
Abstract
Context
Preserving functional connectivity is a conservation priority to secure the long-term viability of geographically dispersed subpopulations, such as the jaguar (Panthera onca) populations in Central America. However, managing connectivity in this region is limited due to the scarcity of local assessments of the connectivity between existing populations, some of which exhibit low levels of gene flow and genetic admixture.
Objectives
We selected the jaguar as a model species to understand how gene flow of large carnivores is shaped in a heavily human-impacted landscape in the Neotropics. We assessed the impact of landscape features and spatial scale on jaguar gene flow across northern Central America, predicted synoptic, landscape-wide functional connectivity across the region; and compared connectivity predictions based on models of gene flow, habitat suitability, and expert knowledge.
Methods
We employed genetic data based on 335 faecal samples collected from 72 individual jaguars. We parameterized gene flow resistance surfaces using linear mixed effects models and the maximum likelihood population-effects method. We evaluated nine landscape variables at six spatial scales, selecting the optimal scale and transformation for each variable according to univariate models and AIC. To predict jaguar gene flow, we developed multivariate models and implemented resistant kernels to forecast functional connectivity between jaguar populations across the study region under three dispersal distance scenarios. Furthermore, we compared the connectivity estimates based on gene flow against those based on habitat suitability and the corridors delineated by expert knowledge in the region.
Results
Low resistance to jaguar gene flow was associated with greater tree cover and vegetation, lower areas of built-up, and intermediate distances from water bodies. Notably, tree cover affected jaguar gene flow on a smaller scale compared to the rest of variables. Higher connectivity, indicated by lower resistance, was found within and around the largest jaguar conservation units (JCUs) such as the Reserva-de-Biosfera-Transfronteriza, Selva-Maya and Maya-Mountains. This contrasted with the smallest JCUs, like Sierra-Santa-Cruz and Sierra-de-las-Minas in eastern Guatemala, and Cordillera-Nombre-de-Dios in Honduras. Across the region, lower connectivity was observed in the Caribbean connection between eastern Guatemala and midwestern Honduras, as well as in the Honduran and Nicaraguan Miskito area. Models based on gene flow and habitat suitability were similar in their predictions of areas of high connectivity; however, the habitat suitability models predicted larger areas of low connectivity than did the gene flow models. Moreover, the expert knowledge corridors were consistent with areas of high and medium connectivity as predicted by the gene flow model.
Conclusions
Gene flow of jaguars is positively driven by the presence of forest and water sources, while human impact (built-up and non-vegetation areas) has a negative effect. Areas of lowest resistance largely correspond to the location of JCUs, which serve as crucial reservoirs of high-quality jaguar habitat within the region. While the largest JCU in the region displayed high connectivity, the low connectivity between the smallest JCUs (i.e. Sierra-Santa-Cruz, Sierra-de-las-Minas and Cordillera-Nombre-de-Dios) underscores the need for conservation attention in these areas. Conservation and management actions such as habitat loss-prevention/restoration and anthropogenic impact mitigation should be prioritized in the binational region of Guatemala-Honduras, a key connectivity bottleneck between the species’ northern and southern ranges. Similarly, attention is warranted in the Honduran-Nicaraguan Miskito area.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dispersal and gene flow between wildlife populations are the cornerstones for maintaining genetic diversity and ensuring the long-term viability of these populations (Bonte et al. 2012). Preserving the exchange of individuals and genes becomes more important than ever in the Anthropocene, as habitat loss and fragmentation threaten most of the world’s vertebrates (Haddad et al. 2015; Tilman et al. 2017; Díaz et al. 2019). In fragmented and degraded landscapes, animals often exhibit altered movement behaviours in response to land cover change and human development (Doherty and Driscoll 2018; Tucker et al. 2018), with consequences that can have cascading effects on populations and increase their extinction risk (Kramer-Schadt et al. 2005; Thatte et al. 2018). Decreased population persistence can occur via direct mortality resulting from unsuitable areas (e.g., anthropogenic barriers; Kramer-Schadt et al. 2004; Schwab and Zandbergen 2011), persecution associated with human-wildlife conflict exacerbation (Vickers et al. 2015), reduced gene flow due to dispersal barriers (Dixon et al. 2007), and ultimately, inbreeding depression and demographic decline (Roelke et al. 1993).
The permeability of landscapes to the movement of animals is defined as connectivity (Taylor et al. 1993). Landscape connectivity is the result of the individuals’ response to the landscape structure in terms of movement and survival ability (Tischendorf and Fahrig 2000). The functional aspect of connectivity can be better understood as successful gene flow occurring across the landscape (Baguette and Van Dyck 2007; Pe’er et al. 2011). Functional connectivity enables populations to adapt to changing conditions, thus its accurate assessment can help inform effective wildlife management strategies (Auffret et al. 2015; Schmidt et al. 2020).
Several models have been developed and adapted to provide insights into the connectivity of populations across various species. These models have been derived either directly from movement data (Richard and Armstrong 2010; Elliot et al. 2014), or indirectly from occurrence data (Mohammadi et al. 2022) or genetic data and associated patterns (Kimmig et al. 2020; Lecis et al. 2022; Portanier et al. 2022). While movement-based models provide detailed information about individuals’ real-time movement decisions, they require measuring animal dispersal at relevant spatial and temporal scales (Tesson and Edelaar 2013). Measuring animal dispersal is challenging, particularly for wide-ranging species of conservation concern, such as large carnivores, where studies require extensive spatial scales that can be difficult to implement due to economic and logistical limitations. Alternatively, occurrence data has been used to create habitat suitability models to link landscape structure to animal movement (Mohammadi et al. 2022), whereas genetic data has been utilized to associate landscape structure to gene flow patterns (Holderegger and Wagner 2008). Nonetheless, habitat selection and movement may be impacted by different factors, leading habitat models to potentially underestimate species’ movement capabilities through low suitability areas, as well as overlook infrequent dispersal events which can have strong implications for populations’ genetic structure and long-term persistence (Mateo-Sánchez et al. 2015). In comparison to occurrence and habitat suitability, genetic data and associated patterns provide further insights, as they reveal more information on how landscapes affect individual movement, dispersal, and breeding over multiple generations (Lowe and Allendorf 2010). Therefore, genetic data and associated analyses present valuable and cost-effective approaches to modelling gene flow and functional connectivity (Zeller et al. 2012; Tesson and Edelaar 2013), shedding light on population processes that cannot be exclusively assessed using movement data alone (e.g., effective population sizes [Ne], bottlenecks, inbreeding [FIS], population structure, contemporary versus historic genetic isolation; Willi et al. 2022).
Gene flow of several large carnivore populations is negatively impacted by human development, including that of pumas (Puma concolor; Ernest et al. 2014) and American black bears (Ursus americanus; Cushman and Lewis 2010) in North America, tigers and leopards in India (Panthera tigris and Panthera pardus; Thatte et al. 2020), Eurasian lynx in Europe (Lynx; Bull et al. 2016), and jaguars in Mexico and Brazil (Panthera onca; Roques et al. 2016). In this study, we used the jaguar as a model species to investigate how gene flow is shaped in a heavily human-impacted landscape in the Neotropics. We aim to map the anticipated functional connectivity of jaguar populations in northern Central America. The jaguar is an ideal model species for studying functional connectivity due to its large area requirements (Thompson et al. 2021), high level of conservation concern (91% of subpopulations show medium to high threat; de la Torre et al. 2017), and reduced genetic exchange in landscapes heavily influenced by human activities (Roques et al. 2016; Wultsch et al. 2016a). Source jaguar populations (Fig. 1, jaguar conservation units—JCUs) and corridors to facilitate their connectivity have been delineated across the species’ range with expert input (Rabinowitz and Zeller 2010; Petracca et al. 2018), aiming to improve jaguar population connectivity on a continental scale, however, these corridors are not based on impartial quantitative analyses.

