1.2 million premature deaths annually [2]. Sidestream smoke typically contains even higher concentrations of ammonia (40- to 170-fold), nitrogen oxides (fourfold to tenfold) and chemical carcinogens (e.g., benzene, tenfold; N-nitrosamines, sixfold to 100-fold; and aniline, 30-fold) than does mainstream smoke, additionally augmenting tobacco’s detrimental health effects [3].
Cigarette smoke (CS) impacts on a myriad of signaling pathways and immune responses of the innate and adaptive immunity [8]. CS effects are diverse and of dual nature—pro-inflammatory and immunosuppressive [8]. Age, sex, ethnic origin, socioeconomic status, and smoking pattern may underlie CS’s differential effects. Moreover, CS’s chemical heterogeneity, individual genetic susceptibility, and the variability in experimental methodologies (e.g., time, frequency, and mode of exposure) may complicate our understanding [8]. As an additional parameter, CS’s total particulate matter (TPM) concentration elicits differing effects, whereby low TPM activates xenobiotic and detoxification mechanisms while high TPM concentrations drive additional inflammatory responses [9]. Airway inflammation is promoted by a manyfold increase in neutrophil, macrophage, and dendritic cell presence, leading to the aggravation of inflammatory processes, release of oxygen species, induction of pro-inflammatory cytokine and chemokine production, and activation of proteases [8]. Further complicating the picture, CS appears to suppress monocytes’ ability to release both pro-inflammatory and immunoregulatory cytokines [10] and decreases alveolar macrophage M1-related inflammatory genes while shifting toward M2 polarization [11]. Moreover, nicotine, and other substances in CS, appears to inhibit the secretion of pro-inflammatory cytokines and chemokines, paradoxically promoting anti-inflammatory benefits [8, 12, 13]. In addition, CS suppresses local innate host defense in the airway through decreases in surfactant proteins [8]. This dysfunctional innate immune function promotes the pathogenesis of pulmonary injury induced by cigarette smoking and increases susceptibility to respiratory pathogens, asthma, allergies, and chronic obstructive pulmonary disease (COPD) [8].
Despite this incontrovertible evidence of causing significant health damage, some studies have previously suggested that tobacco smoking is paradoxically associated with a better survival after acute myocardial infarction as well as protection from preeclampsia (PE) [14,15,16,17]. A number of epidemiological studies have to date demonstrated the shortcomings of the smoker’s paradox theory, indicating a greatly increased risk of future cardiovascular events, including mortality, in current and former smokers of both sexes compared with never-smokers [18, 19]. Pharmacogenetic studies have, however, brought insight into the pathophysiological mechanisms underpinning a possible paradox [20,21,22]. Current smoking (> 0.5 pack/day) induces hepatic cytochrome P450 (CYP450) activation of CYP1A2 and CYP2B6, resulting in enhanced clopidogrel responsiveness and therapeutic benefits, while a greater risk of high platelet reactivity and thrombotic episodes has been noted after smoking cessation [20,21,22].
In line with the above observations, several reports have similarly claimed a smoker’s paradox engendering protection against severe acute respiratory coronavirus 2 (SARS-CoV-2)/coronavirus disease 2019 (COVID-19) [23, 24]. Gene–environmental interactions comparable to tobacco smoke and CYP450 operative in the cardiovascular system may be instrumentally engendering SARS-CoV-2 protection [25,26,27,28,29,30].
We conducted a PubMed literature search for publications in the English language since the start of the pandemic until June 2022, using combinations of the keywords: “smoker’s paradox” and “SARS-CoV-2” and/or “COVID-19”; “nitric oxide (NO)”; “endothelial nitric oxide (NO) synthase (eNOS)”; “renin–angiotensin–aldosterone system (RAAS)”; “angiotensin-converting enzyme 2 (ACE2)”; aryl-hydrocarbon receptor (AHR); and microRNA-155 (miR-155). We noticed a veritable dearth of publications, when “smoker’s paradox” or “paradox” and “smoking” were combined with any of the other search terms. We could not locate any publication when all search terms were combined.
In this review, we aim therefore to present known and novel pathophysiological explanations accounting for the paradoxical observation that current smokers might enjoy a serendipitous protection from contracting a SARS-CoV-2 infection [31,32,33,34]. Despite this initial paradox, once infected, both current and even more so, former smokers, are at a considerably higher risk for fatal COVID-19 complications due to smoking-related associated comorbidities [31]. The review will initially focus on NO bioavailability in the respiratory tract, the port of entry for SARS-CoV-2, and how eNOS genetic polymorphisms and tobacco smoke interactions may influence NO production and bioavailability. Novel potentially protective pathways will also be discussed, including how tobacco smoke influences CYP450, the erythropoietin receptor (EPOR), miRNA-155 (miR-155), and the aryl-hydrocarbon receptor (AHR).
Epidemiological data regarding smoking and SARS-CoV-2 infection
Several studies and systematic meta-analyses have reported a surprising but significant lower prevalence of current tobacco smoking in receiving a positive SARS-CoV-2 nasopharyngeal swab test, reporting a SARS-CoV-2 infection, and risk of hospitalization due to COVID-19 but a higher risk ratio toward adverse COVID-19 disease outcomes [24, 31,32,33, 35]. A dose–response relationship for smoking status, intensity, and duration has been described [34]. A significant, increased risk of hospitalizations, disease severity, and mortality from severe COVID-19 in former smokers compared to never-smokers and current smokers has been described and appears to be driven by the effect of age and comorbidities [33, 35, 36]. Former smokers potentially constitute an aged and burdened patient group, often forced to quit due to clinical manifestations of their smoking habit’s detrimental effects. In an intriguing study on a French aircraft carrier’s isolated environment, 71% of current smokers contracted SARS-CoV-2 vs. 80% of former and never-smokers [37]. It is thus naïve to say that smoking strongly protects smokers against COVID-19 infection, but the significantly lower prevalence of infection, especially in heavy current smokers, justifies the effort to explain the pathophysiology behind tobacco smoke’s paradoxical protection [37].
