
Abstract
The lysosomal endoprotease legumain (asparaginyl endoprotease) has been proposed as a putative biomarker in prostate tumours, in which the enzyme is markedly overexpressed. Overexpression, coupled with highly selective specificity for cleavage of substrates at the C-terminus of asparagine (Asn) residues, make legumain an attractive biochemical target for potential diagnosis, prognosis and treatment. We report the design, synthesis, characterisation and preliminary evaluation of a new rhodamine-B (Rho-B)-labelled legumain peptide substrate probe 5 [Rho-Pro-Ala-Asn-PEG-AQ(4-OH)] and its selective targeting to lysosomes in PC3 prostate cancer cells. Probe 5 was efficiently activated by recombinant human legumain to afford the high quantum yield reporter fluorophore tripeptide 4b (Rho-Pro-Ala-Asn-OH) with concomitant release of intense fluorescence. Furthermore, probe 5 was activated upon incubation with homogenates derived from fresh-frozen tissue material of prostatectomy specimens. Probe 5 represents a new viable biochemical tool for probing the activity of legumain with the potential to be used in ex vivo diagnostics in the cancer pathology laboratory.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer (Pca) is one of the most prevalent cancers and its incidence is increasing in Western society (Siegel et al. 2016). The early diagnosis of pre-invasive and metastatic tumour cells in patients is pivotal for the judicious choice of treatment and success of therapies and for the improvement of survival rates. Early diagnosis of Pca is usually reliant on techniques that include digital rectal examination, magnetic resonance imaging and transrectal ultrasound-guided biopsy in conjunction with screening for serum levels of prostate specific antigen (PSA) which remains the principal biomarker for Pca (Zhang et al. 2017). A challenge across the spectrum of cancers is to identify and incorporate into clinical practice, well-defined and validated diagnostic and predictive biomarkers of disease (Dijkstra et al. 2012; Hemminki et al. 2012; Selvolini and Marrazza 2017; Ueland 2017).
Despite its widespread use, the lack of tumour specificity of PSA can lead to misdiagnosis and over diagnosis of low-risk Pca (Dijkstra et al. 2012). Such over diagnosis is a consequence particularly in the so-called ‘grey area’ of PSA levels between 4.0 and 10.0 ng/mL. Therefore, new biomarkers with ease of detection, increased sensitivity and specificity are required to advance diagnosis of Pca. Potential alternative biomarkers have been proposed and are the subject of recent review (Saini 2016), however, to date no individual marker has replaced the routine focus on PSA.
Recent studies have highlighted that opportunities to exploit biomarker-drug co-development can aid the successful application of molecular marker-based cancer therapies (Twomey et al. 2017). Novel markers including those obtained from the blood or tumour tissue are being evaluated to provide either earlier or more accurate detection or prediction. Given that cancer development and metastasis depend on the function of many proteins, many metabolic enzymes and proteases provide biomarker candidates for cancer early detection (Wang et al. 2017). Tumour-associated proteases are targets for small molecular fluorescent imaging probes and fluorescent theranostics. Progress in chemical probe design and fluorescence imaging is a rapidly developing area and the subject of recently published reviews (Garland et al. 2016; Gao et al. 2017; Lee et al. 2018).
In this study, we focus on the cancer-associated protease and putative tissue biomarker legumain in prostate cancer. Legumain or asparaginyl endoprotease (AEP) [EC.3.4.22.34] has been proposed not only as a putative biomarker in prostate (Ohno et al. 2013), but breast (Gawenda et al. 2007; Liu et al. 2003), ovary (Wang et al. 2012) and colon (Haugen et al. 2015) cancers, in which the enzyme is markedly overexpressed. In colorectal cancer patients, reduced expression of legumain has been related with better prognosis (Murthy et al. 2005).
Legumain is a unique lysosomal member of the clan CD (C13) protease family and is proteolytically active at acidic pH (4.5–5.0) and has selective substrate specificity of cleavage of amide bonds adjacent to C-termini of asparagine residues (Chen et al. 1997; Dall and Brandstetter 2013, 2016). The enzyme is synthesised as a zymogen (56 kDa) and the mechanisms of its lysosome-mediated processing via an intermediate 47 kDa form and a catalytically active 46 kDa form to the mature form of legumain (36 kDa) have been fully described (Dall and Brandstetter 2013, 2016). Furthermore, legumain has been reported to regulate the expression of other cancer-associated proteases including cathepsins B, H and L (Shirahama-Noda et al. 2003) and is involved in extracellular matrix turnover including conversion of metalloproteinase 2 (MMP2) zymogen into the active form of MMP2, a known contributor to cancer invasion and metastasis, by proteolytic removal of an N-terminal propeptide (Chen et al. 2001). Although principally found in lysosomes and late endosomes (Chen et al. 1998), legumain is highly expressed by tumour associated macrophages (TAMs) (Luo et al. 2006) and colocalises with integrins at the invading edge of tumours which correlates with increased invasiveness (Gawenda et al. 2007; Lewis and Murdoch 2005; Liu et al. 2012; Shih et al. 2006; Wu et al. 2006). Legumain has recently been reported to be present in the nucleus and it is suggested that it may contribute to degradation of histones (Dall and Brandstetter 2016).
The prevalence of legumain in the tumour microenvironment has prompted the design of a limited number of prodrugs for targeting its proteolytic activity (Liu et al. 2003; Luo et al. 2006; Smith et al. 2014; Stern et al. 2009). Also, studies of the functional imaging of legumain and other cysteine proteases in cancer have been undertaken using activity-based probes that function as covalent inhibitors in the active site of the enzyme (Edgington et al. 2011, 2013), as an alternative to the substrate approach adopted here. Increased expression in prostate cancer has been correlated with raised invasiveness and aggressiveness (Ohno et al. 2013).
The overexpression of legumain in Pca, combined with its unique restricted substrate specificity makes legumain a smart candidate for tumour-specific detection and in this study have afforded the motivation to exploit these properties in the design of a novel diagnostic substrate probe that could be activated in the tumour microenvironment and to preliminarily evaluate its potential application as a supportive assay in the diagnosis of prostate acinar adenocarcinoma in prostate core biopsy and resection samples.
We report here the design, synthesis and characterisation of a novel intramolecularly quenched, fluorogenic legumain peptide substrate probe 5 [Rho-Pro-Ala-Asn-PEG-AQ(4-OH)], its selective lysosomal targeting in live PC3 prostate cancer cells and activation in both recombinant human legumain and fresh-frozen tissue material from prostatectomy specimens.
Materials and Methods
Chemistry
Common solvents (including HPLC grade) were purchased from Fisher Scientific (Loughborough, UK) and used without further purification. Aluminium plates (Kieselgel 60 F254 silica plates) were used for thin-layer chromatography (TLC) analysis. TLC solvent systems were as follows: (A) CHCl3–MeOH (9:1, v/v), (B) n-BuOH–AcOH–H2O (4:5:1, v/v/v). Davisil Kieselgel LC60A 40–63 µm silica was used for column chromatography (Fisher Scientific; UK). Fmoc-Asn (Trt)-NovaSyn®TGT and peptide derivatives Fmoc-Asn (Trt)-OH, Fmoc-Ala-OH, H-Pro-OtBu·HCl were purchased from Novabiochem® Merck Millipore. All other reagents were obtained from Sigma-Aldrich Merck (Gillingham, UK) Purity of compounds was assessed as ≥ 95% by RP-HPLC on a Waters X-bridge Shield RP18 column (2.1 mm × 30 mm); flow rate 0.2 mL/min) using a one step linear gradient mobile phase A (100% ACN with 0.1% TFA and B (H2O with 0.08% TFA (B). Ratios (A:B) were changed from 25:75 to 50:50 over 10 min; total run time 15 min; injection volume 10.0 µL; column temperature 25º C. 1H and 13C NMR spectra were recorded on either a Bruker AC300 or a Bruker AC400 NMR spectrometer at 25οC at 300.1 MHz and 75.47 MHz, respectively, from samples dissolved either in deuterated DMSO or deuterated chloroform. In 13C, a DEPT (Distortionless Enhancement by Polarization Transfer) experiment was conducted to differentiate between quaternary, methylene carbons, methine and methyl carbons. High resolution mass spectra with electrospray ionization (HRMS-ESI) were recorded using a ThermoFisher LTQ Orbitrap XL or a Waters ZQ4000 instrument. UV–vis spectra were measured on a Beckman DU800 spectrophotometer; scan speed 1200 nm/min and wavelength range 400 to 800 nm. Full characterisation data for all new compounds, including 1H and 13C NMR spectra; UV/visible absorption spectra; and HPLC characterisation chromatograms are presented in the accompanying Supplementary Information file Sections 1, 3–7 and 9).