Study region encompassed by Belize, Guatemala, and Honduras in Central America, showing sampling locations and habitat suitability for jaguars. Main map: location of the collected scat samples (light blue and grey circles), sampling size n (number of individuals) of each sampling group, Jaguar Conservation Units (black dashed borders), and jaguar habitat suitability (areas of high habitat suitability in green, and low suitability in red; Calderón et al. 2022). Insert in right upper corner: Genetic structure of jaguar populations, as reported by Wultsch et al. (2016a, b), within the study region, illustrating genetic differentiation existing among jaguars from Belize, Guatemala, and Honduras, despite their close geographical proximity. The pie charts represent the genetic clusters corresponding to groups of jaguar individuals studied across sampling sites. The colours within these pie charts represent the proportions of membership for each of these sampled groups
Jaguar populations have shown moderate to high levels of genetic diversity and a lack of strong population structure across most of their range (Eizirik et al. 2001; Ruiz-Garcia et al. 2006; Roques et al. 2014). However, there are small, isolated jaguar populations existing in fragmented areas within the South American range, which currently face bottlenecks, loss of genetic diversity, and heightened genetic differentiation due to pronounced signals of anthropogenic-driven drift (Haag et al. 2010; Lorenzana et al. 2020). Likewise, indications of restricted gene flow and anthropogenic impact, particularly between Guatemala and Honduras, were identified in a previous study (Wultsch et al. 2016a).
We focus this study in northern Central America for three primary reasons. Firstly, this region holds the largest stronghold of jaguars after the Amazon (Rabinowitz and Zeller 2010). Secondly, prime jaguar habitat in this region faces serious threats from land use change (Powers and Jetz 2019) and deforestation (Olsoy et al. 2016). And lastly, jaguar populations in this region already exhibit low levels of gene flow and heightened genetic differentiation (Wultsch et al. 2016a). Previous research indicates that while gene flow is high among jaguars in the Selva-Maya (northern Guatemala and central Belize), geographically close jaguar populations in the Selva Maya and Honduras display differences in genetic structure (Wultsch et al. 2016a). Therefore, it is likely that jaguar gene flow may be impacted not only by geographic distance but also by the dynamic landscape changes experienced across the region.
To our knowledge, this is the first study to assess jaguar functional connectivity using empirical data at a regional extent. Previous research in the region has been based solely on literature or local/expert input, either to assess structural connectivity at a large regional scale (Rabinowitz and Zeller 2010; Petracca et al. 2018), or functional (de la Torre et al. 2017) and habitat connectivity (Menchaca et al. 2019) at small, local scales. Furthermore, the present research has the overarching goal to respond to regional jaguar conservation priorities, by “genetic ground-truthing” (assess functional connectivity) of dispersal corridors for jaguars, to improve and maintain population interconnectedness, particularly in northern Central America (Wultsch et al. 2016a; de la Torre et al. 2017). Here, we determine jaguar gene flow using microsatellite data and address four main objectives: (i) identify landscape variables that facilitate or hinder gene flow between jaguar populations in northern Central America, (ii) assess functional connectivity of jaguar populations in the study landscape, (iii) compare spatial predictions of functional connectivity based on gene flow versus expert input (Rabinowitz and Zeller 2010; Petracca et al. 2018) and jaguar habitat suitability data (Calderón et al. 2022); and (iv) identify priority areas for corridor planning that promote genetic exchange among Central American jaguar populations.
We hypothesized that jaguar gene flow would be positively influenced by the availability of shelter habitat (i.e. sites for resting, nursing, protection from humans, and cover for ambush hunting), such as forest cover and water sources (de la Torre and Rivero 2019; Nuñez-Perez and Miller 2019), as well as areas with high net primary productivity (as a proxy for prey biomass). Conversely, we anticipated that gene flow would be negatively impacted by high elevation, extensive human development, areas devoid of vegetation (non-vegetation zones) and the presence of roads (Haag et al. 2010; Valdez et al. 2015; Kantek et al. 2021; Thompson et al. 2021).
Methods
Study area
Central America is a biodiversity hotspot (Myers et al. 2000), serves as the primary stronghold for jaguar populations following the Amazon (trinational ‘Selva-Maya’), and connects the two largest jaguar populations in the region (‘Selva-Maya’ and ‘Reserva-Biosfera-Transfronteriza’; Fig. 1). This region is characterized by a highly diverse pattern of land uses and a notably higher mean human density of 94 ind./km2compared to other Latin American countries, (World Bank 2020). The landscape predominantly consists of broadleaved evergreen and deciduous trees (61%) along with a mosaic of natural vegetation with croplands (21%). The remaining land uses encompass a diverse mix, including croplands (6%), secondary growth forests (4%), grasslands (3%), wetlands (1%) and urban areas (0.5%, ESA 2015). Due to land use change, Central America holds the highest predicted loss of suitable vertebrate’s habitat in the continent (Powers and Jetz 2019), and the highest deforestation rates and the forest degradation worldwide proportionally (Redo et al. 2012). We focus our study on northern Central America, specifically Belize, Guatemala, and Honduras (Fig. 1).
Genetic data and measures
We worked with a dataset of 335 jaguar faecal samples that had been previously collected across northern Central America during the period of 2003–2012 (Wultsch et al. 2016a), timespan within a generation length in our region (Harmsen et al. 2017). Sampling sites were predominantly located in lowland regions characterized by subtropical and tropical climates and a diverse range of land cover types within Belize, Guatemala, and Honduras. The samples were opportunistically collected in the field (Fig. 1) and genotyped using 12 microsatellite loci (Wultsch et al. 2016a, b; Menchaca et al. 2019; Appendix S1). Seventy-two jaguar individuals were identified, comprising 59 males, 3 females, and 10 individuals with unknown sex. Population genetic summary statistics (Appendix S2), gene flow and genetic structure assessments for these sites were published by Wultsch et al. (2016a, b). To assess genetic differentiation between jaguar populations across northern Central America, we calculated pairwise genetic distances based on Factorial Correspondence Analysis (FCA; She et al. 1987) in two steps. First, we performed FCA on a multi-contingency table of the microsatellite genotypes with the software GENETIX v. 4.05.2 (Belkhir et al. 2001). Second, we generated a distance matrix by computing Euclidean distances between the first two FCA axes (the only significant) using the R package ‘ecodist’ v.2.0.9 (Goslee and Urban 2007).
Landscape covariates, resistance surface generation
We selected nine environmental and anthropogenic covariates that influence jaguar habitat use, density, movement, and potentially gene flow (Jȩdrzejewski et al. 2018; Thompson et al. 2021; Calderón et al. 2022). These covariates include distance from water bodies, distance from rivers, distance from roads, elevation, ruggedness, net primary productivity, percentage of built-up, percentage of non-vegetation, and percentage of tree cover (Table S3). We projected all datasets to the Universal Transverse Mercator coordinate system (EPSG: 32,616) and resampled to a 1000 m resolution.
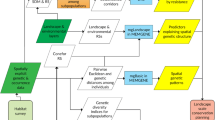
We modelled genetic distances between individuals as a function of resistance and Euclidean distance by following the method provided by Zeller et al. 2023 (Fig. 2, Appendix S4): We first assessed the scale of effect of each of our variables (Jackson and Fahrig 2015; Mcgarigal et al. 2016) on jaguar gene flow, given that gene flow processes and functional connectivity are dependent on spatial scale (Cushman and Landguth 2010; Cushman et al. 2016). To identify the optimal scale for each variable, we smoothed each variable raster using a Gaussian kernel and varied its bandwidth across six scales: 1000 m; 3000 m; 6000 m; 8000 m; 16,000 m; 32,000 m. At each scale, landscape variables were transformed into a gene flow resistance surface hypothesis, converting the values of the raw variables into resistance values using seven transformational forms using fixed values (Fig. 2a; Appendix S5), namely linear, monomolecular convex and monomolecular concave functions, in both their positive and negative forms, as well as the inverse Ricker transformation (Peterman 2018). Lastly, resistance surfaces were re-scaled from 1 to 100 using the raster calculator in ArcGIS 10.8. (ESRI 2022).