NO and SARS-CoV-2 infection
The vascular endothelium with its large surface area is the site of numerous, critical, and opposing processes where renin–angiotensin–aldosterone system’s (RAAS) central effector molecule, Angiotensin II (Ang II), regulates the expression of endothelial NO synthase (eNOS/NOS3) and NO production, whereas NO downregulates the mediator of Ang II, the Ang II type I receptor (AT1R) [38]. Ang II and NO antagonize each other in numerous vascular processes, and their mutual regulation intricately upholds normal and balanced vascular hemodynamic function and barrier integrity [38, 39]. Imbalances between vasodilation (NO) and vasoconstriction (Ang II/AT1R) impair vascular tone and disturb vascular endothelial function, predating cardiovascular and renal pathology [39, 40]. NO is fundamentally involved in maintaining this vasculoprotective balance, sustaining a normal vascular tone, and preserving a normal endothelial function [39, 40]. Moreover, bioavailable NO reduces leukocyte adhesion to the endothelium and displays important antiproliferative, antithrombotic, antioxidative, antimicrobial, and immunoregulatory properties [39]. Apart from eNOS and RAAS interactions, L-arginine substrate availability, and circulating levels of asymmetric dimethylarginine (ADMA: a natural analogue of L-arginine that acts as an endogenous NOS inhibitor) also contribute to the NO homeostasis in healthy endothelium [39, 40]. Furthermore, numerous environmental, genetic, and immunological factors interact at the endothelial level by modulating RAAS and vasculoprotective NO levels to mitigate or worsen endothelial pathology [39,40,41,42,43]. It is, thus, evident that RAAS and the NO system play very important roles in regulating cardiovascular physiology and pathology, and disturbances in their homeostasis will have severe consequences [44].
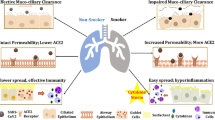
SARS-CoV-2 primarily infects numerous cell types in the respiratory tract (e.g. alveolar epithelial cells, macrophages, and endothelial cells (ECs)) [45]. The virus gains entry into the host cells via its cognate receptor, the angiotensin-converting enzyme 2 (ACE2) receptor, which is highly expressed in the respiratory epithelium [41, 46]. SARS-CoV-2 infection downregulates ACE2 and impairs eNOS activity, thereby leading to reduced NO bioavailability and endotheliitis [42]. Crucially, ACE2 occupies a prominent position within the RAAS mediating its cardioprotective, vasodilatory, and anti-inflammatory effects through Angiotensin (Ang) 1–7 and the Ang II type 2 receptor (AT2R), thus, effectively counterbalancing the vasoconstrictive and pro-inflammatory actions of ACE/Ang II/AT1R axis [47]. As ACE2 is also extensively expressed in the heart, kidney, intestine, and the vascular endothelium that virtually traverses every organ in the human body, SARS-CoV-2, thus, displays significant vascular tropism and avidly infects ECs everywhere, rendering COVID-19 a disease of the vasculature and endothelium [41, 46]. A dysregulated RAAS due to SARS-CoV-2-induced ACE2 reduction and unfettered Ang II/AT1R axis action, along with decreased NO-mediated endothelial protection, will have widespread cardiovascular implications in the form of endotheliitis, endothelial dysfunction, and vasculopathy, hyperinflammation, and cytokine storm in COVID-19 [41, 42, 48]. NO’s robust viricidal properties, as described in SARS-CoV-1/2, will be impeded when its generation and bioavailability is impaired [49, 50]. Thus, increased NO bioavailability in the airways has the mechanistic potential to inhibit SARS-CoV-1/2 infection [49]. In vitro studies during the first SARS epidemic showed that S-nitroso-N-acetylpenicillamine (SNAP), an NO donor compound, inhibited SARS-CoV-1 infection, while similar results were observed for SARS-CoV-2 with mitigation of its replication [49, 51]. It has been reported that NO mechanistically inhibits i) fusion of the nascently-expressed SARS-CoV-1/2 spike (S)-protein to ACE 2 by decreasing its palmitoylation and ii) hinders the early production of viral RNA, both processes being critical in controlling membrane fusion and virion infectivity [49, 50]. Increased NO bioavailability locally in the respiratory tract could be mediated transiently via inhaled tobacco smoke [52] and/or through tobacco smoke modulation of endogenous NO generation through eNOS induction (vide infra, Fig. 1) [53]. The magnitude of this effect will depend on smoking habit duration and intensity, as well and on eNOS polymorphism status, comorbidities, and their pharmacological treatment [53].

Angiotensin-converting enzyme (ACE) 2; angiotensin II type 1 receptor (AT1R); arginase 2 (Arg2); aryl-hydrocarbon receptor (AHR); cigarette smoke extract (CSE); cytochrome P450 1A2 (CYP1A2); endothelial nitric oxide (NO) synthase (eNOS); erythropoietin receptor (EPOR); microRNA-155 (miR-155); nitric oxide (NO); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); polycyclic aromatic hydrocarbons (PAH); and severe acute respiratory coronavirus 2 (SARS-CoV-2). Downward pointing arrows and boxes denote downregulation and inhibition. Upward pointing arrows denote upregulation
Schematic representation of tobacco smoking effects on pathophysiological pathways with potential to engender protection against SARS-CoV-2 infection.
Acute tobacco smoke inhalation effects on pulmonary NO
Tobacco smoking due to smoke inhalation is intrinsically associated with immediate and delayed effects on pulmonary physiology such as increased susceptibility to bacterial, fungal, and viral infections, chronic obstructive pulmonary disease, and neoplasia [2]. Cigarette/tobacco smoke is one of the strongest oxidants known and induces complex defense mechanisms in the respiratory tract to adapt and survive both the acute and the chronic oxidative stress imposed by habitual smoking. Chambers et al. investigated the concentration of NO in the lower respiratory tract (LRT) of smokers and reported consistent NO increases at 1 and 10 min after smoking a cigarette, probably due to trapping in the epithelial lining fluid (ELF) [52]. Both existing and exogenously added NO in the LRT will more avidly react in the ELF with oxygen and thiols to form peroxynitrite and S-nitrosothiols, respectively, before being taken up by the pulmonary capillaries [52]. Moreover, other researchers have reported that inhaled NO is not systemically absorbed from the inhaled tobacco smoke but indeed stays entrapped in the ELF of the LRT contributing to the increase in NO bioequivalent forms [54]. Those NO bioequivalent forms are much less reactive than NO but still serve as sources of NO, retain NO-like biological activity, and are similarly microbicidal and bioavailable [39]. NO bioequivalent forms in the respiratory tract epithelium formed after tobacco smoke inhalation could thus serendipitously shield the host from SARS-CoV-2 particles in aerosol droplets (vide supra), possibly engendering protection from infection [54]. It is unclear whether differences in NO yields of different tobacco products are of importance [55]. Moreover, smokers tend to either smoke in groups in designated enclosed smoking areas, where sidestream and second-hand smoke is also inhaled, or alone outdoors indirectly enforcing social distancing [37, 56,57,58]. The former practice might lead to additional increases in inhaled NO, while the latter may reduce SARS-CoV-2 exposure.