Abbreviations: AEP, asparaginyl endoprotease; AMC, 7-amino-4-methylcoumarin; AQ, anthraquinone; Boc, tert-butoxycarbonyl; Boc2O, di-tert-butyl dicarbonate; CBZ, benzyloxycarbonyl; DCM, dichloromethane; DIPEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; FRET, fluorescence resonance energy transfer; HOBt, 1-hydroxybenzotriazole; MES, 2-(N-morpholino)ethanesulfonic acid; MMP, matrix metalloproteinase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PEG, polyethyleneglycol; PyBop, benzotriazole-1-yl-oxy-tris-pyrrolidinophosphonium hexafluorophosphate; PyClock, 6-chloro-benzotriazole-1-yloxy-tris-pyrrolidinophosphonium hexafluorophosphate; Rho, rhodamine; TAM, tumour associated macrophage; TBTU, 2-(1H-benzotriazole-1-yl)-1,1,3,3,-tetramethylaminium tetrafluoroborate; TFA, trifluoroacetic acid; Trt, triphenylmethyl (trityl).
Experimental Synthesis Protocols
(2-{2-[2-(4-Hydroxy-9,10-dioxo-9,10-dihydro-anthracen-1-ylamino)-ethoxy]-ethoxy}-ethyl)-carbamic Acid Tert-butyl Ester (2)
To a solution of leucoquinizarin 1 (1 g, 4.1 mmol) in DCM (10 mL) was added 2,2′-(ethylenedioxy)bis(ethylamine) (2.4 mL, 16.4 mmol) and the reaction mixture was refluxed for 1 h. Et3N (100 µL) was added and the solution was aerated at rt. After 2 h, the solvent was evaporated in vacuo and the crude 1-{2-[2-(2-aminoethoxy)-ethoxy]ethylamino}-4-hydroxy-anthracene-9,10-dione was dissolved in MeOH (2.5 mL). Boc2O (3.6 g, 16.5 mmol) in MeOH (4 mL) was added dropwise and the mixture was stirred at 0 °C for 2 h. The solvent was evaporated and the residue was dissolved in CHCl3 (100 mL). The organic layer was washed with water (3 × 100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo and purified by silica-gel column chromatography [CHCl3 and EtOAc (4:1) plus MeOH (1–10%)]. The fractions containing the major product were combined, filtered, evaporated in vacuo to dryness to give compound 2 as a purple solid (0.82 g, 42%). Rf 0.5 (solvent B). 1H NMR (CDCl3, 300 MHz): δ (ppm) 1.41 [s, 9H, C(CH3)3], 3.36 (m, 2H, CH2–NHCO), 3.55 (m, 4H, CH2–O–CH2–CH2–O–CH2–), 3.70 (m, 4H, –O–CH2–CH2–O–), 3.78 (t, 2H, Aq-NH–CH2), 5.28 (s, 1H, NHCO), 7.14 (m, 2H), 7.70 (m, 2H), 8.24 (m, 2H), 10.30, (t, 1H, Aq-NH, J = 6 Hz) 13.59 (s, 1H, OH). 13C NMR (CDCl3,75.5 MHz): δ (ppm) 28.4 (C(CH3)3), 40.5 (CH2), 42.6 (CH2), 69.6 (CH2–O), 70.2 (CH2–O), 70.4 (CH2–O), 70.7 (CH2–O), 79.0 C(CH3)3, 108.6 (CAR), 113.6 (CAR), 123.8 (CHAR), 126.2 (CHAR), 126.7 (CHAR), 128.6 (CHAR), 132.4 (CHAR), 132.5 (CAR), 134.0 (CHAR), 135.2 (CAR), 147.3 (CAR), 156.0 (CAR), 156.6 (C=O, Boc), 181.9 (C=O, Aq), 187.3 (C=O, Aq). HRMS (ESI) m/z: [M + H]+ Calcd for [C25H31N2O7]+ 471.2126; Found 471.2117.
2-{2-[2-(4-Hydroxy-9,10-dioxo-9,10-dihydro-anthracen-1-ylamino)-ethoxy]-ethoxy}-ethylammonium Trifluoroacetate (3)
Compound 2 (0.8 g, 1.69 mmol) was dissolved in TFA (2 mL) at rt. After 30 min, the solvent was evaporated and the residual solid re-evaporated with ethanol (3 × 10 mL) before dissolving in a minimum volume of ethanol (3 mL). Addition of ether (100 mL) gave a precipitate of the deprotected anthraquinone spacer-linked amino acid conjugate as the N-terminal trifluoroacetate salt 3 which was filtered off and dried in vacuo (0.6 g, 95%). Rf 0.4 (solvent B). RP-HPLC tR 4.2 min. 1H NMR (DMSO-d6, 300 MHz): δ (ppm) 3.02 (t, 2H, CH2–NH3+), 3.52 (m, 2H, Aq-NH–CH2), 3.68 (m, 8H, CH2–O–CH2–CH2–O–CH2–), 7.21 (d, 1H, J = 9 Hz), 7.37 (d, 1H, J = 9 Hz), 7.81 (m, 2H), 8.00 (s, 3H, NH +3 ), 8.11 (m, 2H), 10.21, (t, 1H, Aq-NH), 13.54 (s, 1H, OH). 13C NMR (DMSO-d6, 75.5 MHz): δ (ppm) 38.9 (CH2), 42.0 (CH2), 66.7 (CH2–O), 68.9 (CH2–O), 69.6 (CH2–O), 69.6 (CH2–O), 107.2 (CAR), 112.7 (CAR), 115.2 (CAR), 119.2 (CAR), 125.3 (CHAR), 125.8 (CHAR), 126.2 (CHAR), 128.4 (CHAR), 131.7 (COOH), 132.7 (CHAR), 134.4 (CHAR), 147.0 (CAR), 156.0 (CAR), 158.1 to 158.5 (q, CF3), 180.6 (C=O, Aq), 186.5 (C=O, Aq). HRMS (ESI) m/z: [M-C2O2F3]+ Calcd for [C20H23N2O5]+ 371.1601; Found 371.1598.
{9-[2-(2-{1-[1-Carboxy-2-(trityl-carbamoyl)-ethylcarbamoyl]-ethylcarbamoyl}-pyrrolidine-1-carbonyl)-phenyl]-6-diethylamino-xanthen-3-ylidene}-diethylammonium Trifluoroacetate (4a)
Fmoc-Asn(Trt)-NovaSyn®TGT (1.0 g) resin was swelled with DCM for 1 h. The resin-bound Fmoc-protected peptide was treated with 20% (v/v) piperidine in DMF (2 × 10 mL) for 15 min (2×) followed by washing with DMF (3×). A solution of Fmoc-Xaa-OH (2 eq), TBTU (1.9 eq), HOBt (1.9 eq), DIPEA (4 eq.) in DMF (8–10 mL) was added to the resin bound free amine peptide and shaken for 1 h at rt. The resin-bound Fmoc-protected peptide was treated with 20% piperidine in DMF (v/v) for 15 min (2×). The resin was washed with DMF (3×). A solution of Rho-B (0.22 g, 1.5 eq.), Pyclock (0.25 g, 1.5 eq.), DIPEA (0.15 mL, 3 eq) in DMF (4 mL) was added and shaken for 1 h. The coupling was repeated another times and the resin was washed with DMF (3×). The resin was washed with DCM (10×) and cleaved from the resin by repeated treatment with a cleavage solution of 0.5% TFA in 100 mL DCM for 1 h. The combined, filtered extracts were evaporated in vacuo to dryness and the resulting residue was triturated with ether to give compound 4a as a pink solid (0.29 g). Rf 0.5 (solvent B). RP-HPLC tR 11.2 min.
1H NMR (DMSO-d6, 300 MHz): δ (ppm) 1.06 (d, 3H, CH3-Ala, J = 6 Hz), 1.14 (t, 12H, 4 × CH3-Rho, J = 6.9 Hz), 1.52 (m, 1H, β-CH-Pro), 1.72 (m, 2H, γ-CH2-Pro); 1.97 (m, 1H, β-CH′-Pro), 2.49 (β-CH2-Asn, obscured by solvent), 2.99 (q, 8H, 4 × CH2-N-Rho), 3.47 (m, 2H, δ-CH2-Pro), 4.09 (t, 1H, α-CH-Pro), 4.28 (m, 1H, α-CH-Asn), 4.45 (m, 1H, α-CH-Ala), 6.94 (d, 2H, H-4, 5, J = 3 Hz); 7.09–7.32 [m, unresolved, 19H (H-1, 2, 7, 8 and Trt)], 7.53 (m, 1H, H-6′), 7.75 (s, 1H, NHCO), 7.77 (m, 3H, H-3′, 4′, 5′), 7.85 (d, 1H, NHCO), 7.91 (d, 1H, NHCO). HRMS (ESI) m/z: [M-C2O2F3]+ Calcd for [C59H63N6O7]+ 967.4753; Found 967.4747.