Conceptual flow diagram for analysis of jaguar gene flow and functional connectivity in northern Central America. For details on the methods, refer to Appendix S4
To optimize landscape resistance values and evaluate how landscape features affect jaguar gene flow in our study region, we evaluated scenarios of isolation by distance (IBD; Wright 1942) and isolation by resistance (IBR; McRae 2006; Peterman et al. 2014). To this end, we identified the optimal scale and transformation for each covariate by running univariate models across all scales and transformational forms. We evaluated these models using Akaike’s Information Criterion corrected for small sample sizes AICc (Burnham and Anderson 2002) and retained the model with the lowest AICc value for each covariate (Fig. 2a). We then combined all non-correlated covariates at their optimal scale and transformation to create all plausible multi-variable, multi-scale models of resistance. Correlations among predictors were assessed by running partial Mantel correlations between the covariates’ resistance distance matrices using the ‘vegan’ R-package v.2.6-2 (Oksanen et al. 2022). Non-correlated covariates (Pearson’s moment correlation; |r|< 0.6) at their optimal scale and transformation were combined to create multi-variable, multi-scale models of resistance (Fig. 2b, Table S8). For these multivariate models, we summed all variables in each model to create a single composite resistance surface across which resistance distances were calculated (i.e. for each multivariate model there is one composite resistance surface). We did so because animals do not experience elements separately while moving through a landscape, thus composite surfaces better reflect animal’s responses to aggregated landscapes (Peterman and Pope 2021). All composite surfaces were rescaled from 1 to 100. Functional connectivity of jaguar populations was assessed using cumulative resistant kernels (Compton et al. 2007), a synoptic modelling approach that predicts and maps expected functional connectivity as a continuous surface, accounting for species’ dispersal abilities (Fig. 2c). Resistant kernels were calculated using source locations obtained by sampling the top resistance surface with the ‘spatially balanced point tool’ in ArcGIS v.10.8.0 (ESRI 2022). We applied this last step to the connectivity surfaces based on gene flow, and for comparison, to a recently developed habitat suitability model (Calderon et al. 2022). Given the lack of information on dispersal distances of jaguars in Central America, we developed three connectivity scenarios based on estimates of dispersal distances of females (75 km; Bernal-Escobar et al. 2015), males (250 km; Bernal-Escobar et al. 2015; Wultsch et al. 2016b), as well as the maximum average dispersal distance based on Sutherland et al. (2000) and the average body size reported for male jaguars in Central America (90 km. for an average weight of 56 kg; Aranda 1992; de la Torre and Rivero 2017; Nuñez-Perez and Miller 2019). For further details on the methods, refer to Appendix S4.
Lastly, we compared the connectivity predictions obtained using our genetic model (this study) with two other models: (i) the habitat suitability model described earlier (raster data; Calderón et al. 2022), and (ii) the model derived from expert input (vector data; Rabinowitz and Zeller 2010; Petracca et al. 2018). For the first comparison, we calculated the difference between the inverse habitat suitability-based (i.e. 1—Habitat Suitability) and gene flow-based connectivity surfaces with the ‘raster calculator’ in ArcGIS v.10.8.0 (ESRI 2022). For the second comparison, we used our gene flow connectivity surface (this study) and compared the values of 200 randomly selected points located within the expert-based corridors (Rabinowitz and Zeller 2010; Petracca et al. 2018) with 1,701 randomly selected points spanning the jaguar extent of occurrence within our study region (see Fig S7). We compared these two random samples of gene flow connectivity points using a two-sided Mann–Whitney U test with R package ‘stats’ v.4.0.4. We expected that the sample from the expert-based corridors would exhibit a higher median connectivity compared to the random sample from across the full extent of our study region.
Results
Landscape resistance and connectivity modelling
Resistance to jaguar gene flow decreased with higher levels of tree cover and primary productivity, while it increased with higher values of built-up areas, non-vegetation, elevation, ruggedness, and proximity to roads and rivers (Fig. S6). Notably, gene flow resistance was lowest at intermediate distances from water bodies, progressively increasing with greater distances from these water sources, and being highest in close proximity to them. Tree cover and ruggedness had the smallest scale of effect for jaguars (16,000 m; Table 1), while the other variables had a scale of effect of 32,000 m. All univariate models outperformed the model of isolation by distance (IBD; Euclidean distance).
All competitive resistance surfaces (i.e. within Δ4 AIC) included tree cover and built-up area. The top composite resistance surface additionally included distance from water bodies and non-vegetation (AICc = − 1084.168, BIC = − 1054.961) and explained 83% of the variance observed in the genetic differences between individuals (mR2 = 0.69, cR2 = 0.83; Table 2; complete model list in Appendix S6; results of Partial Mantel tests in Table S7). Therefore, we proceed to describe and discuss the results derived from this top model.
Lower resistance was found within and around the largest JCU in the region (i.e. Reserva-de-Biosfera-Transfronteriza), in comparison to the smallest JCUs (i.e. eastern-Guatemala: Sierra-Santa-Cruz and Sierra-de-las-Minas; and Honduras: Cordillera-Nombre-de-Dios; Fig. 1a, 3). Across all dispersal distance scenarios (Fig. 1b–d, 3), connectivity was highest around the JCU Reserva-de-Biosfera-Transfronteriza, lower along the Caribbean edge of the Honduran-Nicaraguan Miskito region (northeastern border of Honduras and Nicaragua); and lowest across all the Pacific area in the south of the study region. Only in the 250 km scenario, high connectivity expanded from the Reserva-de-Biosfera-Transfronteriza towards the Caribbean coast between eastern Guatemala and midwestern Honduras (Fig. 1d, 3).

Resistance (a) and functional population connectivity surfaces based on dispersal distance scenarios (b–d) for jaguars in northern Central America obtained using genetic data (1. Gene flow) and habitat use data based on the inverse of the habitat suitability map of Calderón et al. 2022 (2. Habitat suitability). Red polygons depict expert-based delineated corridors proposed by Petracca et al. (2018) and Rabinowitz and Zeller (2010). Text labels represent the following JCUs: Selva Maya (SM), Maya Mountains (MM), Reserva de Biosfera Transfronteriza (RBT)
The resistance and kernel connectivity surfaces obtained from the inverse of the jaguar habitat suitability map showed lowest resistance (Fig. 2a, 3) and higher connectivity in the east of the study region across all dispersal distance scenarios (Fig. 2a–d, 3). Predictors indicated very limited connectivity for most areas in western Honduras as well as eastern and central Guatemala. The highest connectivity was primarily predicted in eastern Honduras and northeastern Nicaragua, and to a lesser degree between Guatemala and Belize; these areas encompass the three largest JCUs in the region (i.e. Reserva-de-Biosfera-Transfronteriza, Selva-Maya and Maya-Mountains).
Comparison of spatial predictions of functional connectivity: gene flow vs habitat suitability vs expert input
Overall, the gene flow model displayed more distinct movement pathways throughout the region and across all dispersal scenarios, even in areas of lower predicted connectivity, such as eastern Guatemala and western Honduras. Across various dispersal scenarios, there was consistency between the gene flow and habitat suitability-based connectivity predictions around the largest JCU in the region (i.e. Reserva-de-Biosfera-Transfronteriza), as well as some areas in Guatemala and Belize (i.e. Selva-Maya and Maya-Mountains; Fig. 4a–c: turquoise areas). However, the habitat suitability-based model predicted much lower levels of connectivity in comparison to gene flow (Fig. 4a–c: blue areas) across dispersal scenarios and across the study region, particularly outside JCUs and within the region between Guatemala and Honduras. In the latter case, the habitat suitability data alone did not reveal clear connectivity pathways. Lastly, the habitat suitability model predicted higher connectivity than gene flow along the Caribbean edge of the Honduran-Nicaraguan Miskito, in Guatemala’s Lacandon region, the northern tip of Belize, and central Honduras (Fig. 4a–c: green areas).

Difference between jaguar population connectivity estimates based on habitat use versus gene flow across a 75 km, b 90 km, and c 250 km dispersal distance scenarios. Values range from positive, for areas where habitat suitability estimated higher connectivity than gene flow (green), zero for areas where habitat suitability and gene flow predicted similar connectivity (turquoise), and negative for areas where gene flow predicted higher connectivity than habitat suitability (blue)
Connectivity values within the corridors for jaguar movement delineated based on expert knowledge (Rabinowitz and Zeller 2010; Petracca et al. 2018) were significantly higher than those calculated across the jaguar range in the study region across all dispersal scenarios (W75km = 170,538, P75km = 0.039; W90km = 173,216, P90 km = 0.014; W250km = 191,685, P250 km = < 0.0001), evidencing that expert-based corridors are located within the areas with higher predicted gene flow and connectivity (Fig. 3). However, our genetic model revealed alternative areas of connectivity, which are not included in the expert-based corridor delineation. Specifically, in the region between Honduras and Guatemala, our genetic model identified higher gene flow-based connectivity extending south of the expert-based corridor along the Caribbean Coast between these two countries.