The above process may be a highly efficient mechanism by which NOs modulate their biological effects through more stable bioequivalents in the acute setting. Furthermore, it is believed that the magnitude of the change in the LRT NO concentration following acute oxidant stress (between cigarettes) reflects the pulmonary antioxidant capacity to restore normal pulmonary homeostasis [52].
Canonical NO generation via the eNOS: genetic polymorphism effects
With chronic tobacco smoke exposure in habitual healthy smokers other mechanisms involving upregulation and induction of numerous antioxidant enzymes, NO-generating pathways and detoxification systems are called into play to withstand the continuous and chronic oxidant challenge smoking represents. A critically important cellular response to tobacco smoke is through its effect on the eNOS enzyme.
Three distinct NO synthases (NOS) encoded by their respective genes, neuronal NOS (NOS1 or nNOS), inducible NOS (NOS2 or iNOS), and endothelial NOS (NOS3 or eNOS: major isoform regulating vascular function) catalyze the conversion of L-arginine to citrulline and produce NO [39]. For the NOS enzyme to be functional, enzyme dimerization, in the presence of various co-factors including flavin adenine dinucleotide, flavin mononucleotide, calmodulin, tetrahydrobiopterin (BH4), zinc, and iron protoporphyrin IX (heme), is required [40]. Tobacco smoke can disturb some of these co-factors resulting in eNOS uncoupling and reduced NO generation in the long term [59].
An additional layer of complexity in NO generation is added by the presence of NOS3 genetic polymorphisms [60]. Commonly researched eNOS polymorphisms include rs1799983 (G894-T, Glu298Asp), rs2070744 in the promoter region (T786-C), and rs61722009 with a rare 4 × 27 bp repeat and a common 5 × 27 bp repeat allele in intron 4 (VNTR4a/4b) (Table 1) [43, 60].
Genotype-based simulations have indicated that the combined effect of NOS3 genetic polymorphisms contribute to a 30.5% variability in NO production [61]. Haplotype-based association studies more appropriately evaluate inter-locus interaction effects present in in vivo human phenotypic expression, demonstrating up to fourfold NO generation variability, are modifiable by ethnicity, pharmacological interventions, and additionally influenced by cigarette smoke (CS) [60]. eNOS-mediated NO effects through induction of gene expression would be requiring a longer time frame as maximal induction takes hours, and necessitate habitual tobacco smoking [52].
Delayed pleiotropic tobacco smoke effects on eNOS: the role of genetic polymorphisms
Functional in vitro assays of different NOS3 genetic polymorphisms have revealed differential effects in eNOS transcription efficiency both as single alleles and as haplotypes [53]. Those variations were further increased when cigarette smoke extract (CSE) was added to the culture media of the single rs2070744 T- and C-alleles [53]. CSE doubled the transcription efficiency of the rs2070744 T-allele versus itself without CSE and versus the C-allele [53]. Furthermore, CSE significantly increased eNOS promoter rs2070744 T-allele transcription efficiency, especially in the presence of the rare rs61722009 4 × 27 bp repeats [53]. A marginal, non-significant decrease was observed when the rs2070744 C-allele was combined with either 4 × or 5 × 27 bp repeats in the presence of CSE [53]. It appears thus that tobacco smoke may affect the functionality of the eNOS promoter and stimulate increased transcription efficiency possibly leading to increases in NO generation, at least when the eNOS has not yet been uncoupled or otherwise adversely affected by a smoking habit of long duration. If we extrapolate Wang et al. in vitro findings into in vivo human phenotypic expression, a non-smoking rs2070744 T/rs61722009 5 × 27 bp homozygote would have between 2 and 5 times lower eNOS levels compared to any other non-smoking or smoking homozygotes, respectively [53]. The greatest return of tobacco smoke effect with doubling of eNOS levels was observed with the haplotype combination rs2070744 T-allele/rs61722009 4 × 27 bp repeats. However, eNOS transcription efficiency and eNOS protein levels do not always lead to an expected eNOS activity increase as demonstrated in an earlier study by Wang et al. [62]. While they could not identify any interaction with the tobacco smoke and rs1799983 or rs2070744, they found that smoking modestly increased eNOS enzyme activities in the 5/5 × 27bps homozygotes and downregulated them in the rare 4 × 27 bp repeats. It remains to be explained how protein levels and enzyme activities are related [62]. Additional studies have demonstrated significant combined effects of smoking, drinking, and the rs1799983 (T-allele) polymorphism of the NOS3 gene on blood pressure in Chinese male hypertensive subjects [29]. These observations, albeit contradictory at times, provide important evidence that tobacco smoking may significantly modify molecular and genetic mechanisms to generate pleiotropic biochemical effects that potentially impact on the risk for SARS-CoV-2 infection in smokers. Haplotype-based association studies and eNOS enzyme activity measurements will be needed to confirm possible associations.
Alternative (non-canonical) NO generation pathways relevant to tobacco smoke
The endothelium-dependent, CYP450-mediated NO release from nitrate may serve as a compensatory mechanism to restore NO availability during endothelial dysfunction when the canonical eNOS pathway is compromised [39]. The CYP1A2 comprises 10% of the liver CYP450 [20]. The CYP1A2 (− 163C > A) A allele (AA and AC genotypes) is prone to induction through tobacco smoke, resulting in higher CYP1A2 metabolic activity and is responsible for increased clopidogrel responsiveness, thus accounting for a smoker’s paradox in certain smokers [20]. It is conceivable that increased NO generation and bioavailability through this non-canonical pathway could contribute to and enhance an NO-mediated SARS-CoV-2 protection (Fig. 1). As approximately 80% of the world population carries the AA + AC genotypes, it is expected that a great majority of individuals would be susceptible to the effect of smoking [20].