[9-(2-{2-[1-(2-Carbamoyl-1-carboxy-ethylcarbamoyl)-ethylcarbamoyl]-pyrrolidine-1-carbonyl}-phenyl)-6-diethylamino-xanthen-3-ylidene]-diethylammonium Trifluoroacetate (4b)
Compound 4a (0.1 g, 92.5 mmol) was dissolved in TFA (3 mL). After 3 h, the solution was evaporated in vacuo and the residue triturated with diethyl ether to give compound 4b as a pink solid (0.069 g, 89%). Rf 0.53 (solvent A). RP-HPLC tR 4.60 min.
1H NMR (DMSO-d6, 300 MHz): δ (ppm) 1.05 (d, 3H, CH3-Ala, J = 6 Hz), 1.22 (t, 12H, 4 × CH3-Rho, J = 6.9 Hz), 1.57 (m, 1H, β-CH-Pro), 1.80 (m, 2H, γ-CH2-Pro); 1.96 (m, 1H, β-CH′-Pro), 2.42 (β-CH2-Asn), 3.55 (m, 2H, δ-CH2-Pro), 3.66 (q, 8H, 4 × CH2-N-Rho), 4.05 (m, 1H, α-CH-Pro), 4.23 (m, 1H, α-CH-Asn), 4.38 (m, 1H, α-CH-Ala), 6.94 (d, 2H, H-4, 5, J = 3 Hz); 7.14 (m, 4H, H-1, 2, 7, 8), 7.52 (m, 1H, H-6′), 7.75 (s, 1H, NHCO), 7.77 (m, 3H, H-3′, 4′, 5′), 7.90 (d, 1H, NHCO). HRMS (ESI) m/z: [M-C2O2F3]+ Calcd for [C40H49N6O7]+ 725.3657; Found 725.3653.
{9-[2-(2-{1-[2-Carbamoyl-1-(2-{2-[2-(4-hydroxy-9,10-dioxo-9,10-dihydro-anthracen-1-ylamino)-ethoxy]-ethoxy}-ethylcarbamoyl)-ethylcarbamoyl]-ethylcarbamoyl}-pyrrolidine-1-carbonyl)-phenyl]-6-diethylamino-xanthen-3-ylidene}-diethylammonium Trifluoroacetate (5)
To a solution of compound 3 (0.1 g, 0.20 mmol) in DMF (2 mL) was added compound 4a (0.18 g, 0.18 mmol) in the presence of PyBOP (0.161 g, 0.30 mmol), HOBt (0.042 g, 0.31 mmol) and DIPEA (0.165 mL, 0.93 mmol) in DMF (4 mL) and at rt. After 2 h, the organic layer was washed with water (3 × 100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo and purified by silica-gel column chromatography [CHCl3 and EtOAc (4:1) plus MeOH (1–10%)]. The fractions containing the major product were combined, filtered, evaporated in vacuo to dryness. The residue was treated with TFA (1 mL) for 3 h. The solution was evaporated in vacuo to dryness. The residue was triturated with ether to give compound 5 as a purple solid (0.1 g, 31.2%). Rf 0.5 (solvent A). RP-HPLC tR 5.2 min.
1H NMR (DMSO-d6, 300 MHz): δ (ppm) 1.07 (d, 3H, CH3-Ala, J = 6 Hz), 1.20 (t, 12H, 4 × CH3-Rho, J = 6.9 Hz), 1.55 (m, 1H, β-CH-Pro), 1.65 (m, 2H, γ-CH2-Pro), 1.95 (m, 1H, β-CH′-Pro), 2.41 (d, 2H, β-CH2-Asn, J = 6 Hz), 3.12 (m, 2H, CH2–NHCO) 3.32–3.75 (m, 20H, δ-CH2-Pro, CH2–O–CH2–CH2–O–CH2, 4 × CH2-N-Rho, Aq-NH–CH2), 3.80–3.99 (m, 2H, α-CH-Pro, α-CH-Asn), 4.32 (m, 1H, α-CH-Ala), 6.87 (d, 2H, H-4, 5, Rho, J = 3 Hz), 7.08 (m, 4H, H-1, 2, 7, 8, Rho), 7.32 (m, 1H, H-2, Aq), 7.51 (d, 2H, H-3, Aq; H-6′, Rho), 7.65 (s, 1H, NHCO), 7.76 (3H, H-3′, H-4′, H-5′), 7.83 (s, 1H, NHCO), 7.86 (m, 2H, H-6, 7, Aq), 8.19 (d, 1H, NHCO), 8.22 (m, 2H, H-5, -8, Aq), 10.35 (t, 1H, Ar–NH), 13.57 (s, 1H, OH). 13C NMR (DMSO-d6, 75.5 MHz): δ (ppm) 12.4 (4 × CH3-Rho), 17.2 (CH3-Ala), 25.1 (γ-CH2-Pro), 29.0 (β-CH2-Pro), 38.6 (β-CH2-Asn, obscured by solvent), 40.3 (CH2-N, obscured by solvent), 42.1 (CH2-N), 45.3 (CH2-N), 48.7 (α-CH), 49.5 (α-CH), 49.8 (CH2-N), 58.9 (α-CH), 68.8 (CH2–O), 68.0 (CH2–O), 69.6 (CH2–O), 69.7 (CH2–O), 95.7 [CHAR (C-4,-5, Rho)], 107.3 (CAR), 112.6 (CAR), 112.8 (CAR), 113.3 (CAR), 113.8 (CHAR), 114.2 (CHAR), 125.7 (CHAR), 125.9 (CHAR), 126.3 (CHAR), 127.3 (CHAR), 128.6 (CHAR), 129.8 (CHAR), 130.2 (CHAR), 130.3 (CAR), 131.0 (CHAR), 131.8 (CHAR), 132.9 (CHAR), 134.7 (CHAR), 136.5 (CAR), 147.3 (CAR), 154.9 (CAR), 155.0 (CAR), 155.9 (CAR), 156.1 (CAR), 157.0 (CAR), 157.2 (CAR), 166.0 (CAR), 170.6 (NHCO), 170.7 (NHCO), 171.4 (NHCO), 171.6 (NHCO), 171.7 (NHCO), 180.7 (C=O, Aq), 186.7 (C=O, Aq). HRMS (ESI) m/z: [M-C2O2F3]+ Calcd for 1077.5080 [C60H69N8O11]+; Found 1077.5065.
[9-(2-{2-[1-(2-Carbamoyl-1-carboxy-ethylcarbamoyl)-ethylcarbamoyl]-ethylcarbamoyl}-phenyl)-6-diethylamino-xanthen-3-ylidene]-diethylammonium Trifluoroacetate (6)
Compound 6 was synthesised by directly analogous procedures to those for compounds 4a and 4b, using Fmoc-Asn(Trt)-NovaSyn®TGT resin (1.0 g), as a pink solid (0.152 g; 74%). Rf 0.4 (solvent A). RP-HPLC tR 4.08 min. 1H NMR (DMSO-d6, 300 MHz): δ (ppm) 1.08–1.21(m, unresolved, 15H, CH3-Ala, 4 × CH3-Rho), 2.10 (m, 2H, CH2–CONH), 2.49 (d, 2H, β-CH2-Asn), 3.18 (t, 2H, CH2–NHCO, J = 6.9 Hz), 3.37 (q, 8H, 4 × CH2-N-Rho), 4.17 (m, 1H, α-CH-Asn), 4.45 (m, 1H, α-CH-Ala), 6.29–6.45 (m, 5H, unresolved, H-2, 4, 5, 7 and NHCO), 6.87 (s, 1H, NHCO), 6.90 (d, 1H, H-6′), 7.34 (s, 1H, NHCO), 7.49 (m, 2H, H-1, 8), 7.78 (m, 1H, H-4′), 8.02 (t, 2H, H-3′, 5′). HRMS (ESI) m/z: [M-C2O2F3]+ Calcd for 657.1838 [C33H29N8O11]+; Found 657.1824.
Rho-Pro-Ala-Ala-PEG-AQ(4-OH) (7)
Compound 7 (control peptide) was synthesised by an analogous procedure to that used for probe 5, described above, except alanine (Ala) was substituted for Asparagine (Asn) in the peptide sequence. To a solution of compound H-Pro-Ala-Ala-PEG-AQ(4-OH) (0.2 g, 0.27 mmol) in DMF (3 mL) was added Rho-B (0.16 g, 0.33 mmol), PyBOP (0.172 g, 0.33 mmol), HOBt (0.04 g, 0.32 mmol) and DIPEA (0.22 mL, 1.2 mmol) in DMF (4 mL) at rt to give 7 (following method A) as a purple solid (0.1 g, 32%). Rf 0.4 (Solvent System B). HRMS (ESI) m/z: [M-C2O2F3]+ Calcd for 1034.5022 [C59H68N7O10]+; Found 1034.5021.
Fluorescence Studies: pH Dependence of Fluorescence of Rhodamine-Labelled Tripeptides 4b and 6
Fluorescence emission spectra were measured on a Perkin Elmer LS50B luminescence spectrometer; scan speed 500 nm/min, λex 562 nm, λem 570–700 nm, excitation slit and emission slit widths were 4.5 nm and 5.5 nm, respectively. For the experiments, compounds 4b and 6 were mixed with Mcllvaine assay buffer (McIlvaine 1921) to produce a final concentration in a quartz cuvette (3 mL) of 10 μM and 40 μM, respectively, across pH 3–8 range.