Discussion
Utilizing recent advancements in resistance surface estimation and conducting genetic sampling across international boundaries, our study stands as the first to model jaguar gene flow and connectivity across a large, heterogeneous, regional extent of conservation concern in Central America. Our top gene flow resistance model predicted lowest resistance in areas with higher tree cover and vegetation, lower built-up areas, and intermediate distances from water sources. These areas of lowest resistance closely align with the locations of the JCUs, which constitute the main reservoirs of high-quality jaguar habitat in the region (Calderón et al. 2022). Tree cover exhibited a smaller scale effect on jaguar gene flow in comparison to the other variables. This might imply that tree cover may be mediating gene flow most strongly through local breeding or short-distance dispersal (as exhibited by philopatric female jaguars; Kantek et al. 2021), while built-up areas, non-vegetation, and water sources may play a role in long-distance dispersal. Similar findings have been reported for cougars in North America (Zeller et al. 2023).
Jaguar gene flow appears to be primarily driven by availability of shelter habitat. However, the high resistance posed by built-up and non-vegetation areas to gene flow may also encompass the effects of direct human-jaguar interactions that are prevalent in anthropogenic landscapes. In such landscapes, jaguar populations exhibit lower levels of gene flow (Roques et al. 2016) and increased human-caused mortality due to human-related causes, including hunting (Jędrzejewski et al. 2017a, b; Arias et al. 2021), vehicle collisions (Srbek-Araujo et al. 2015), and retaliatory killing (Garcia-Alaniz et al. 2010; Moreno et al. 2015; Jędrzejewski et al. 2017a, b); the latter being a significant driver of jaguar extirpation (Jędrzejewski et al. 2017a, b; Villalva and Palomares 2022). The impact of human-caused mortality on population viability can be significantly large, especially when it disproportionally affects important demographic groups (i.e. juveniles), and when limited connectivity restricts demographic rescue (Lamb et al. 2020). Therefore, securing connectivity between jaguar populations in anthropogenic landscapes is currently a pivotal conservation priority for the populations’ long-term viability (de la Torre et al. 2016).
Our model-based predictions across all dispersal scenarios showed similar general patterns and produced plausible scenarios of jaguar population connectivity. Nevertheless, we believe that those based on 75 km and 90 km dispersal distances are the most plausible for our study region, as they were most consistent with the current extent of jaguar occurrence (Quigley et al. 2017) and with previous studies focused on landscape connectivity and gene flow within our study area (Wultsch et al. 2016a; Petracca et al. 2018). Accordingly, we believe the results from the 75 km and 90 km dispersal distance scenarios are robust enough to inform current conservation efforts, and we refer specifically to these scenarios from here on unless otherwise stated.
Functional connectivity for jaguar populations was found to be concentrated within and around the JCUs in the region, decreasing further from these areas, which is likely caused by the high concentration of source points selected in those regions of high habitat quality. The highest connectivity was observed along and around the biggest JCU (i.e. Reserva-Transfronteriza in Nicaragua; Fig. 1), followed by the next largest JCUs in Belize and northern Guatemala (i.e. Selva-Maya and Maya-Mountains). These findings support previous research on jaguar genetic diversity and population structure in the region (Wultsch et al. 2016a). Areas of lower population connectivity were predicted along the Caribbean coast between Guatemala and Honduras, as well as in the Honduran-Nicaraguan Miskito region. The former area is consistent with high human development and forest loss (Aide et al. 2013; Olsoy et al. 2016), high loss of suitable jaguar habitat (Calderón et al. 2022), extremely low jaguar occurrence (Hoskins et al. 2018), and jaguar populations exhibiting low levels of genetic diversity (Wultsch et al. 2016a). Similarly, the Honduran-Nicaraguan Miskito region has also undergone habitat degradation (Mora et al. 2016), resulting in low densities of jaguars (Gonthier and Castañeda 2013) and high levels of retaliatory killing of jaguars due to cattle predation (Chinchilla et al. 2022). Our findings in the Miskito area are further supported by previous studies of landscape connectivity based on camera trap records in the region (Hernández Potosme 2019).
Our study builds upon previous connectivity assessments in the region in two significant ways. First, it employs a rigorous statistical modelling approach that explicitly takes into account the spatial scale dependence in jaguar-landscape interactions. This approach enables us to identify the appropriate functional scale for each landscape predictor studied. This step often overlooked, is critical as it ensures robust inference and improves the effectiveness of management inputs that support habitat and population connectivity efforts on the ground (Jackson and Fahrig 2015; Krishnamurthy et al. 2016). Second, we provide spatially explicit, synoptic connectivity maps for jaguar populations in the region, accounting for the species’ dispersal ability.
We believe that our model-based predictions are robust enough to support current and future conservation and management efforts for this species of conservation concern, living in a hotspot of range loss for vertebrate species (Powers and Jetz 2019). However, there are three potential sources of uncertainty that need consideration. First, despite our extensive sampling efforts and research collaborations across Central America, we were unable to obtain samples from the entire extent of jaguar occurrence in our study region. We collected data on 72 individual jaguars primarily from lowland subtropical and tropical areas. Nonetheless, our individual-based approach for analysis (i.e. analysing all individuals within sampling sites instead of a selection of individuals, see: Seaborn et al. 2019) remains a robust method for studying connectivity in mobile species, even when dealing with samples as small as < 5 individuals per sampling site (Prunier et al. 2013). Additionally, the microsatellites used in this study exhibited moderate to high polymorphism between populations (Wultsch et al. 2016a), enhancing their ability to detect population differentiation and structure (Pritchard and Rosenberg 1999). Therefore, while our sample size might seem small, it allowed us to effectively examine genetic patterns in jaguar populations within this region. Nevertheless, the genetic differentiation patterns reported here may be influenced by our clustered sampling design and the data gaps in central Guatemala and southern Honduras. Hence, we emphasize the importance of conducting continuous genetic sampling in these areas in the future to provide a more comprehensive understanding of functional connectivity patterns across this region. Second, due to our limited sample size, we were unable to validate our model using internal cross-validation folds (Row et al. 2017) or using an external dataset. While this prevented us from quantitatively assessing the uncertainty of our model predictions, we are confident that we have addressed most potential sources of uncertainty by: (i) using resistance surfaces that correspond to ecological processes (Peterman et al. 2014) and are parameterized with empirical data (Clevenger et al. 2002; Shirk et al. 2010; Cushman et al. 2013), (ii) using genetic data that focuses solely on movements relevant for populations’ viability (i.e. successful breeding), representing the long-term average of effective dispersal without being limited to observational periods (Spear et al. 2010); (iii) accounting for spatial-scale dependence and species’ dispersal ability in the assessment of connectivity; and (iv) considering different scenarios of dispersal distance based on allometric scaling and field data (Sutherland et al. 2000; Bernal-Escobar et al. 2015; Wultsch et al. 2016b). Third, we were also unable to explicitly account for human-caused mortality, a decisive factor impacting gene flow resistance with important implications for functional connectivity. There is scarce data on anthropogenic jaguar mortality across the species range (Altrichter et al. 2006; Jędrzejewski et al. 2017a, b; Romero-Muñoz et al. 2019), especially in Central America (Moreno et al. 2015). We rely on built-up area and tree cover maps to encompass the effects of this anthropogenic-driven mortality in the absence of appropriate data, supported by the fact that jaguar extirpation is mostly driven by anthropogenic activities, such as deforestation, cattle breeding, and retaliatory killing (Jędrzejewski et al. 2017a, b).
We emphasize two key findings arising from the connectivity patterns derived from gene flow, habitat suitability, and expert-input data, which have important implications for guiding future work. First, we highlight that gene flow connectivity differs from habitat connectivity and one may not be used as a proxy for the other (Sartor et al. 2022), since habitats suitable for permanent establishment do not necessarily correspond to those facilitating dispersal (Reding et al. 2013; Mateo-Sánchez et al. 2015; Keeley et al. 2017). Regarding expert input, we found a general alignment between the proposed expert knowledge-based corridors and our gene flow-predicted high and medium connectivity areas. We highlight that empirical data tend to offer more optimal parameterization of environmental variables in comparison to expert input (Clevenger et al. 2002; Shirk et al. 2010; Cushman et al. 2013). However, the expert-input corridors for jaguar movement evaluated in this study have been systematically updated using field data and robust inference methods (Zeller et al. 2011; Petracca et al. 2018) subsequent to their first delineation (Rabinowitz and Zeller 2010). Accordingly, we agree that expert-input, supported by field data and robust statistical methods proves valuable for synthesizing knowledge about complex animal-habitat relationships, particularly in situations with limited empirical data available.