Erythropoietin (EPO) and its receptor (EPOR) in smokers
Tobacco smoke’s carbon monoxide hypoxia-inducing capacity is well-known and despite lower EPO levels compared to non-smokers, current smokers exhibit secondary polycythemia due to increased expression of the EPOR [63, 64]. Together with the β-common receptor (βcR), the EPOR is also an integral part of the tissue protective receptor (TPR) through which EPO exerts its non-hematopoietic tissue protective actions via eNOS [65]. Increased shearing stress (known to activate eNOS [39]) on the endothelium due to increased blood viscosity in secondary polycythemia along with EPOR-mediated eNOS activation might result in increased SARS-CoV-2 protective NO generation and bioavailability (Fig. 1).
The importance of the EPOR-mediated eNOS activation is exemplified in schizophrenia (SCZ) where up to 75% of patients are smokers and smoke more cigarettes per day than do smokers in the general population [66]. However, a fourfold increase in COVID-19 mortality has been reported in SCZ, thus contradicting a smoker’s paradox [67]. We have postulated that this great increase in COVID-19 mortality may be due to β-cytokine and βcR polymorphisms, previously described in SCZ [67]. Perturbations in the βcR would impair and negate EPO/EPOR’s neuro-, cardio-, reno-, and potentially SARS-CoV-2-protective effects through the TPR. This is because the βcR and the EPOR are integral parts of the TPR and essential in the formation of an active βcR-EPOR-eNOS complex needed to result in protective increased NO generation and bioavailability [65].
microRNA-155
microRNAs (miRNAs) are 18–25 nucleotide long, small, non-coding one-stranded RNA molecules that can target and silence around 60% of all human genes through translational repression [68]. miRNA-155 (miR-155) is an ancient, evolutionarily well-conserved miRNA and a key modulator of both innate and adaptive immune responses, with critical roles in viral and parasitic infections [69]. miR-155 targets over 140 genes involved in numerous physiological and pathological processes including hematopoietic lineage differentiation, immunity, inflammation, cancer, cardiovascular diseases, diabetes, and particularly viral infections [69]. Increased expression of miR-155 in the lungs of smokers without airflow limitation and patients with mild-to-moderate COPD as well as in the lungs of CS-exposed mice has been reported [70]. Interestingly, miR-155 targets the AGTR1 gene that encodes the AT1R, the key pro-inflammatory receptor in the RAAS, whereby its repression can mitigate Ang II’s pro-inflammatory actions and its cytokine storm causing potential (Fig. 1) [69, 71]. The carriers of the AT1R + 1166C-allele of the rs5186 polymorphism—that is unresponsive to miR-155’s AT1R repressive effects—demonstrate increased oxygen dependency and severity of COVID-19 compared to the A allele carriers [72]. Importantly, SARS-CoV-2-spike 1 protein (S1)-ACE2 complex is internalized through an AT1R-dependent endocytosis, thus reduced AT1R membrane presence through miR-155-induced AT1R repression could theoretically directly inhibit further SARS-CoV-2 cell entry [73]! Elevated basal miR-155 levels in smokers and COPD potentially create a more advantageous environment at the time of the initiation of SARS-CoV-2 infection, in theory creating a SARS-CoV-2 cell entry barrier. Moreover, arginase 2 (Arg2), another direct target for miR-155, when repressed prevents the depletion of L-arginine, the obligate substrate of eNOS, leading to improved substrate availability and additional increases in NO production and NO bioavailability, further aiding the above-mentioned NO antiviral actions [74].
Aryl‑hydrocarbon receptor (AHR) signal mediated ACE2 expression modulation
Among the multitude of gases and chemical compounds in cigarette smoke are polycyclic aromatic hydrocarbons (PAHs) that can bind to and activate the AHR (Fig. 1) [17, 75]. PAHs may also explain the paradoxical effects of cigarette use on PE through AHR-dependent immunosuppressive effects on the mother and the placenta [17]. Furthermore, PAH-induced AHR activation significantly reduces ACE2 expression (both at the mRNA and protein level) in vitro in numerous CSE-treated cell lines and COPD, resulting in suppression of internalization and decreased replication of SARS-CoV-2 [75,76,77]. Concurrent increase in CYP1A1, a well-known AHR target gene was noted after treatment with CSE and AHR agonists confirming AHR activation [75]. Other researchers have demonstrated contradictory findings resulting from the observed AHR upregulation in SARS-CoV-2 infection [78, 79]. SARS-CoV-2 appears to employ AHR induction as a means to evade host immune response and contribute to the lung pathogenesis of COVID-19, possibly through upregulation of ACE2 expression [78, 79]. Clearly, more studies are needed to elucidate AHR’s role in SARS-CoV-2. Finally, interactions between AHR and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a known regulator of miR-155 expression, may also mediate associations to miR-155 and hint to the possible involvement of NF-κB/miR-155 axis in SARS-CoV-2 infection and COVID-19 immunopathology (Fig. 1) [78, 80].
Conclusion
Our review highlights known environmental (NO bioavailability in the LRT) and novel genetic mechanisms (tobacco smoke effects on eNOS and CYP450 polymorphisms, EPOR activation, miR-155 and AHR) that may independently and/or jointly contribute to a transient and serendipitous SARS-CoV-2 protection (Fig. 1). Despite an eNOS induction in genetically predisposed smokers, and initially increased NO bioavailability, chronic tobacco use will ultimately lead to eNOS uncoupling [59]. Uncoupled eNOS markedly contributes to oxidative stress in vascular tissue through generation of reactive oxygen species instead of protective NO [39, 40, 59]. In addition, miR-155’s pleiotropic effects, potentially transiently protective against SARS-CoV-2, will ultimately turn detrimental as its effect in COPD is pro-inflammatory and promotes emphysematous changes [70]. The net effects on NO bioavailability and the immune system through the above described acute and delayed mechanisms are short-lived and will gradually become irrelevant, as smoking, being the most potent common oxidant challenge encountered by the human respiratory tract, will always damage the arterial endothelium and wall, and invariably result in dismal cardiovascular, hematological, and respiratory pathologies, ultimately predisposing for a lethal COVID-19 disease course [64, 81].