Determination of Relative Fluorescence Quantum Yields
N-Rhodamine labelled tripeptides 4b and 6 and the reference standard rhodamine B were prepared as stock solutions (1 mg/mL) in ethanol. The absorbance spectra (see Supplementary Information file) of the compounds in ethanol (pH 5) were recorded on a Beckman DU800 spectrophotometer with a scan speed 1200 nm/min and wavelength range 400 to 800 nm in 1 cm path length quartz cuvettes. The Beer–Lambert law (A = εcl) was used to determine the extinction coefficients of test samples. Extinction coefficients were determined at λmax. Fluorescence emission spectra were recorded by diluting the reference standard and test samples x 10 in ethanol, scan speed 500 nm/min; λex 535 nm, λem 540–700 nm; excitation slit and emission slit widths were 4.5 nm and 5.5 nm. The quantum yield was calculated using the following equation:
where m is the slope of the line obtained from the plot of the integrated fluorescence versus absorbance; n is the refractive index of the solvent and subscript R refers to the reference fluorophore of known quantum yield (rhodamine B, Φ = 0.73 ± 0.02) (Rurack 2008). Given that ethanol was used as the solvent for the reference and each of compounds 4b and 6, the term \(\left( {{{\text{n}}^2}/{\text{n}}_{\text{R}}^2} \right) \, = \, 1\).
Activation of Probe 5 by Recombinant Human Legumain in Vitro
Human recombinant legumain was obtained from R&D Systems (Abingdon, UK). A FLUOstar Omega multi-mode microplate reader (BMG Labtech, Ortenberg, Germany) was used to measure fluorescence. Stock solutions of probe 5 and fluorophore 4b were prepared at 1 mg/mL in DMSO. Legumain activation and assay buffers were prepared as follows: activation buffer (50 mM sodium acetate, 100 mM NaCl, pH 4.0); assay buffer [50 mM MES hydrate (Sigma) 250 mM NaCl, pH 5.0]. Recombinant human legumain (10.0 µg) was diluted with activation buffer (100 µL, pH 4.0) and frozen at − 80 °C. An aliquot (476.2 ng per 5 µL) of de-frosted foregoing solution was incubated at 37 °C for 2 h, followed by dilution with assay buffer to 1 ng/μL. The final concentrations of compounds 5 and 4b were each 10 µM in total reaction volumes of 100 µL in assay buffer (pH 5.0) either with or without rh-legumain (40.0 ng). Fluorescence of probe 5 was measured using a microplate reader (λex—540 nm; λem—590 nm) at 37 °C for 2 h using F16 black maxisorp 96 well plates. The continuous increase in fluorescence intensity was monitored by repeated measurements as a measure of enzyme-mediated probe activation. Mean relative fluorescence intensity was plotted against time with subtraction of background fluorescence.
Substrate Kinetic Assay
Pre-activated rh-legumain was added to legumain assay buffer (final volume 100 μL) with substrate probe 5 at concentrations ranging from 1 to 40 μM in a 96-well plate (n = 3) at a final legumain concentration of 0.4 ng/μL. Hydrolysis of substrate probe 5 was monitored fluorimetrically (λex 540 nm; λem 590 nm, based on the fluorescence properties of cleavage product and reporter fluorophore 4b) using a microplate reader at 2 min intervals at 37 °C for 2 h. Relative fluorescence intensity data of substrate probe 5 was processed by using enzyme kinetics wizard in SigmaPlot 12 software to fit the data to a non-linear regression curve. Km was determined using a Michaelis–Menten plot of initial enzyme velocity versus substrate concentration. Km ± 1.5 μM for probe 5 (activated by rh-legumain).
Distribution Coefficient Assay
The distribution coefficient of compounds 3, 5 and 4b was determined by the ‘shake-flask’ method (Leo et al. 1971). PBS (0.01 M phosphate buffer; 0.0027 M potassium chloride; 0.137 M sodium chloride) and 1-octanol were from Sigma-Aldrich Merck (Gillingham, UK). Compounds (1 mg) were suspended in PBS buffer (pre-saturated for 24 h with 1-octanol) (900 µL) and 1-octanol (pre-saturated for 24 h with PBS buffer) (900 µL), mixed by vortex and shaken at rt for 24 h to allow the compound to partition between the two phases. The layers were separated by centrifugation at 2000 g for 5 min and the concentration in each phase was determined from calibration curves of absorbance versus concentration. The experiments were performed in triplicate. The distribution coefficients were calculated according to the following equation:
Cell Culture
PC-3 prostate cancer cells were obtained from the American Type Culture Collection (ATCC). PC-3 cells were grown in T75 tissue culture flasks using RPMI 1640 medium (Gibco; ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Sigma-Aldrich Merck, Gillingham, UK) in a humidified incubator (5% CO2, 95% air) at 37 °C.
In Vitro Chemosensitivity Assay
Cell viability was determined by an MTT-based method (Plumb et al. 1989). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H tetrazolium bromide] was obtained from Sigma-Aldrich Merck (Gillingham, UK). Briefly, PC-3 cells in exponential growth phase were seeded in 96-well plates to achieve a final density of 3 × 104 cells per well. After 24 h incubation at 37 °C (5% CO2, 95% air), the cells were treated with final drug concentrations ranging from 100 to 0.01μM. Following 96 h continuous exposure to drug at 37 °C, cells were incubated with fresh drug-free medium immediately prior to addition of MTT solution (5 mg/mL in PBS). Medium and MTT were removed after 4 h and DMSO (150 μL) was added. The absorbance was measured at 550 nm, using a Labsystem Multiskan microplate reader. IC50 values were obtained from growth curves of drug concentration against % survival, using SigmaPlot 12 software, and are expressed in μM.
Confocal Microscopy Studies in PC3 Prostate Cancer Cells
For live cell imaging, PC-3 cells (1.5 × 105) were seeded in a 35 mm glass bottomed micro well dish (Ibidi/Thistle Scientific, Glasgow, UK) and cultured for 24 h. For lysosome co-staining, cells were washed x 3 with PBS. LysoTracker Green DND 26 [(60 nM) Invitrogen, Paisley, UK] was added to the cell culture medium and incubated at 37 °C for 30 min prior to addition of probe 5 (1 µM) and imaging. For mitochondria co-staining, cells were washed x 3 with PBS, MitoTracker Green FM [(50 nM) Invitrogen, Paisley, UK] was added to the cell culture medium and incubated at 37 °C for 30 min prior to addition of probe 5 (1 µM) and imaging. Cells were imaged using a Zeiss LSM800 confocal laser scanning microscope with a HCX PL APO lambda blue 63.0 × 1.4 oil objective, using Diode (405 nm), Argon (458, 488, 514 nm), HeNe (543 nm), HeNe (594 nm) and HeNe (633 nm) lasers. Image analyses were performed using Image J software.
Fresh-Frozen Tissue Material
Fresh-frozen tissue curls were prepared during diagnostic frozen section sampling of 2 fresh prostatectomy specimens. Frozen curls were collected in sterile Eppendorf tubes individually per each frozen section. Tubes were transferred immediately from − 20 °C into − 80 °C. Haematoxylin and eosin (H & E) frozen section slides were examined by pathologists to confirm that the section from the first specimen (PO1) contained benign prostatic tissue which showed no evidence of malignancy, whereas the section from the second specimen (PO2) contained a small foci of prostatic adenocarcinoma.
Both prostatectomy specimens were processed into formalin-fixed paraffin embedded tissue sections for diagnostic histological examination; H & E slides were examined by pathologists. On histological examination, sections from the first specimen (PO1) of the prostate showed a 3 mm focus of Gleason grade 3 + 3 = 6, adenocarcinoma in the right lobe more towards the apical side. There were foci of high grade PIN in adjacent slices. No other tumour was identified elsewhere. The tumour replaced < 1% of the prostatic tissue. Background prostate showed nodular and numerous foci of stromal hyperplasia. Mild chronic inflammation was also present.
Sections from the second prostatectomy specimen (PO2) showed bilateral multifocal prostatic adenocarcinoma of acinar type. The tumour was present as two large and two small nodules, the larger nodules measured 1.5 × 1 cm, which showed Gleason grade of 3 + 4 = 7 (95:5). The tumour replaced approximately up to 5% of the prostate volume. There were foci of high grade PIN and there was diffuse perineural invasion. There was no definite evidence of extra prostatic extension and no involvement of seminal vesicle or vas by the tumour. The background prostate showed nodular hyperplasia and mild chronic inflammation.
PO1 specimen was from a 71 y old male with raised PSA—5.2, DRE—Benign, and Radiological progression.
PO2 specimen was from a 52 y old male with raised PSA 4.2, DRE—Benign, and Radiological progression.