Our second recommendation for future research is that the effect of landscape covariates on gene flow may be assessed across a wide range of multiple spatial scales (Jackson and Fahrig 2015; Moraga et al. 2019). We found the strongest support for broader scale effects, which varied depending on the type of landscape feature. This is not surprising, as the spatial scales of gene flow are typically much larger than those for abundance or occurrence (Jackson and Fahrig 2014), with the latter two being the usual inputs for habitat suitability models. Future research incorporating spatial-scale dependence across the species’ range will yield important management implications for regional jaguar conservation and management. Such studies will provide insights on whether jaguar-landscape interactions are dependent on the environmental context (see Jackson and Fahrig 2015) or whether they can be generally applied to management actions across the entire geographical range of the species.
Conservation implications
Our jaguar model highlights key areas for population connectivity, while informing concrete conservation actions for securing the long-term persistence of the species in the region. Among the highlighted areas, the binational area between Guatemala-Honduras as well as the Miskito region may be considered a priority for management actions. The former region, identified as having low connectivity by both the habitat suitability and expert-based models, not only holds the smallest JCU in the region but also serves as a critical connection point between jaguar populations from North America and the rest of the continent. In the short and medium terms, maintaining jaguar genetic connectivity in this region could involve preventing habitat loss and degradation and implementing restoration efforts, along with mitigating anthropogenic impacts. One approach to achieving these goals is the adoption of sustainable land management practices compatible with jaguar conservation, such as community managed reserves (Mena et al. 2020) or forestry concessions (Tobler et al. 2018) in the buffer zones of the JCUs. This would be especially useful for the smallest JCUs in eastern Guatemala and Honduras, preventing the loss of the last jaguar source populations in this area. Similarly, this strategy would apply to the Miskito Caribbean slope, avoiding the expansion of habitat degradation towards the region’s largest JCU, the Reserva-de-Biosfera-Transfronteriza. Additionally, to reduce habitat loss between Honduras and Guatemala, there is a significant opportunity for governments, projects, and organizations to support community-based initiatives that seek to officially declare watersheds and corridors in this binational area. Furthermore, mitigation of anthropogenic pressure could be achieved by two key actions: (a) the implementation of integral wildlife-human coexistence approaches (e.g. König et al. 2020) that promote jaguar-human coexistence and minimize jaguar anthropogenic mortality; and (b) fostering deeper engagement of local communities in conservation goals through community-based projects (e.g. community monitoring; Valsecchi et al. 2022) that promote the social acceptance of conservation efforts (e.g. inclusive participation and decision-making in conservation solutions; Volski et al. 2021). In the long term, however, achieving effective carnivore conservation in this region of increasing human development will require the implementation of holistic conservation approaches, grounded not only in wildlife-human coexistence principles and participatory bottom-up approaches but also in socio-environmental justice for the local communities inhabiting these landscapes (Büscher and Fletcher 2020; Toncheva et al. 2022).
Data availability
In the Supplementary information we provide the microsatellite data (Table S1) as well as a sample code of our core analysis for running the Maximum likelihood population effects mixed model (Appendix S9).
References
Aide TM, Clark ML, Grau HR, López-Carr D, Levy MA, Redo D, Bonilla-Moheno M, Riner G, Andrade-Núñez MJ, Muñiz M (2013) Deforestation and reforestation of Latin America and the Caribbean (2001–2010). Biotropica 45(2):262–271
Altrichter M, Boaglio G, Perovic P (2006) The decline of jaguars Panthera onca in the Argentine Chaco. Oryx 40(3):302–309
Aranda JM (1992) El jaguar (Panthera onca) en la Reserva Calakmul, Mexico: morfología, hábitos alimentrarios y densidad de población. In: Fudeci, Clemente R (eds) Felinos de Venezuela, Biología, Ecología y Conservacón. Raul Clemente Editores, Valencia, pp 235–274
Arias M, Hinsley A, Nogales-Ascarrunz P, Carvajal-Bacarreza PJ, Negroes N, Glikman JA, Milner-Gulland EJ (2021) Complex interactions between commercial and noncommercial drivers of illegal trade for a threatened felid. Anim Conserv 24(5):810–819
Auffret AG, Plue J, Cousins SAO (2015) The spatial and temporal components of functional connectivity in fragmented landscapes. Ambio 44(1):51–59
Baguette M, Van Dyck H (2007) Landscape connectivity and animal behavior: functional grain as a key determinant for dispersal. Landsc Ecol 22(8):1117–1129
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2001) GENETIX 4.05, logiciel sous Windows TM pour la genetique des populations (4.05). nome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II,. https://kimura.univ-montp2.fr/genetix/
Bernal-Escobar A, Payan E, Cordovez JM (2015) Sex dependent spatially explicit stochastic dispersal modeling as a framework for the study of jaguar conservation and management in South America. Ecol Model 299(March):40–50
Bonte D, Van Dyck H, Bullock JM, Coulon A, Delgado M, Gibbs M, Lehouck V, Matthysen E, Mustin K, Saastamoinen M, Schtickzelle N, Stevens VM, Vandewoestijne S, Baguette M, Barton K, Benton TG, Chaput-Bardy A, Clobert J, Dytham C et al (2012) Costs of dispersal. Biol Rev 87(2):290–312
Bull JK, Heurich M, Saveljev AP, Schmidt K, Fickel J, Förster DW (2016) The effect of reintroductions on the genetic variability in Eurasian lynx populations: the cases of Bohemian-Bavarian and Vosges-Palatinian populations. Conserv Genet 17(5):1229–1234
Burnham KP, Anderson DR (2002) Model Selection and multimodel inference—a practical information-theoretic approach, 2nd edn. Springer, New York
Büscher B, Fletcher R (2020) The conservation revolution: radical ideas for saving nature beyond the anthropocene. Verso Trade, London
Calderón AP, Louvrier J, Planillo A, Araya-Gamboa D, Arroyo-Arce S, Barrantes-Núñez M, Carazo-Salazar J, Corrales-Gutiérrez D, Doncaster CP, Foster R, García MJ, Garcia-Anleu R, Harmsen B, Hernández-Potosme S, Leonardo R, Trigueros DM, McNab R, Meyer N, Moreno R et al (2022) Occupancy models reveal potential of conservation prioritization for Central American jaguars. Anim Conserv 25:680–691
Chinchilla S, van der Berghe E, Polisar J, Arevalo C, Bonacic C (2022) Livestock—carnivore coexistence: moving beyond. Animals 12:1–15
Clevenger AP, Wierzchowski J, Chruszcz B, Gunson K (2002) GIS-generated, expert-based models for identifying wildlife habitat linkages and planning mitigation passages. Conserv Biol 16(2):503–514
Compton BW, McGarigal K, Cushman SA, Gamble LR (2007) A resistant-kernel model of connectivity for amphibians that breed in vernal pools. Conserv Biol 21(3):788–799
Cushman SA, Landguth EL (2010) Scale dependent inference in landscape genetics. Landsc Ecol 25(6):967–979
Cushman SA, Lewis JS (2010) Movement behavior explains genetic differentiation in American black bears. Landsc Ecol 25(10):1613–1625
Cushman SA, Mcrae B, Adriaensen F, Beier P, Shirley M, Zeller K (2013) Biological corridors and connectivity. Key Top Conserv Biol 2:384–404