Haplotype-based eNOS genetic studies and eNOS activity measurements would be required to elucidate the effect of smoking in SARS-CoV-2 infected patients. In addition, genetic polymorphism studies of relevant RAAS molecules and CYP450 would be necessary as those genetic variants also significantly modify eNOS effects, pharmacological interventions in smoking-related comorbidities, and ultimately COVID-19 disease prognosis [47]. AHR and miR-155 studies are clearly needed to further elucidate their interactions. Innovative SARS-CoV-2 treatments involving AHR agonism and miRNA modulation could also be developed.
In the end, there’s no free lunch. Life choices always matter, and tobacco smoking remains the leading cause of death, illness, and impoverishment. Responsibility must be accepted for mistreating our endothelial cells with tobacco smoke, high sugar, high lipids, high blood pressure, and a sedentary lifestyle devoid of physical exercise [82]. While NO bioavailability increase and beneficial immunoregulatory modulations that arise through exogenous, endogenous, genetic, and/or therapeutic modalities may have direct and specific SARS-CoV-2 viricidal effects, NO supply from tobacco smoke inhalation equals self-harm [83]. A serendipitous paradoxical SARS-CoV-2 avoidance of infection in smokers will ultimately set the stage for future lethal pulmonary, cardiovascular, and neoplastic sequelae. The smoker’s paradox is in believing that smokers can avoid tobaccos’ lethality.
Data availability
All data analysed during this narrative review are included in this published article.
References
Worldodometer. https://www.worldometers.info/coronavirus/. Accessed 2023-01-31 2022
Tobacco. World Health Organization. 2021. https://www.who.int/news-room/fact-sheets/detail/tobacco. Accessed 2023-01-31 2022
15th Report on Carcinogens. National Toxicology Program. 2021. https://ntp.niehs.nih.gov/go/roc15. Accessed 2023-01-31 2022
Centers for Disease C, Prevention, National Center for Chronic Disease P, Health P, Office on S, Health. Publications and Reports of the Surgeon General. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Atlanta (GA): Centers for Disease Control and Prevention (US); 2010.
Zhou G (2019) Tobacco, air pollution, environmental carcinogenesis, and thoughts on conquering strategies of lung cancer. Cancer Biol Med 16(4):700–713. https://doi.org/10.20892/j.issn.2095-3941.2019.0180
Le Foll B, Piper ME, Fowler CD, Tonstad S, Bierut L, Lu L et al (2022) Tobacco and nicotine use. Nat Rev Dis Primers 8(1):19. https://doi.org/10.1038/s41572-022-00346-w
Smoking & Tobacco Use: Fast Facts. Center for Disease Control and Prevention. 2021. https://www.cdc.gov/tobacco/data_statistics/fact_sheets/fast_facts/index.htm#diseases. Accessed 2023-01-31 2022
Strzelak A, Ratajczak A, Adamiec A, Feleszko W (2018) Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: a mechanistic review. Int J Environ Res Public Health. https://doi.org/10.3390/ijerph15051033
Dvorkin-Gheva A, Vanderstocken G, Yildirim A, Brandsma CA, Obeidat M, Bossé Y et al (2016) Total particulate matter concentration skews cigarette smoke’s gene expression profile. ERJ Open Res. https://doi.org/10.1183/23120541.00029-2016
da Silva CO, Gicquel T, Daniel Y, Bártholo T, Vène E, Loyer P et al (2020) Alteration of immunophenotype of human macrophages and monocytes after exposure to cigarette smoke. Sci Rep 10(1):12796. https://doi.org/10.1038/s41598-020-68753-1
Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O’Connor TP et al (2009) Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 183(4):2867–2883. https://doi.org/10.4049/jimmunol.0900473
Valdez-Miramontes CE, Trejo Martínez LA, Torres-Juárez F, Rodríguez Carlos A, Marin-Luévano SP, de Haro-Acosta JP et al (2020) Nicotine modulates molecules of the innate immune response in epithelial cells and macrophages during infection with M. tuberculosis. Clin Exp Immunol 199(2):230–43. https://doi.org/10.1111/cei.13388
Mahmoudzadeh L, Abtahi Froushani SM, Ajami M, Mahmoudzadeh M (2023) Effect of nicotine on immune system function. Adv Pharm Bull 13(1):69–78. https://doi.org/10.34172/apb.2023.008
Bundhun PK, Wu ZJ, Chen MH (2015) Impact of modifiable cardiovascular risk factors on mortality after percutaneous coronary intervention: a systematic review and meta-analysis of 100 studies. Medicine 94(50):e2313. https://doi.org/10.1097/md.0000000000002313
Chen KY, Rha SW, Li YJ, Jin Z, Minami Y, Park JY et al (2012) “Smoker’s paradox” in young patients with acute myocardial infarction. Clin Exp Pharmacol Physiol 39(7):630–635. https://doi.org/10.1111/j.1440-1681.2012.05721.x
Ekblad MO, Gissler M, Korhonen PE (2022) New theory about the pathophysiology of preeclampsia derived from the paradox of positive effects of maternal smoking. J Hypertens 40(6):1223–1230. https://doi.org/10.1097/hjh.0000000000003142
Holme JA, Valen H, Brinchmann BC, Vist GE, Grimsrud TK, Becher R et al (2022) Polycyclic aromatic hydrocarbons (PAHs) may explain the paradoxical effects of cigarette use on preeclampsia (PE). Toxicology. 473:153206. https://doi.org/10.1016/j.tox.2022.153206
Bouabdallaoui N, Messas N, Greenlaw N, Ferrari R, Ford I, Fox KM et al (2021) Impact of smoking on cardiovascular outcomes in patients with stable coronary artery disease. Eur J Prev Cardiol 28(13):1460–1466. https://doi.org/10.1177/2047487320918728