Fresh-Frozen Tissue Assay
Materials: Assay buffer [50 mM MES hydrate (Sigma) 250 mM NaCl, pH 5.0]. Test compounds: legumain substrate probe 5 (10 µM) and negative control, legumain non-substrate peptide 7 (10 µM); iodoacetamide (protease inhibitor, Sigma) (1 mM); F16 black maxisorp 96 well plates. Fluorescence was measured using a Perkin Elmer Enspire Multimode plate reader (λex 540 nm; λem 590 nm).
Method: fresh-frozen prostate tissue curls kept in the freezer were removed and placed on ice, weighed (60 mg sample mass) and added to the assay buffer at pH 5.0. Tissues were homogenised using a precooled spatula and blended in Eppendorf tubes on ice (4 ºC). Following homogenisation, the tissue preparation was centrifuged for 15 min at 10,000 g. The supernatant was removed and used to incubate test compounds (96 well plates); the incubates were shaken for 5 min prior to recording the fluorescence emission spectra. Relative fluorescence intensity was monitored for a period of 210 min at 37 ºC. Preincubation of homogenates with iodoacetamide (1 mM for 15 min) prior to addition of test compound(s) was performed to abrogate protease activity. Mean relative fluorescence intensity was plotted against time, with subtraction of (any) background fluorescence.
Results and Discussion
Probe Design
Biomolecular applications of fluorogenic molecular probes demand design features that permit selective activation, a substantial increase in emission intensity upon reaction with the target protease, efficiency and stability. A fluorescence resonance energy transfer (FRET) system was designed to provide for continuous reaction monitoring and rapid determination of enzyme activity and that could be envisaged as a potential simple tool for use in the cancer pathology laboratory, requiring inexpensive instrumentation; notably, to detect legumain biomarker activity in prostate tissue samples. The design of probe 5 [Rho-Pro-Ala-Asn-PEG-AQ(4-OH)] (Fig. 1) as a new FRET peptide substrate of legumain, incorporating a rhodamine B derivative modified at the 2´-carboxylic acid group was selected on the basis of its favourable photostability, high relative fluorescence quantum yield, long emission wavelength, low cost and ease of access by chemical synthesis. The rhodamine donor fluorophore was demonstrated to be part of an efficient FRET pair with a novel PEG-spaced 1-amino-4-hydroxyanthraquinone derivative; the latter quencher (acceptor) was selected on the prediction that its absorption spectrum would significantly overlap the emission spectrum of the fluorophore, thereby functioning as an effective ‘black hole’ quencher, effectively conferring no background fluorescence. Related 1,4-diaminoanthraquinones are non-fluorescent quenchers with applications that include real-time PCR (Jernigan and Lawrence 2013; May et al. 2005). For strong recognition by legumain, the substrate probe 5 satisfied the stringent requirement for Asn in the P1 position; whereas Pro was selected in the P3 position based on a previously intimated best fit in an activity based probe (Sexton et al. 2007). Furthermore, coupling rhodamine B to the N-terminus of Pro ensured that the fluorophore was ‘locked’ into a ring-opened form by tertiary amide formation, thereby preventing cyclisation to non-fluorescent lactam forms, analogous to the properties of piperazine derivatives of Rho-B (Beija et al. 2009). Inclusion of a short PEG- or polyethyleneglycol-like linker, between the C-terminus of Asn and the amino-hydroxyanthraquinone quencher, enhanced aqueous solubility and could substitute for a P1′ amino acid residue. Upon activation by legumain, the probe 5 was cleaved at the C-terminal side of the key Asn residue to release the strongly fluorescent tripeptide reporter fluorophore 4b (Rho-Pro-Ala-Asn-OH). Control compounds 7 and 6 were synthesised to complement the probe 5 and its fluorophore product 4b, respectively. Tripeptide 6 (which contained β-Ala in place of Pro) was prepared to affirm that it was important to retain the secondary amino acid Pro adjacent to the rhodamine label for efficient fluorescence and stability. Compound 7, the quenched analogue of probe 5 was prepared as a negative control peptide for legumain activation (containing P1-Ala in place of the key Asn residue) given the absence of a cleavage ‘hotspot’.

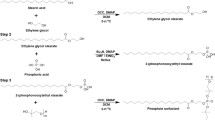
Synthesis of the Fluorogenic Legumain Peptide Substrate probe 5a. aa 2,2′-(Ethylenedioxy)bis(ethylamine), DCM, 40 °C, 1 h; b Boc2O 20%, MeOH, 0 °C 2 h. c TFA, RT, 30 min. d PyBOP, HOBt, DIPEA, DMF, rt, 2 h. e TFA, rt, 2 h. Compounds 4a, 4b and 6 were synthesised by solid phase peptide synthesis. Compound 7 [Rho-Pro-Ala-Asn-PEG-AQ(4-OH)] (negative control peptide) was synthesised by an analogous procedure to that used for probe 5, described above, except Ala was substituted for Asn at P1 in the peptide sequence
Synthesis
Figure 1 shows the synthetic procedures for the preparation of the novel legumain-activatable, FRET probe 5, its quencher moiety and associated rhodamine labelled tripeptides. The fluorogenic legumain substrate 5 was synthesised by a convergent method. Controlled mono-amination (Kikuchi et al. 1982) of leucoquinizarin 1 with 2,2′-(ethylenedioxy)bis(ethylamine) in DCM, followed by air oxidation of the intermediate leuco-form of the product in the presence of Et3N afforded 2-{2-[2-(4-hydroxy-9,10-dioxo-9,10-dihydro-anthracen-1-ylamino)-ethoxy]-ethoxy}-ethyl-ammonium in its crude form. Facile chromatographic purification of the latter was achieved by temporary protection of the primary amino group using Boc2O in MeOH to afford compound 2. Treatment of compound 2 with TFA afforded the stable, conveniently stored amine trifluoroacetate salt 3. The rhodamine B-labelled tripeptide Rho-Pro-Ala-Asn(Trt)-OH, 4a was synthesised on Fmoc-Asn(Trt)-NovaSyn®TGT resin. The coupling reactions were performed with a twofold molar excess of Fmoc-Ala-OH and Fmoc-Pro-OH, consecutively, using a 1.9-fold molar excess of activating agents (TBTU/HOBt) in the presence of DIPEA in DMF. Attachment of the rhodamine B fluorophore was achieved via PyClock/DIPEA-mediated coupling on resin. The resin-bound, N-labelled, Asn-protected tripeptide was cleaved with TFA in DCM (0.5%) at room temperature to afford compound 4a. Subsequently, the fluorescent tripeptide 4a was coupled to the amino-hydroxyanthraquinone PEG-like spacer compound 3 (fluorescence quencher), using PyBOP, HOBt and DIPEA, to afford a trityl protected rhodamine-anthraquinone conjugate intermediate that upon TFA-mediated trityl deprotection, gave the novel peptide-linked fluorogenic rhodamine-anthraquinone conjugate probe 5.
Separately, TFA removal of the trityl group in compound 4a gave Rho-Pro-Ala-Asn-OH 4b; an authentic sample of the predicted fluorescent product from legumain-mediated cleavage of fluorogenic substrate probe 5 at the C-terminus of its Asn residue.
For fluorescence studies, comparator rhodamine-labelled negative control tripeptide, Rho-β-Ala-Ala-Asn-OH 6, in which rhodamine-B was attached to the primary amino group of a β-Alanine residue (in contrast to the secondary amino group of Pro in the equivalent P3 position in probe 5) was synthesised by analogous SPPS procedures to those used for probe 5.
Fluorescence Properties of Rho-Labelled Tripeptides 4b and 6; pH Dependence
We have shown previously that oligopeptide substrates, fluorescently labelled with fluorescein derivatives from the xanthene class of dyes, can be efficiently activated by the cancer-associated endoprotease MMP-9 in the microenvironment of myeloma (Valckenborgh et al. 2005) however, fluoresceins suffer from undesirable decrease in fluorescence intensity on lowering the pH (Cook and Le 2006), which makes them unsuitable for applications in the acidic environment of the lysosomes. To overcome this limitation, in this study, the xanthene class of rhodamine-based compounds offered a more robust platform for the design of efficient fluorogenic substrates of the endoprotease legumain. The fluorescent cleavage product expected from the legumain-mediated activation of probe 5 was the rhodamine B-labelled, Rho-Pro-Ala-Asn-OH 4b. An authentic sample of fluorophore 4b was synthesised and characterised to determine whether or not its photophysical properties would be suitable to facilitate continuous reaction monitoring of activation of probe 5 by legumain. As shown in Fig. 2a, Rho-labelled tripeptide 4b was strongly fluorescent across the tested pH (3.0–8.0) range; thus making it a suitable fluorophore for biological studies under physiological conditions, including the highly acidic environment of the lysosome and the pH at which the lysosomal legumain is catalytically active (Chen et al. 1997).