Cushman SA, Elliot NB, Macdonald DW, Loveridge AJ (2016) A multi-scale assessment of population connectivity in African lions (Panthera leo) in response to landscape change. Landsc Ecol 31(6):1337–1353
de la Torre JA, Rivero M (2017) A morphological comparison of jaguars and pumas in southern Mexico. Therya 8(2):117–122
de la Torre JA, Rivero M (2019) Insights of the movements of the jaguar in the tropical forests of Southern Mexico. In: Reyna-Hurtado R, Chapman CA (eds) Movement ecology of neotropical forest mammals: focus on social animals. Springer, Cham, pp 217–241
de la Torre JA, Ceballos G, Chávez C, Zarza H, Medellín RA (2016) Prioridades y recomendaciones ¿Cuáles son las necesidades para el jaguar en los próximos años? In: Medellín R, de la Torre A, Zarza H, Chávez C, Ceballos G (eds) El jaguar en el siglo XXI. La perspectiva continental. Ediciones Científicas Universitarias, pp 495–505
de la Torre JA, Núñez JM, Medellín RA (2017) Habitat availability and connectivity for jaguars (Panthera onca) in the Southern Mayan Forest: conservation priorities for a fragmented landscape. Biol Conserv 206:270–282
Díaz S, Settele J, Brondízio ES, Ngo HT, Agard J, Arneth A, Balvanera P, Brauman KA, Butchart SHM, Chan KMA, Lucas AG, Ichii K, Liu J, Subramanian SM, Midgley GF, Miloslavich P, Molnár Z, Obura D, Pfaff A et al (2019) Pervasive human-driven decline of life on Earth points to the need for transformative change. Science 366(1327):1–10
Dixon JD, Oli MK, Wooten MC, Eason TH, McCown JW, Cunningham MW (2007) Genetic consequences of habitat fragmentation and loss: the case of the Florida black bear (Ursus americanus floridanus). Conserv Genet 8(2):455–464
Doherty TS, Driscoll DA (2018) Coupling movement and landscape ecology for animal conservation in production landscapes. Proc Biol Sci 285(1870):20172272
Eizirik E, Kim JH, Menotti-Raymond M, Crawshaw PG, O’Brien SJ, Johnson WE (2001) Phylogeography, population history and conservation genetics of jaguars (Panthera onca, Mammalia, Felidae). Mol Ecol 10(1):65–79
Elliot NB, Cushman SA, Macdonald DW, Loveridge AJ (2014) The devil is in the dispersers: predictions of landscape connectivity change with demography. J Appl Ecol 51(5):1169–1178
Ernest HB, Vickers TW, Morrison SA, Buchalski MR, Boyce WM (2014) Fractured genetic connectivity threatens a Southern California puma (Puma concolor) population. PLoS ONE. https://doi.org/10.1371/journal.pone.0107985
ESRI (2022) ArcGIS Desktop (10.8.0). Environmental Systems Research Institute
Garcia-Alaniz N, Naranjo EJ, Mallory FF (2010) Human-felid interactions in three mestizo communities of the Selva Lacandona, Chiapas, Mexico: benefits, conflicts and traditional uses of species. Hum Ecol 38(3):451–457
Gonthier DJ, Castañeda FE (2013) Large- and medium-sized mammal survey using camera traps in the sikre river in the Río Plátano biosphere reserve, Honduras. Trop Conserv Sci 6(4):584–591
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22(7):1–19
Haag T, Santos AS, Sana DA, Morato RG, Cullen L, Crawshaw PG, De Angelo C, Di Bitetti MS, Salzano FM, Eizirik E (2010) The effect of habitat fragmentation on the genetic structure of a top predator: loss of diversity and high differentiation among remnant populations of Atlantic Forest jaguars (Panthera onca). Mol Ecol 19(22):4906–4921
Haddad NM, Brudvig LA, Clobert J, Davies KF, Gonzalez A, Holt RD, Lovejoy TE, Sexton JO, Austin MP, Collins CD, Cook WM, Damschen EI, Ewers RM, Foster BL, Jenkins CN, King AJ, Laurance WF, Levey DJ, Margules CR et al (2015) Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci Adv 1(2):1–10
Harmsen BJ, Foster RJ, Sanchez E, Gutierrez-González CE, Silver SC, Ostro LET, Kelly MJ, Kay E, Quigley H (2017) Long term monitoring of jaguars in the Cockscomb Basin Wildlife Sanctuary, Belize; Implications for camera trap studies of carnivores. PLoS ONE 12(6):e0179505
Hernández Potosme SM (2019) Factores del paisaje que influyen en la distribución de jaguares: Contribución de línea base para proponer una Unidad de Conservación del Jaguar, Nicaragua. Centro Agronómico Tropical de Investigación y Enseñanza, Turrialba
Holderegger R, Wagner HH (2008) Landscape genetics. Bioscience 58(3):199–207
Hoskins HMJ, Burdekin OJ, Dicks K, Slater KY, McCann NP, Jocque M, Castañeda F, Reid N (2018) Non-volant mammal inventory of Cusuco National Park, northwest Honduras: reporting the presence of Jaguar, Panthera onca (Linnaeus, 1758), and demonstrating the effects of zonal protectioon mammalian abundance. Check List 14(5):877–891
Jackson ND, Fahrig L (2014) Landscape context affects genetic diversity at a much larger spatial extent than population abundance. Ecology 95(4):871–881
Jackson HB, Fahrig L (2015) Are ecologists conducting research at the optimal scale? Glob Ecol Biogeogr 24(1):52–63
Jędrzejewski W, Boede EO, Abarca M, Sánchez-Mercado A, Ferrer-Paris JR, Lampo M, Velásquez G, Carreño R, Viloria ÁL, Hoogesteijn R, Robinson HS, Stachowicz I, Cerda H, Weisz MM, Barros TR, Rivas GA, Borges G, Molinari J, Lew D et al (2017a) Predicting carnivore distribution and extirpation rate based on human impacts and productivity factors; assessment of the state of jaguar (Panthera onca) in Venezuela. Biol Conserv 206:132–142
Jędrzejewski W, Carreño R, Sánchez-Mercado A, Schmidt K, Abarca M, Robinson HS, Boede EO, Hoogesteijn R, Viloria ÁL, Cerda H, Velásquez G, Zambrano-Martínez S (2017b) Human-jaguar conflicts and the relative importance of retaliatory killing and hunting for jaguar (Panthera onca) populations in Venezuela. Biol Conserv 209(March):524–532
Jȩdrzejewski W, Robinson HS, Abarca M, Zeller KA, Velasquez G, Paemelaere EAD, Goldberg JF, Payan E, Hoogesteijn R, Boede EO, Schmidt K, Lampo M, Viloria ÁL, Carreño R, Robinson N, Lukacs PM, Nowak JJ, Salom-Pérez R, Castañeda F et al (2018) Estimating large carnivore populations at global scale based on spatial predictions of density and distribution—application to the jaguar (Panthera onca). PLoS ONE 13(3):1–25
Kantek DLZ, Trinca CS, Tortato F, Devlin AL, de Azevedo FCC, Cavalcanti S, Silveira L, Miyazaki SS, Junior PGC, May-Junior JA, Fragoso CE, Sartorello LR, Rampim LE, Haberfeld MB, de Araujo GR, Morato RG, Eizirik E (2021) Jaguars from the Brazilian Pantanal: low genetic structure, male-biased dispersal, and implications for long-term conservation. Biol Conserv 259:109153
Keeley ATH, Beier P, Keeley BW, Fagan ME (2017) Habitat suitability is a poor proxy for landscape connectivity during dispersal and mating movements. Landsc Urban Plan 161:90–102
Kimmig SE, Beninde J, Brandt M, Schleimer A, Kramer-Schadt S, Hofer H, Börner K, Schulze C, Wittstatt U, Heddergott M, Halczok T, Staubach C, Frantz AC (2020) Beyond the landscape: resistance modelling infers physical and behavioural gene flow barriers to a mobile carnivore across a metropolitan area. Mol Ecol 29(3):466–484
König HJ, Kiffner C, Kramer-Schadt S, Fürst C, Keuling O, Ford AT (2020) Human–wildlife coexistence in a changing world. Conserv Biol 34(4):786–794
Kramer-Schadt S, Revilla E, Wiegand T, Breitenmoser U (2004) Fragmented landscapes, road mortality and patch connectivity: modelling influences on the dispersal of Eurasian lynx. J Appl Ecol 41(4):711–723
Kramer-Schadt S, Revilla E, Wiegand T (2005) Lynx reintroductions in fragmented landscapes of Germany: projects with a future or misunderstood wildlife conservation? Biol Conserv 125(2):169–182
Krishnamurthy R, Cushman SA, Sarkar MS, Malviya M, Naveen M, Johnson JA, Sen S (2016) Multi-scale prediction of landscape resistance for tiger dispersal in central India. Landsc Ecol 31(6):1355–1368