Mohammadi SS, Zibaeenezhad MJ, Sayadi M, Khorshidi S, Hadiyan E, Razeghian-Jahromi I (2021) The impact of smoking on clinical outcomes after percutaneous coronary intervention in women compared to men. J Interv Cardiol 2021:6619503. https://doi.org/10.1155/2021/6619503
Park KW, Park JJ, Jeon K-H, Kang S-H, Oh I-Y, Yang H-M et al (2011) Enhanced clopidogrel responsiveness in smokers. Arterioscler Thromb Vasc Biol 31(3):665–671. https://doi.org/10.1161/ATVBAHA.110.217182
Ramotowski B, Gurbel PA, Tantry U, Budaj A (2019) Smoking and cardiovascular diseases: paradox greater than expected? Pol Arch Intern Med 129(10):700–706. https://doi.org/10.20452/pamw.14931
Gurbel PA, Bliden KP, Logan DK, Kereiakes DJ, Lasseter KC, White A et al (2013) The Influence of smoking status on the pharmacokinetics and pharmacodynamics of clopidogrel and prasugrel: the PARADOX study. J Am Coll Cardiol 62(6):505–512. https://doi.org/10.1016/j.jacc.2013.03.037
Usman MS, Siddiqi TJ, Khan MS, Patel UK, Shahid I, Ahmed J et al (2021) Is there a smoker’s paradox in COVID-19? BMJ Evid Based Med 26(6):279–284. https://doi.org/10.1136/bmjebm-2020-111492
Meini S, Fortini A, Andreini R, Sechi LA, Tascini C (2021) The paradox of the low prevalence of current smokers among COVID-19 patients hospitalized in nonintensive care wards: results from an italian multicenter case-control study. Nicotine Tob Res 23(8):1436–1440. https://doi.org/10.1093/ntr/ntaa188
Ljungberg L, Alehagen U, Länne T, Björck H, De Basso R, Dahlström U et al (2011) The association between circulating angiotensin-converting enzyme and cardiovascular risk in the elderly: a cross-sectional study. J Renin Angiotensin Aldosterone Syst 12(3):281–289. https://doi.org/10.1177/1470320310391326
Laustiola KE, Lassila R, Nurmi AK (1988) Enhanced activation of the renin-angiotensin-aldosterone system in chronic cigarette smokers: a study of monozygotic twin pairs discordant for smoking. Clin Pharmacol Ther 44(4):426–430. https://doi.org/10.1038/clpt.1988.175
Nadalin S, Ristic S, Rebic J, Sendula Jengic V, Kapovic M, Buretic-Tomljanovic A (2017) The insertion/deletion polymorphism in the angiotensin-converting enzyme gene and nicotine dependence in schizophrenia patients. J Neural Transm 124(4):511–518. https://doi.org/10.1007/s00702-016-1670-y
Nadalin S, Flego V, Pavlić SD, Volarić D, Radojčić Badovinac A, Kapović M et al (2020) Association between the ACE-I/D polymorphism and nicotine dependence amongst patients with lung cancer. Biomed Rep 13(6):58. https://doi.org/10.3892/br.2020.1365
Hong Z, Pan L, Ma Z, Zhu Y, Hong Z (2019) Combined effects of cigarette smoking, alcohol drinking and eNOS Glu298Asp polymorphism on blood pressure in Chinese male hypertensive subjects. Tob Induc Dis 17:59. https://doi.org/10.18332/tid/110678
Cheon KT, Choi KH, Lee HB, Park SK, Rhee YK, Lee YC (2000) Gene polymorphisms of endothelial nitric oxide synthase and angiotensin-converting enzyme in patients with lung cancer. Lung 178(6):351–360. https://doi.org/10.1007/s004080000039
Farsalinos K, Barbouni A, Poulas K, Polosa R, Caponnetto P, Niaura R (2020) Current smoking, former smoking, and adverse outcome among hospitalized COVID-19 patients: a systematic review and meta-analysis. Ther Adv Chronic Dis 11:2040622320935765. https://doi.org/10.1177/2040622320935765
Vardavas CI, Nikitara K (2020) COVID-19 and smoking: a systematic review of the evidence. Tob Induc Dis 18:20. https://doi.org/10.18332/tid/119324
Puebla Neira D, Watts A, Seashore J, Polychronopoulou E, Kuo Y-F, Sharma G (2021) Smoking and risk of COVID-19 hospitalization. Respir Med 182:106414. https://doi.org/10.1016/j.rmed.2021.106414
Prinelli F, Bianchi F, Drago G, Ruggieri S, Sojic A, Jesuthasan N et al (2021) Association between smoking and SARS-CoV-2 infection cross-sectional study of the EPICOVID19 internet-based survey. JMIR Public Health Surveill 7(4):e27091-e. https://doi.org/10.2196/27091
Arbel Y, Fialkoff C, Kerner A, Kerner M (2022) Can smoking prevalence explain COVID-19 indicators (cases, mortality, and recovery)? A comparative study in OECD countries. Environ Sci Pollut Res. https://doi.org/10.1007/s11356-022-21240-8
Simons D, Shahab L, Brown J, Perski O (2021) The association of smoking status with SARS-CoV-2 infection, hospitalization and mortality from COVID-19: a living rapid evidence review with Bayesian meta-analyses (version 7). Addiction 116(6):1319–1368. https://doi.org/10.1111/add.15276
Paleiron N, Mayet A, Marbac V, Perisse A, Barazzutti H, Brocq F-X et al (2021) Impact of tobacco smoking on the risk of COVID-19: a large scale retrospective cohort study. Nicotine Tob Res 23(8):1398–1404. https://doi.org/10.1093/ntr/ntab004
Yan C, Kim D, Aizawa T, Berk BC (2003) Functional interplay between angiotensin II and nitric oxide: cyclic GMP as a key mediator. Arterioscler Thromb Vasc Biol 23(1):26–36. https://doi.org/10.1161/01.atv.0000046231.17365.9d
Zhao Y, Vanhoutte PM, Leung SWS (2015) Vascular nitric oxide: beyond eNOS. J Pharmacol Sci 129(2):83–94. https://doi.org/10.1016/j.jphs.2015.09.002
Förstermann U, Münzel T (2006) Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113(13):1708–1714. https://doi.org/10.1161/circulationaha.105.602532