a Fluorescence spectra of compound 4b (Rho-Pro-Ala-Asn-OH; 10 µM) in Mcllvaine buffer; pH range 3.0–8.0, λex 562 nm, λem 583 nm. b Fluorescence spectra of compound 6 (Rho-β-Ala-Ala-Asn-OH; 40 µM) in Mcllvaine buffer pH range 3.0–8.0, λex 562 nm, λem 583 nm
The importance of the tertiary amide group at the Rho-Pro junction was underlined by comparing the properties of fluorophore 4b with a control peptide 6 in which Rho-β-Ala was substituted. As shown in Fig. 2b, compound 6 was minimally fluorescent at pH 3.0–4.0 and barely fluorescent, even at fourfold greater concentration than fluorophore 4b, at the lysosomal pH range (5.0–6.0) of active legumain, making it unsuitable for cell studies and imaging. These observations were consistent with the known formation of non-fluorescent spirolactam forms of secondary amide-linked rhodamines (Beija et al. 2009). Furthermore, the selection of fluorophore 4b as a component of the FRET substrate probe 5, was based upon its invariant fluorescence emission wavelengths or intensity of emission at the analytical wavelength (λem 583 nm) with varying pH and corresponding constant absorption bands in the visible spectrum (see Supplementary Information Section 5 Fig. S4). The selection of the quencher moiety (acceptor) compound 3, in probe 5, comprised of a purple 1-amino-substituted-4-hydroxyanthraquinone chromophore, was made on the basis of its strong visible absorption (see Supplementary Information Section 4 Fig. S3) spectral overlap with the fluorescence emission of fluorophore 4b, fulfilling a fundamental requirement for an efficient FRET pair (Sapsford et al. 2006).
Furthermore, the efficient silencing of rhodamine fluorescence in FRET probe 5 is shown over a range of concentrations when compared with compound 4b, its legumain-released cleavage product fluorophore (Fig. 3).

Comparison of variation in Relative Fluorescence Intensity with concentration for probe 5 and fluorophore tripeptide 4b (Rho-Pro-Ala-Asn-OH), λem 583 nm; (n = 3)
Determination of Relative Fluorescence Quantum Yields and Substrate Kinetic Assay
The most widely used, commercially available, fluorogenic (non-FRET-based) substrate probe for legumain, CBZ-Ala-Ala-Asn-AMC (Km 80 ± 6 μM) (Mathieu et al. 2002) consists of the fluorogenic chromophore 7-amino-4-methylcoumarin (AMC) and a legumain-recognised peptide. Biological applications of this substrate are limited because its fluorescence is pH-dependent and it can incur interference from other molecules in a biological matrix due to its short emission wavelength. Conversely, we have shown that the rhodamine B-based fluorophore 4b in addition to pH-independent fluorescence, has a higher quantum yield, is excited and emits at longer wavelengths (Table 1), making it more suitable for biological applications than several commercially available fluorophores. Rhodamine B was used as a reference standard for relative fluorescence quantum yield determinations. The fluorescence quantum yield of the Rho-Pro-linked fluorophore 4b (ΦF 0.95) was higher than the Rho-β-Ala-linked analogue 6 (ΦF 0.11) due to the latter adopting ring-closed non-fluorescent forms at low pH. The ability to control the fluorescent properties of the rhodamine label in fluorophore 4b facilitated the design of probe 5 prompting our experiments to evaluate its potential application in profiling protease activity in live cancer cells and lysosome targeting. Legumain substrate probe 5 was shown to have a lower Km value (Km ± 1.5 μM) and greater specific activity (960 pmol/min/µg) than CBZ-Ala-Ala-Asn-AMC (Mathieu et al. 2002).
In Vitro Chemosensitivity
In order for the novel legumain probe 5 to have potential future applications in in vivo models, it was important to assess its effect on the growth of cancer cells. The cancer cell growth inhibitory properties (Table 1) of the legumain substrate probe 5 [Rho-Pro-Ala-Asn-PEG-AQ(4-OH)] and its legumain mediated cleavage fragments: the quencher 3 [PEG-AQ(4-OH)] and fluorescent tripeptide 4b (Rho-Pro-Ala-Asn-OH) were determined by a modification (Plumb et al. 1989) of the colourimetric-based MTT assay in PC3 prostate cancer cells (96 h incubation). (See Supplementary Information Section 2 Fig. S1 for MTT cytotoxicity assay data). Fluorescent tripeptide reporter 4b was non-toxic at all concentrations tested (up to 100 µM) and fluorogenic substrate probe 5 had an IC50 value > 100 µM. The amino-hydroxyanthraquinone quencher 3 was also poorly cytotoxic (IC50 23.27 ± 0.04 µM) at the highest test concentration. The data indicated that probe 5 (and its legumain-induced cleavage products) should be relatively non-toxic to the normal cellular environment.
Activation of Probe 5 by Recombinant Human Legumain In Vitro
To assess the ability of the fluorogenic FRET probe 5 [(Rho-Pro-Ala-Asn-PEG-AQ(4-OH)] to act as a legumain substrate, an in vitro fluorimetric assay was developed using rh-legumain at 37 °C in MES assay buffer (pH 5.0). Incubation of probe 5 (10 µM) with rh-legumain (0.40 ng/μL) at 37 °C for 2 h, afforded a time-dependent increase in relative fluorescence intensity, monitored at the emission wavelength (λem 583 nm) of rhodamine-labelled peptide 4b, as shown in (Fig. 4). Incubation of probe 5 in assay buffer in the absence of enzyme resulted in no fluorescence release. Furthermore, pre-incubation of rh-legumain with the cysteine protease inhibitor iodoacetamide (1 mM; 15 min) completely abolished fluorescence release. The designed probe 5 was determined to be an efficient FRET substrate affording good restoration of fluorescence upon activation by legumain under acidic pH conditions that mimic those typically observed in lysosomes.

Variation in relative fluorescence intensity released with time for probe 5 (10 µM) upon incubation with rh-legumain (0.40 ng/μL) in legumain assay buffer (pH 5.0) with (red dots) and without (green dots) pre-incubation of legumain with iodoacetamide (1 mM, 15 min), λex 544 nm, λem 583 nm; enzyme-free probe 5 only (blue dots) (n = 3)
Subcellular Localisation of Probe 5 to Lysosomes of Live PC3 Prostate Cancer Cells
The acidity of the lysosome (pH 4.5–5.5) is critical to the optimal activity of legumain. Confocal imaging was performed to analyse the cellular uptake and localisation of legumain probe 5 in live PC-3 prostate cancer cells in the presence of commercially available, lysosomal-targeting LysoTracker Green DND 26 probe. Legumain-expressing (Ohno et al. 2013), PC-3 cells were treated with a co-stain solution of LysoTracker Green DND 26 (60 nM) and probe 5 (1 µM). Co-stain was applied to the cell culture medium and incubated at 37 °C for 30 min prior to imaging. Figure 5a shows the cellular uptake of probe 5 in PC-3 cells at imaging time points 0, 30, 60 and 120 min. Legumain substrate probe 5 was detected in the lysosomes, but not in the nucleus. Probe 5 showed a punctate staining pattern near the perinuclear regions of the live cells (panels E, H and K) and clear colocalisation with stained lysosomes (panels F, I and L). The cellular uptake of probe 5 was facilitated by its cationic lipophilic character (Log D: + 1.78 ± 0.29).

a Confocal microscopy live cell images of PC-3 cells treated with probe 5 (1 μM) for up to 2 h (scale bar 20 μm). Lysosomes were stained with LysoTracker Green DND 26 (60 nM). Areas where probe 5 and lysosomes colocalize appear as yellow. Images were captured at 63 x magnification. b Degree of colocalization of probe 5 and LysoTracker Green DND 26
The colocalization of probe 5 with LysoTracker Green DND 26, is consistent with reports that rhodamine-based compounds prefer the acidic environment of lysosomes (Yu et al. 2015) over other organelles (probe 5 did not colocalize with MitoTracker Green FM-treated PC-3 cells; (data available in Supplementary Information Section 8 Fig. S7). Probe 5 was observed in the lysosomes within 30 min of treatment confirming that it can reach the environment of its intended activating enzyme; it is well established that the target protease legumain is overexpressed within the acidic environment of the lysosomes (Halfon et al. 1998; Kembhavi et al. 1993). Pearson’s correlation coefficient and Mander’s overlap coefficient were used for quantifying the degree of co-localisation of probe 5 and LysoTracker Green DND 26 (Fig. 5b), showing good agreement between methods across the 2 h incubation.
Prostate Cancer Fresh-Frozen Tissue Studies
Given the limitations of serum PSA levels as a biomarker, histological evaluation of prostate core biopsies and resection samples is often more reliable, but the use of a histological diagnosis only as a sole marker, in the decision of treatment and prognosis, may not be appropriate, as it is not always predictive of which lesion will remain dormant for a substantial number of years, and which one will become aggressive in a short period of time. This emphasises the need for additional assays and technologies that could be used as diagnostic and predictive assays of prostate cancer development and which could help inform or advance further therapies.