Lamb CT, Ford AT, McLellan BN, Proctor MF, Mowat G, Ciarniello L, Nielsen SE, Boutin S (2020) The ecology of human–carnivore coexistence. Proc Natl Acad Sci USA 117(30):17876–17883
Lecis R, Dondina O, Orioli V, Biosa D, Canu A, Fabbri G, Iacolina L, Cossu A, Bani L, Apollonio M, Scandura M (2022) Main roads and land cover shaped the genetic structure of a Mediterranean island wild boar population. Ecol Evol 12(4):1–13
Lorenzana G, Heidtmann L, Haag T, Ramalho E, Dias G, Hrbek T, Farias I, Eizirik E (2020) Large-scale assessment of genetic diversity and population connectivity of Amazonian jaguars (Panthera onca) provides a baseline for their conservation and monitoring in fragmented landscapes. Biol Conserv 242:108417
Lowe WH, Allendorf FW (2010) What can genetics tell us about population connectivity ? Mol Ecol 19:3038–3051
Mateo-Sánchez MC, Balkenhol N, Cushman S, Pérez T, Domínguez A, Saura S (2015) A comparative framework to infer landscape effects on population genetic structure: are habitat suitability models effective in explaining gene flow? Landsc Ecol 30(8):1405–1420
Mcgarigal K, Yi H, Zeller KA, Timm BC, Cushman SA (2016) Multi-scale habitat selection modeling: a review and outlook. Landsc Ecol 31(6):1161–1175
McRae BH (2006) Isolation by resistance. Evolution 60(8):1551
Mena JL, Yagui H, Tejeda V, Cabrera J, Pacheco-Esquivel J, Rivero J, Pastor P (2020) Abundance of jaguars and occupancy of medium- and large-sized vertebrates in a transboundary conservation landscape in the northwestern Amazon. Global Ecol Conserv 23:e01079
Menchaca A, Rossi NA, Froidevaux J, Dias-Freedman I, Caragiulo A, Wultsch C, Harmsen B, Foster R, de la Torre JA, Medellin RA, Rabinowitz S, Amato G (2019) Population genetic structure and habitat connectivity for jaguar (Panthera onca) conservation in Central Belize. BMC Genet 20(1):100
Myers N, Russell AM, Mittermeier CG, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403(6772):853–858
Mohammadi A, Almasieh K, Nayeri D, Adibi MA, Wan HY (2022) Comparison of habitat suitability and connectivity modelling for three carnivores of conservation concern in an Iranian montane landscape. Landscape Ecol 37(2):411–430
Mora JM, Polisar J, Portillo H, Castañeda F (2016) Estado de conservación del jaguar (Panthera onca) en Honduras. In: Medellín R, de la Torre A, Zarza H, Chávez C, Ceballos G (eds) El jaguar en el siglo XXI La perspectiva continental. Ediciones Científicas Universitarias, pp 137–167
Moraga AD, Martin AE, Fahrig L (2019) The scale of effect of landscape context varies with the species’ response variable measured. Landsc Ecol 34(4):703–715
Moreno R, Meyer N, Olmos M, Hoogesteijn R, Hoogesteijn A (2015) Causes of jaguar killing in Panama—a long term survey using interviews. Catnews 62(July):40–42
Nuñez-Perez R, Miller B (2019) Movements and home range of Jaguars (Panthera onca) and mountain lions (Puma concolor) in a tropical dry forest of Western Mexico. In: Reyna-Hurtado R, Chapman CA (eds) Movement ecology of neotropical forest mammals: focus on social animals. Springer, Cham, pp 243–262
Oksanen JARI, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MH H (2022) vegan: Community Ecology Package. R package version 2.5–7
Olsoy PJ, Zeller KA, Hicke JA, Quigley HB, Rabinowitz AR, Thornton DH (2016) Quantifying the effects of deforestation and fragmentation on a range-wide conservation plan for jaguars. Biol Conserv 203:8–16
Peer G, Henle K, Dislich C, Frank K (2011) Breaking functional connectivity into components: A novel approach using an individual-based model, and first outcomes. PLoS ONE 6(8):e22355
Peterman WE (2018) ResistanceGA: an R package for the optimization of resistance surfaces using genetic algorithms. Methods Ecol Evol 9(6):1638–1647
Peterman WE, Pope NS (2021) The use and misuse of regression models in landscape genetic analyses. Mol Ecol 30(1):37–47
Peterman WE, Connette GM, Semlitsch RD, Eggert LS (2014) Ecological resistance surfaces predict fine-scale genetic differentiation in a terrestrial woodland salamander. Mol Ecol 23(10):2402–2413
Petracca LS, Frair JL, Cohen JB, Calderón AP, Carazo-Salazar J, Castañeda F, Corrales-Gutiérrez D, Foster RJ, Harmsen B, Hernández-Potosme S, Herrera L, Olmos M, Pereira S, Robinson HS, Robinson N, Salom-Pérez R, Urbina Y, Zeller KA, Quigley H (2018) Robust inference on large-scale species habitat use with interview data: the status of jaguars outside protected areas in Central America. J Appl Ecol 55(2):723–734
Portanier E, Léger F, Henry L, Gayet T, Queney G, Ruette S, Devillard S (2022) Landscape genetic connectivity in European wildcat (Felis silvestris silvestris): a matter of food, shelters and demographic status of populations. Conserv Genet 23(3):653–668
Powers RP, Jetz W (2019) Global habitat loss and extinction risk of terrestrial vertebrates under future land-use-change scenarios. Nat Clim Chang 9(4):323–329
Pritchard JK, Rosenberg NA (1999) Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 65:220–228
Prunier JG, Kaufmann B, Fenet S, Picard D, Pompanon F, Joly P, Lyon D, Ecologie UMR (2013) Optimizing the trade-off between spatial and genetic sampling efforts in patchy populations : towards a better assessment of functional connectivity using an individual-based sampling scheme. Mol Ecol. https://doi.org/10.1111/mec.12499
Quigley H, Foster R, Petracca L, Payan E, Salom R, Harmsen B (2017) Panthera onca. Panthera Onca (Errata Version Published in 2018). https://www.iucnredlist.org/es/species/15953/123791436#bibliography
Rabinowitz A, Zeller KA (2010) A range-wide model of landscape connectivity and conservation for the jaguar, Panthera onca. Biol Conserv 143(4):939–945
Reding DM, Cushman SA, Gosselink TE, Clark WR (2013) Linking movement behavior and fine-scale genetic structure to model landscape connectivity for bobcats (Lynx rufus). Landsc Ecol 28(3):471–486
Redo DJ, Grau HR, Aide TM, Clark ML (2012) Asymmetric forest transition driven by the interaction of socioeconomic development and environmental heterogeneity in Central America. Proc Natl Acad Sci USA 109(23):8839–8844
Richard Y, Armstrong DP (2010) Cost distance modelling of landscape connectivity and gap-crossing ability using radio-tracking data. J Appl Ecol 47(3):603–610
Roelke ME, Martenson JS, O’Brien SJ (1993) The consequences of demographic reduction and genetic depletion in the endangered Florida panther. Curr Biol 3(6):340–350
Romero-Muñoz A, Torres R, Noss AJ, Giordano AJ, Quiroga V, Thompson JJ, Baumann M, Altrichter M, McBride R, Velilla M, Arispe R, Kuemmerle T (2019) Habitat loss and overhunting synergistically drive the extirpation of jaguars from the Gran Chaco. Divers Distrib 25(2):176–190
Roques S, Furtado M, Jácomo ATA, Silveira L, Sollmann R, Tôrres NM, Godoy JA, Palomares F (2014) Monitoring jaguar populations Panthera onca with non-invasive genetics: a pilot study in Brazilian ecosystems. Oryx 48(3):361–369
Roques S, Sollman R, Jácomo A, Tôrres N, Silveira L, Chávez C, Keller C, do Prado DM, Torres PC, dos Santos CJ, da Luz XBG, Magnusson WE, Godoy JA, Ceballos G, Palomares F (2016) Effects of habitat deterioration on the population genetics and conservation of the jaguar. Conserv Genet 17(1):125–139
Row JR, Knick ST, Oyler-McCance SJ, Lougheed SC, Fedy BC (2017) Developing approaches for linear mixed modeling in landscape genetics through landscape-directed dispersal simulations. Ecol Evol 7(11):3751–3761
Ruiz-Garcia M, Payán E, Murillo A, Alvarez D (2006) DNA microsatellite characterization of the jaguar (Panthera onca) in Colombia. Genes Genet Syst 81(2):115–127