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395(10234):1417–1418. https://doi.org/10.1016/s0140-6736(20)30937-5
Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H et al (2021) SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res 128(9):1323–1326. https://doi.org/10.1161/CIRCRESAHA.121.318902
Cotta Filho CK, Oliveira-Paula GH, Rondon Pereira VC, Lacchini R (2020) Clinically relevant endothelial nitric oxide synthase polymorphisms and their impact on drug response. Expert Opin Drug Metab Toxicol 16(10):927–951. https://doi.org/10.1080/17425255.2020.1804857
Stegbauer J, Kuczka Y, Vonend O, Quack I, Sellin L, Patzak A et al (2008) Endothelial nitric oxide synthase is predominantly involved in angiotensin II modulation of renal vascular resistance and norepinephrine release. Am J Physiol Regul Integr Comp Physiol 294(2):R421–R428. https://doi.org/10.1152/ajpregu.00481.2007
Kobayashi J (2021) Lifestyle-mediated nitric oxide boost to prevent SARS-CoV-2 infection: a perspective. Nitric Oxide 115:55–61. https://doi.org/10.1016/j.niox.2021.08.001
Turner AJ, Nalivaeva NN (2022) Angiotensin-converting enzyme 2 (ACE2): two decades of revelations and re-evaluation. Peptides 151:170766. https://doi.org/10.1016/j.peptides.2022.170766
Papadopoulos KI, Papadopoulou A, Aw TC (2023) A protective erythropoietin evolutionary landscape, NLRP3 inflammasome regulation, and multisystem inflammatory syndrome in children. Hum Cell 36(1):26–40. https://doi.org/10.1007/s13577-022-00819-w
Teuwen LA, Geldhof V, Pasut A, Carmeliet P (2020) COVID-19: the vasculature unleashed. Nat Rev Immunol 20(7):389–391. https://doi.org/10.1038/s41577-020-0343-0
Akaberi D, Krambrich J, Ling J, Luni C, Hedenstierna G, Jarhult JD et al (2020) Mitigation of the replication of SARS-CoV-2 by nitric oxide in vitro. Redox Biol 37:101734. https://doi.org/10.1016/j.redox.2020.101734
Akerstrom S, Gunalan V, Keng CT, Tan YJ, Mirazimi A (2009) Dual effect of nitric oxide on SARS-CoV replication: viral RNA production and palmitoylation of the S protein are affected. Virology 395(1):1–9. https://doi.org/10.1016/j.virol.2009.09.007
Keyaerts E, Vijgen L, Chen L, Maes P, Hedenstierna G, Van Ranst M (2004) Inhibition of SARS-coronavirus infection in vitro by S-nitroso-N-acetylpenicillamine, a nitric oxide donor compound. Int J Infect Dis 8(4):223–226. https://doi.org/10.1016/j.ijid.2004.04.012
Chambers DC, Tunnicliffe WS, Ayres JG (1998) Acute inhalation of cigarette smoke increases lower respiratory tract nitric oxide concentrations. Thorax 53(8):677–679. https://doi.org/10.1136/thx.53.8.677
Wang J, Dudley D, Wang XL (2002) Haplotype-specific effects on endothelial NO synthase promoter efficiency: modifiable by cigarette smoking. Arterioscler Thromb Vasc Biol 22(5):e1-4. https://doi.org/10.1161/01.atv.0000016248.51577.1f
Rångemark C, Wennmalm A (1996) Smoke-derived nitric oxide and vascular prostacyclin are unable to counteract the platelet effect of increased thromboxane formation in healthy female smokers. Clin Physiol 16(3):301–315. https://doi.org/10.1111/j.1475-097x.1996.tb00576.x
Borland C, Higenbottam T (1987) Nitric oxide yields of contemporary UK, US and French cigarettes. Int J Epidemiol 16(1):31–34. https://doi.org/10.1093/ije/16.1.31
Zhang Y, Wang J, Lai K, Bian H, Chen H, Gao L (2022) Socializing with smoker and social smoking behavior among Chinese Male smokers with low nicotine dependence: the mediating roles of belief of smoking rationalization and smoker identity. Int J Environ Res Public Health 19(22):14765
Sureda X, Fernández E, Martínez-Sánchez JM, Fu M, López MJ, Martínez C et al (2015) Secondhand smoke in outdoor settings: smokers’ consumption, non-smokers’ perceptions, and attitudes towards smoke-free legislation in Spain. BMJ Open 5(4):e007554. https://doi.org/10.1136/bmjopen-2014-007554
Ko H (2020) The effect of outdoor smoking ban: evidence from Korea. Health Econ 29(3):278–293. https://doi.org/10.1002/hec.3979
Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S et al (2000) Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : evidence for a dysfunctional nitric oxide synthase. Circ Res 86(2):E36-41. https://doi.org/10.1161/01.res.86.2.e36
Oliveira-Paula GH, Lacchini R, Tanus-Santos JE (2016) Endothelial nitric oxide synthase: from biochemistry and gene structure to clinical implications of NOS3 polymorphisms. Gene 575(2 Pt 3):584–599. https://doi.org/10.1016/j.gene.2015.09.061
Katkam SK, Indumathi B, Tasneem FSD, Rajasekhar L, Kutala VK (2018) Impact of eNOS 27-bp VNTR (4b/a) gene polymorphism with the risk of Systemic Lupus Erythematosus in south Indian subjects. Gene 658:105–112. https://doi.org/10.1016/j.gene.2018.03.021
Wang XL, Sim AS, Wang MX, Murrell GA, Trudinger B, Wang J (2000) Genotype dependent and cigarette specific effects on endothelial nitric oxide synthase gene expression and enzyme activity. FEBS Lett 471(1):45–50. https://doi.org/10.1016/s0014-5793(00)01356-9
Eisenga MF, Kieneker LM, Touw DJ, Nolte IM, van der Meer P, Huls G et al (2018) Active smoking and hematocrit and fasting circulating erythropoietin concentrations in the general population. Mayo Clin Proc 93(3):337–343. https://doi.org/10.1016/j.mayocp.2018.01.005
Alkhedaide AQ (2020) Tobacco smoking causes secondary polycythemia and a mild leukocytosis among heavy smokers in Taif City in Saudi Arabia. Saudi J Biol Sci 27(1):407–411. https://doi.org/10.1016/j.sjbs.2019.11.001