To this end, one of our principal objectives was to investigate whether or not the designed fluorogenic FRET substrate probe for legumain 5 would be activated by tissue homogenates from prostate tissue samples; and whether or not this could be attributed to legumain activity, mindful that other proteases in tissue may interact with the probe. In prostate cancer patients all tissue specimens derived either from biopsy or prostatectomy are of diagnostic value. Therefore, it is often difficult to obtain material dedicated purely for research activities. Since for our assay we did not require to maintain tissue morphology; rather, to see what section of tissue was put into homogenates, we therefore opted to test our idea on trimming surplus material that remained from diagnostic frozen sections for two areas: benign and malignant. We selected the first specimen (PO1) with the least amount of tumour in the gland (< 1%), where the frozen section was taken from a clearly defined benign area, and the second specimen (PO2), where the tumour was clearly identified on macroscopic assessment and the frozen section was taken from this malignant area.
As a prelude to incubating the legumain substrate probe 5 with prostate tissue homogenates, it was considered important to prepare a negative control analogue 7 which lacked the cleavage hotspot for legumain. This was achieved by substituting the Asn residue by Ala, to afford the sequence Rho-Pro-Ala-Ala-PEG-AQ(4-OH) in 7. As shown in (Fig. 6), probe 5 was shown to be an efficiently-activated substrate whereas compound 7 was unaffected by rh-legumain. It was recognised that tissue can be expected to possess protease activity other than that due to legumain; however, in the given substrates the N-terminus is protected from possible aminoprotease cleavage by the rhodamine capping group and the C-terminus is protected from carboxypeptidase action by the anthraquinone quencher capping group, conferring resistance to enzyme-mediated reactions at the termini. Potentially, other endoproteases may cleave the control peptide 7 and/or probe 5 and one identifiable class may be the cathepsin family of cysteine proteases that co-occur in the lysosomes, and which catalyse proteolysis of a wide range of peptide and protein substrates (Łęgowska et al. 2014). In order to provide further strong evidence that probe 5 is selectively cleaved by active legumain, incubations of probe 5 were conducted with recombinant cathepsin L, known to be secreted by lysosomal exocytosis by prostate cancer cells (Sudhan and Siemann 2013). Substrate 5 was not activated by cathepsin L (data available in Supplementary Information Section 11 Fig. S14).

Variation in relative fluorescence intensity with time upon incubation of Rho-Pro-Ala-Asn-PEG-AQ(4-OH) (probe 5; 10 µM) and Rho-Pro-Ala-Ala-PEG-AQ(4-OH) (control peptide 7; 10 µM) with rh-legumain (0.40 ng/μL) in legumain assay buffer (pH 5.0) λex 544 nm, λem 583 nm (n = 3)
Prostate Cancer Fresh-Frozen Tissue Patient-Derived Samples: Probe Activation Studies
Prostate tissue samples were homogenised and the resulting supernatants were treated with probe 5 in legumain MES assay buffer at pH 5.0. Representative data from tissue samples from two patients (PO1 and PO2) are presented in Fig. 7a and b, respectively.

a Variation in relative fluorescence intensity released with time for probe 5 (10 µM) with and without pre-incubation with iodoacetamide (1 mM, 15 min), and control peptide 7 (10 µM), upon incubation with tissue homogenate from prostate benign* tissue sample (PO1); λex 544 nm, λem 583 nm, (n = 3); *classified by histological examination. b Variation in relative fluorescence intensity released with time for probe 5 (10 µM) with and without pre-incubation with iodoacetamide (1 mM, 15 min), and control peptide 7 (10 µM), upon incubation with tissue homogenate from prostate cancer tissue sample (PO2); λex 544 nm, λem 583 nm, (n = 3)
In each example, a substantial and sustained increase in relative fluorescence intensity was observed upon incubation with the designed legumain substrate probe 5, whereas no increase occurred with the control non-legumain substrate 7 (wherein the critical Asn residue and cleavage ‘hotspot’ had been replaced by Ala).
Furthermore, in each case, the fluorescence release due to activation of probe 5 was prevented by pre-incubation with the legumain protease inhibitor iodoacetamide.
The tissue sample data closely mirrors that obtained for the activation of probe 5 by recombinant human legumain and is entirely consistent with the predicted response of a legumain-mediated activation.
A large increase in fluorescence upon activation of probe 5 was observed in the case of the malignant tumour sample (PO2) that was abrogated by the legumain inhibitor iodoacetamide. However, for the sample (PO1) classified benign by histological examination, a notable fluorescence release was obtained and similarly abrogated. Further studies are required to assess whether probe 5, as a potential complementary tool, may be able to discern low level malignancy compared to other methods, should expression of legumain prove to be a reliable and validated biomarker.
Conclusions
The fluorogenic probe 5 is an efficient FRET peptide substrate for cancer associated legumain (accessible by a simple and inexpensive synthesis) that affords strong restoration of fluorescence during incubation with rh-legumain. An advantage of probe 5 is that unlike previously reported substrates, the fluorescence intensity of the liberated fluorophore 4b (high fluorescence quantum yield ΦF 0.95) is unaltered over a wide range of pH (3–8), an important factor given the acidic pH environment of many solid tumours, making it an effective and sensitive reporter fluorophore. Furthermore, it is shown that probe 5 is taken up by live legumain-expressing PC3 prostate cancer cells and transported to the lysosomes where putative biomarker legumain is located. In preliminary investigations, the probe 5 approach in frozen tissue samples has afforded some promising results in correlation with reported histological findings, as we have seen a large increase in fluorescence intensity in the sample with foci of prostatic adenocarcinoma compared to the sample with benign prostate glands. In future, more studies using probe 5 need to be done in prostate tissue specimens (ongoing) to validate its application as a new supportive assay; these experiments will be necessary also to interrogate correlation of results with imaging and PSA levels.
Probe 5 represents a new viable biochemical tool for probing the activity of legumain in vitro with the potential to be used ex vivo to characterise prostate biopsy samples in a diagnostic setting in the pathology laboratory, to supplement current tumour characterisation.
References
Beija M, Afonso CAM, Martinho JMG (2009) Synthesis and applications of rhodamine derivatives as fluorescent probes. Chem Soc Rev 38:2410–2433. https://doi.org/10.1039/b901612k
Chen JM, Dando PM, Rawlings ND et al (1997) Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J Biol Chem 272:8090–8098. https://doi.org/10.1074/jbc.272.12.8090
Chen J, Dando P, Stevens R et al (1998) Cloning and expression of mouse legumain, a lysosomal endopeptidase. Biochem J 335:111–117
Chen J-M, Fortunato M, Stevens RAE, Barrett AJ (2001) Activation of progelatinase A by mammalian legumain, a recently discovered cysteine proteinase. Biol Chem 382:777–783. https://doi.org/10.1515/bchm.2001.382.5.777
Cook A, Le A (2006) The effect of solvent and pH on the fluorescence excitation and emission spectra of solutions containing fluorescein. J Phys Chem Lab 10:44–49
Dall E, Brandstetter H (2013) Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc Natl Acad Sci 110:10940–10945. https://doi.org/10.1073/pnas.1300686110
Dall E, Brandstetter H (2016) Structure and function of legumain in health and disease. Biochimie 122:126–150. https://doi.org/10.1016/j.biochi.2015.09.022
Dijkstra S, Hamid ARAH, Leyten GHJM, Schalken JA (2012) Personalized management in low-risk prostate cancer: the role of biomarkers. Prostate Cancer 2012:1–7. https://doi.org/10.1155/2012/327104
Edgington LE, Verdoes M, Bogyo M (2011) Functional imaging of proteases: recent advances in the design and application of substrate-based and activity-based probes. Curr Opin Chem Biol 15:798–805. https://doi.org/10.1016/j.cbpa.2011.10.012
Edgington LE, Verdoes M, Ortega A et al (2013) Functional imaging of legumain in cancer using a new quenched activity-based probe. J Am Chem Soc 135:174–182. https://doi.org/10.1021/ja307083b
Fischer A, Cremer C, Stelzer EHK (1995) Fluorescence of coumarins and xanthenes after two-photon absorption with a pulsed titanium–sapphire laser. Appl Opt 34:1989–2003
Gao M, Yu F, Lv C et al (2017) Fluorescent chemical probes for accurate tumor diagnosis and targeting therapy. Chem Soc Rev 46:2237–2271. https://doi.org/10.1039/C6CS00908E
Garland M, Yim JJ, Bogyo M (2016) A bright future for precision medicine: advances in fluorescent chemical probe design and their clinical application. Cell Chem Biol 23:122–136. https://doi.org/10.1016/j.chembiol.2015.12.003
Gawenda J, Traub F, Lück HJ et al (2007) Legumain expression as a prognostic factor in breast cancer patients. Breast Cancer Res Treat 102:1–6. https://doi.org/10.1007/s10549-006-9311-z
Halfon S, Patel S, Vega F et al (1998) Autocatalytic activation of human legumain at aspartic acid residues. FEBS Lett 438:114–118
Haugen MH, Boye K, Nesland JM et al (2015) High expression of the cysteine proteinase legumain in colorectal cancer—implications for therapeutic targeting. Eur J Cancer 51:9–17. https://doi.org/10.1016/j.ejca.2014.10.020
Hemminki K, Liu H, Heminki A, Sundquist J (2012) Power and limits of modern cancer diagnostics: cancer of unknown primary. Ann Oncol 23:760–764. https://doi.org/10.1093/annonc/mdr369
Jernigan FE, Lawrence DS (2013) A broad spectrum dark quencher: construction of multiple colour protease and photolytic sensors. Chem Commun 49:6728. https://doi.org/10.1039/c3cc42628a
Kembhavi AA, Buttle DJ, Knight CG, Barrett AJ (1993) The two cysteine endopeptidases of legume seeds: purification and characterization by use of specific fluorometric assays. Arch Biochem Biophys 303:208–213. https://doi.org/10.1006/abbi.1993.1274
Kikuchi M, Yamagishi T, Hida M (1982) Kinetic studies on the amination of leucoquinizarin. Bul Chem Soc Jpn 55:1209–1212
Lee MH, Sharma A, Chang MJ et al (2018) Fluorogenic reaction-based prodrug conjugates as targeted cancer theranostics. Chem Soc Rev 47:28–52. https://doi.org/10.1039/C7CS00557A
Łęgowska M, Wysocka M, Burster T et al (2014) Ultrasensitive internally quenched substrates of human cathepsin L. Anal Biochem 466:30–37. https://doi.org/10.1016/j.ab.2014.08.010
Leo A, Hansch C, Elkins D (1971) Partition coefficients and their uses. Chem Rev 71:525–616
Lewis C, Murdoch C (2005) Macrophage responses to hypoxia. Am J Pathol 167:627–635. https://doi.org/10.1016/S0002-9440(10)62038-X
Liu C, Sun C, Huang H (2003) Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res 63:2957–2964
Liu Y, Bajjuri KM, Liu C, Sinha SC (2012) Targeting cell surface alpha(v)beta(3) integrin increases therapeutic efficacies of a legumain protease-activated auristatin prodrug. Mol Pharm 9:168–175. https://doi.org/10.1021/mp200434n
Luo Y, Zhou H, Krueger J et al (2006) Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Investig 116:2132–2141. https://doi.org/10.1172/JCI27648
Mathieu M, Bogyo M, Caffrey C et al (2002) Substrate specificity of schistosome versus human legumain determined by P1-P3 peptide libraries. Mol Biochem Parasitol 121:99–105
May JP, Brown LJ, van Delft I et al (2005) Synthesis and evaluation of a new non-fluorescent quencher in fluorogenic oligonucleotide probes for real-time PCR. Org Biomol Chem 3:2534–2542. https://doi.org/10.1039/b504759e
McIlvaine T (1921) A buffer solution for colorimetric comparision. J Biol Chem 49:183–186
Murthy RV, Arbman G, Gao J (2005) Legumain expression in relation to clinicopathologic and biological variables in colorectal cancer. Clin Cancer Res 11:2293–2299. https://doi.org/10.1158/1078-0432.CCR-04-1642
Ohno Y, Nakashima J, Izumi M et al (2013) Association of legumain expression pattern with prostate cancer invasiveness and aggressiveness. World J Urol 31:359–364. https://doi.org/10.1007/s00345-012-0977-z
Plumb JA, Milroy R, Kaye SB (1989) Effects of the pH dependence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide-formazan absorption on chemosensitivity determined by a novel tetrazolium-based assay. Cancer Res 49:4435–4440
Rurack K (2008) Fluorescence quantum yields: methods of determination and standards. In: Resch-Genger U (ed) Standardization and quality assurance in fluorescence measurements I. Springer, Berlin, pp 101–145
Saini S (2016) PSA and beyond: alternative prostate cancer biomarkers. Cell Oncol 39:97–106. https://doi.org/10.1007/s13402-016-0268-6
Sapsford KE, Berti L, Medintz IL (2006) Materials for fluorescence resonance energy transfer analysis: beyond traditional donor-acceptor combinations. Angew Chem Int Ed 45:4562–4588. https://doi.org/10.1002/anie.200503873
Selvolini G, Marrazza G (2017) MIP-based sensors: promising new tools for cancer biomarker determination. Sensors 17:E718. https://doi.org/10.3390/s17040718
Sexton KB, Witte MD, Blum G, Bogyo M (2007) Design of cell-permeable, fluorescent activity-based probes for the lysosomal cysteine protease asparaginyl endopeptidase (AEP)/legumain. Bioorg Med Chem Lett 17:649–653. https://doi.org/10.1016/j.bmcl.2006.10.100
Shih J-, Yuan A, Chen J-W, Yang P (2006) Tumor-associated macrophage: its role in cancer invasion and metastasis. J Cancer Mol 2:101–106
Shirahama-Noda K, Yamamoto A, Sugihara K et al (2003) Biosynthetic processing of cathepsins and lysosomal degradation are abolished in asparaginyl endopeptidase-deficient mice. J Biol Chem 278:33194–33199. https://doi.org/10.1074/jbc.M302742200
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66:7–30. https://doi.org/10.3322/caac.21332
Smith RL, Åstrand OAH, Nguyen LM et al (2014) Synthesis of a novel legumain-cleavable colchicine prodrug with cell-specific toxicity. Bioorg Med Chem 22:3309–3315. https://doi.org/10.1016/j.bmc.2014.04.056
Stern L, Perry R, Ofek P et al (2009) A novel antitumor prodrug platform designed to be cleaved by the endoprotease legumain. Bioconjug Chem 20:500–510. https://doi.org/10.1021/bc800448u
Sudhan DR, Siemann DW (2013) Cathepsin L inhibition by the small molecule KGP94 suppresses tumor microenvironment enhanced metastasis associated cell functions of prostate and breast cancer cells. Clin Exp Metastasis 30:891–902. https://doi.org/10.1007/s10585-013-9590-9
Twomey JD, Brahme NN, Zhang B (2017) Drug-biomarker co-development in oncology—20 years and counting. Drug Resist Updates 30:48–62. https://doi.org/10.1016/j.drup.2017.02.002
Ueland F (2017) A perspective on ovarian cancer biomarkers: past, present and yet-to-come. Diagnostics 7:E14. https://doi.org/10.3390/diagnostics7010014
Valckenborgh Van, Mincher D, Di Salvo A et al (2005) Targeting an MMP-9-activated prodrug to multiple myeloma-diseased bone marrow: a proof of principle in the 5T33MM mouse model. Leukemia 19:1628–1633. https://doi.org/10.1038/sj.leu.2403866
Wang L, Chen S, Zhang M et al (2012) Legumain: a biomarker for diagnosis and prognosis of human ovarian cancer. J Cell Biochem 113:2679–2686. https://doi.org/10.1002/jcb.24143
Wang DL, Xiao C, Fu G et al (2017) Identification of potential serum biomarkers for breast cancer using a functional proteomics technology. Biomark Res 5:11. https://doi.org/10.1186/s40364-017-0092-9
Wu W, Luo Y, Sun C et al (2006) Targeting cell-impermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms. Cancer Res 66:970–980. https://doi.org/10.1158/0008-5472.CAN-05-2591
Yu K-K, Hou J-T, Li K et al (2015) A single design strategy for dual sensitive pH probe with a suitable range to map pH in living cells. Sci Rep 5:15540. https://doi.org/10.1038/srep15540
Zhang K, Bangma CH, Roobol MJ (2017) Prostate cancer screening in Europe and Asia. Asian J Urol 4:86–95. https://doi.org/10.1016/j.ajur.2016.08.010
Acknowledgements
We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University, Swansea, UK, for mass spectrometric analyses; and Maria Gauci, Edinburgh Napier University, for the development of HPLC methodology; the Histopathology Laboratory at Portsmouth Hospitals NHS Trust, Portsmouth, UK, for providing access to samples; Dr Siavash Rahimi, Mr Lemke Solomon and Mr Nagar Yoodhvir at Portsmouth Hospitals NHS Trust, Portsmouth, UK, for their clinical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Sunil Mathur, Agnes Turnbull, Iolia Akaev, Craig Stevens, Neerja Agrawal, Mridula Chopra and David Mincher declare that they have no conflict of interest.
Ethical Approval
The part of this study on two human tissue samples was recognised as a service development to test available equipment for further set-up of research projects; informed consent was not required as tissue material (consented for clinical-diagnostic procedures) was collected from surplus from histological frozen section techniques.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mathur, S., Turnbull, A., Akaev, I. et al. Design of a New Peptide Substrate Probe of the Putative Biomarker Legumain with Potential Application in Prostate Cancer Diagnosis Ex Vivo. Int J Pept Res Ther 26, 1965–1980 (2020). https://doi.org/10.1007/s10989-019-09994-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-019-09994-1