Su KH, Shyue SK, Kou YR, Ching LC, Chiang AN, Yu YB et al (2011) beta Common receptor integrates the erythropoietin signaling in activation of endothelial nitric oxide synthase. J Cell Physiol 226(12):3330–3339. https://doi.org/10.1002/jcp.22678
Henderson DC, Vincenzi B, Andrea NV, Ulloa M, Copeland PM (2015) Pathophysiological mechanisms of increased cardiometabolic risk in people with schizophrenia and other severe mental illnesses. Lancet Psychiatry 2(5):452–464. https://doi.org/10.1016/s2215-0366(15)00115-7
Papadopoulos KI, Sutheesophon W, Aw T-C (2022) Genetic polymorphisms affecting nitric oxide and β-cytokine pathways may contribute to increased COVID-19 mortality in schizophrenia. Asian J Psychiatry 69:102981. https://doi.org/10.1016/j.ajp.2021.102981
Papadopoulos KI, Wattanaarsakit P, Prasongchean W, Narain R (2016) Gene therapies in clinical trials. In: Narain R (ed) Polymers and nanomaterials for gene therapy. Woodhead Publishing, Sawston, pp 231–256
Faraoni I, Antonetti FR, Cardone J, Bonmassar E (2009) miR-155 gene: a typical multifunctional microRNA. Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease 1792(6):497–505. https://doi.org/10.1016/j.bbadis.2009.02.013
De Smet EG, Van Eeckhoutte HP, Avila Cobos F, Blomme E, Verhamme FM, Provoost S et al (2020) The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol 13(3):423–436. https://doi.org/10.1038/s41385-019-0241-6
Wu Z, Hu R, Zhang C, Ren W, Yu A, Zhou X (2020) Elevation of plasma angiotensin II level is a potential pathogenesis for the critically ill COVID-19 patients. Crit Care 24(1):290. https://doi.org/10.1186/s13054-020-03015-0
Izmailova O, Shlykova O, Vatsenko A, Ivashchenko D, Dudchenko M, Koval T et al (2022) Allele C (rs5186) of at1r is associated with the severity of COVID-19 in the Ukrainian population. Infect Genet Evol 98:105227. https://doi.org/10.1016/j.meegid.2022.105227
Ogunlade BO, Lazartigues E, Filipeanu CM (2021) Angiotensin Type 1 receptor-dependent internalization of SARS-CoV-2 by angiotensin-converting enzyme 2. Hypertension 77(4):e42–e43. https://doi.org/10.1161/HYPERTENSIONAHA.120.16795
Dunand-Sauthier I, Irla M, Carnesecchi S, Seguín-Estévez Q, Vejnar CE, Zdobnov EM et al (2014) Repression of arginase-2 expression in dendritic cells by microRNA-155 is critical for promoting T cell proliferation. J Immunol 193(4):1690–1700. https://doi.org/10.4049/jimmunol.1301913
Tanimoto K, Hirota K, Fukazawa T, Matsuo Y, Nomura T, Tanuza N et al (2021) Inhibiting SARS-CoV-2 infection in vitro by suppressing its receptor, angiotensin-converting enzyme 2, via aryl-hydrocarbon receptor signal. Sci Rep 11(1):16629. https://doi.org/10.1038/s41598-021-96109-w
Tomchaney M, Contoli M, Mayo J, Baraldo S, Li S, Cabel CR et al (2021) Paradoxical effects of cigarette smoke and COPD on SARS-CoV-2 infection and disease. BMC Pulm Med 21(1):275. https://doi.org/10.1186/s12890-021-01639-8
Wang G, Zhao Q, Zhang H, Liang F, Zhang C, Wang J et al (2021) Degradation of SARS-CoV-2 receptor ACE2 by the E3 ubiquitin ligase Skp2 in lung epithelial cells. Front Med 15(2):252–263. https://doi.org/10.1007/s11684-021-0837-6
Giovannoni F, Li Z, Remes-Lenicov F, Dávola ME, Elizalde M, Paletta A et al (2021) AHR signaling is induced by infection with coronaviruses. Nat Commun 12(1):5148. https://doi.org/10.1038/s41467-021-25412-x
Almeida-da-Silva CLC, Matshik Dakafay H, Liu K, Ojcius DM (2021) Cigarette smoke stimulates SARS-CoV-2 internalization by activating AhR and increasing ACE2 expression in human gingival epithelial cells. Int J Mol Sci. https://doi.org/10.3390/ijms22147669
Khokhar M, Tomo S, Purohit P (2022) MicroRNAs based regulation of cytokine regulating immune expressed genes and their transcription factors in COVID-19. Meta Gene 31:100990. https://doi.org/10.1016/j.mgene.2021.100990
Michael PR (2000) Cigarette smoking, endothelial injury and cardiovascular disease. Int J Exp Pathol 81(4):219–230. https://doi.org/10.1046/j.1365-2613.2000.00162.x
Papadopoulos KI, Sutheesophon W, Aw T-C (2022) Too hard to die: Exercise training mediates specific and immediate SARS-CoV-2 protection. World J Virol. https://doi.org/10.5501/wjv.v11.i2.0000
Redaelli S, Magliocca A, Malhotra R, Ristagno G, Citerio G, Bellani G et al (2022) Nitric oxide: clinical applications in critically ill patients. Nitric Oxide 121:20–33. https://doi.org/10.1016/j.niox.2022.01.007
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
KIP conceived and conceptualized the pathophysiology and designed the review, drafted the initial manuscript, and reviewed and revised the manuscript. AP performed the literature search, extracted vital information, contributed to the synthesis of the review, and reviewed and revised the manuscript. TCA coordinated and supervised literature search, made substantial and direct intellectual contributions, and critically reviewed the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Papadopoulos, K.I., Papadopoulou, A. & Aw, T.C. Live to die another day: novel insights may explain the pathophysiology behind smoker’s paradox in SARS-CoV-2 infection. Mol Cell Biochem 478, 2517–2526 (2023). https://doi.org/10.1007/s11010-023-04681-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-023-04681-8