Sartor CC, Wan HY, Pereira JA, Eizirik E, Trigo TC, de Freitas TRO, Cushman SA (2022) Landscape genetics outperforms habitat suitability in predicting landscape resistance for congeneric cat species. J Biogeogr. https://doi.org/10.1111/jbi.14498
Schmidt C, Domaratzki M, Kinnunen RP, Bowman J, Garroway CJ (2020) Continent-wide effects of urbanization on bird and mammal genetic diversity. Proc R Soc B. https://doi.org/10.1098/rspb.2019.2497
Schwab AC, Zandbergen PA (2011) Vehicle-related mortality and road crossing behavior of the Florida panther. Appl Geogr 31(2):859–870
Seaborn T, Hauser SS, Konrade L, Waits LP, Goldberg CS (2019) Landscape genetic inferences vary with sampling scenario for a pond—breeding amphibian. Ecol Evol 9(9):5063–5078
She JX, Autem M, Kotulas G, Pasteur N, Bonhomme F (1987) Multivariate analysis of genetic exchanges between Solea aegyptiaca and Solea senegalensis (Teleosts, Soleidae). Biol J Lin Soc 32(4):357–371
Shirk AJ, Wallin DO, Cushman SA, Rice CG, Warheit KI (2010) Inferring landscape effects on gene flow: a new model selection framework. Mol Ecol 19(17):3603–3619
Spear SF, Balkenhol N, Fortin M-J, Mcrae BH, Scribner K (2010) Use of resistance surfaces for landscape genetic studies: considerations for parameterization and analysis. Mol Ecol 19(17):3576–3591
Srbek-Araujo AC, Mendes SL, Chiarello AG (2015) Jaguar (Panthera onca Linnaeus, 1758) roadkill in Brazilian Atlantic Forest and implications for species conservation. Braz J Biol 75(3):581–586
Sutherland GD, Harestad AS, Price K, Lertzman KP (2000) Scaling of natal dispersal distances in terrestrial birds and mammals. Ecol Soc. https://doi.org/10.5751/es-00184-040116
Taylor PD, Fahrig L, Henein K, Merriam G (1993) Connectivity is a vital element of landscape structure. Oikos 68(3):571
Tesson SVM, Edelaar P (2013) Dispersal in a changing world: opportunities, insights and challenges. Mov Ecol 1(1):1–6
Thatte P, Joshi A, Vaidyanathan S, Landguth E, Ramakrishnan U (2018) Maintaining tiger connectivity and minimizing extinction into the next century: insights from landscape genetics and spatially-explicit simulations. Biol Conserv 218:181–191
Thatte P, Chandramouli A, Tyagi A, Patel K, Baro P, Chhattani H, Ramakrishnan U (2020) Human footprint differentially impacts genetic connectivity of four wide-ranging mammals in a fragmented landscape. Divers Distrib 26(3):299–314
Thompson JJ, Morato RG, Niebuhr BB, Alegre VB, Oshima JEF, de Barros AE, Paviolo A, de la Torre JA, Lima F, McBride RT, Cunha de Paula R, Cullen L, Silveira L, Kantek DLZ, Ramalho EE, Maranhão L, Haberfeld M, Sana DA, Medellin RA et al (2021) Environmental and anthropogenic factors synergistically affect space use of jaguars. Curr Biol 31(15):3457-3466.e4
Tilman D, Clark M, Williams DR, Kimmel K, Polasky S, Packer C (2017) Future threats to biodiversity and pathways to their prevention. Nature 546(7656):73–81
Tischendorf L, Fahrig L (2000) On the usage and measurement of landscape connectivity. Oikos 90(1):7–19
Tobler MW, Garcia Anleu R, Carrillo-Percastegui SE, Ponce Santizo G, Polisar J, Zuñiga Hartley A, Goldstein I (2018) Do responsibly managed logging concessions adequately protect jaguars and other large and medium-sized mammals? Two case studies from Guatemala and Peru. Biol Conserv 220:245–253
Toncheva S, Fletcher R, Turnhout E (2022) Convivial conservation from the bottom up: human-bear cohabitation in the Rodopi mountains of Bulgaria. Conserv Soc 20(2):124–135
Tucker MA, Böhning-Gaese K, Fagan WF, Fryxell JM, Van Moorter B, Alberts SC, Ali AH, Allen AM, Attias N, Avgar T, Bartlam-Brooks H, Bayarbaatar B, Belant JL, Bertassoni A, Beyer D, Bidner L, van Beest FM, Blake S, Blaum N et al (2018) Moving in the Anthropocene: global reductions in terrestrial mammalian movements. Science (new York, N.y.) 359(6374):466–469
Valdez FP, Haag T, Azevedo FCC, Silveira L, Cavalcanti SMC, Salzano FM, Eizirik E (2015) Population genetics of jaguars (Panthera onca) in the Brazilian Pantanal: molecular evidence for demographic connectivity on a regional scale. J Hered 106(S1):503–511
Valsecchi J, Monteiro MCM, Alvarenga GC, Lemos LP, Ramalho EE (2022) Community-based monitoring of wild felid hunting in Central Amazonia. Anim Conserv. https://doi.org/10.1111/acv.12811
Vickers TW, Sanchez JN, Johnson CK, Morrison SA, Botta R, Smith T, Cohen BS, Huber PR, Ernest HB, Boyce WM (2015) Survival and mortality of pumas (Puma concolor) in a fragmented, urbanizing landscape. PLoS ONE 10(7):1–18
Villalva P, Palomares F (2022) A continental approach to jaguar extirpation: a tradeoff between anthropic and intrinsic causes. J Nat Conserv 66:126145
Volski L, McInturff A, Gaynor KM, Yovovich V, Brashares JS (2021) Social effectiveness and human-wildlife conflict: linking the ecological effectiveness and social acceptability of livestock protection tools. Front Conserv Sci 2(August):1–17
Willi Y, Kristensen TN, Sgro CM, Weeks AR, Ørsted M, Hoffmann AA (2022) Conservation genetics as a management tool: the five best-supported paradigms to assist the management of threatened species. Proc Natl Acad Sci USA 119(1):1–10
World Bank (2020) Human population, DataBank Microdata Data Catalog. Food and Agriculture Organization and World Bank Population Estimates. https://data.worldbank.org/indicator/SP.POP.TOTL
Wright S (1942) Isolation by distance. Genetics 38:114–138
Wultsch C, Caragiulo A, Dias-Freedman I, Quigley H, Rabinowitz S, Amato G (2016a) Genetic diversity and population structure of Mesoamerican jaguars (Panthera onca): implications for conservation and management. PLoS ONE 11(10):e0162377
Wultsch C, Waits LP, Kelly MJ (2016b) A comparative analysis of genetic diversity and structure in jaguars (Panthera onca), Pumas (Puma concolor), and Ocelots (Leopardus pardalis) in fragmented landscapes of a critical Mesoamerican linkage zone. PLoS ONE 11(3):e0151043
Zeller KA, Nijhawan S, Salom-Pérez R, Potosme SH, Hines JE (2011) Integrating occupancy modeling and interview data for corridor identification: a case study for jaguars in Nicaragua. Biol Conserv 144(2):892–901
Zeller KA, McGarigal K, Whiteley AR (2012) Estimating landscape resistance to movement: a review. Landsc Ecol 27(6):777–797
Zeller KA, Wultsch C, Welfelt LS, Beausoleil RA, Landguth EL (2023) Accounting for sex-specific differences in gene flow and functional connectivity for cougars and implications for management. Landsc Ecol 38:223–237
Acknowledgements
APC thanks her colleagues Julie Louvrier and Aimara Planillo for their helpful comments on sections of the manuscript text, and to Viktoriia Radchuk for her continuous mentoring support. We thank the American Museum of Natural History, all field and laboratory associates who helped with faecal sample collection and genetic analysis. APC is funded by the German Academic Exchange Service (Program-57381412). APC, VG, SKS are associated with the BioMove research training group DFG-GRK 2118/1. Open Access funding was enabled and organized by Projekt DEAL. The publication of this article was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 491292795. This research was supported in part by the USDA Forest Service, Rocky Mountain Research Station, Aldo Leopold Wilderness Research Institute. The findings and conclusions in this publication are those of the authors and should not be construed to represent any official USDA or US Government determination or policy.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
APC, PL-G, CW, RF, SK-S and KZ conceived the ideas and designed the methodology. RF, BH, OF, RG-A, FC, CW and GA contributed to the genetic sampling and analysis effort. Analyses were performed by APC and PL-G, guided by KZ. APC led the manuscript writing supported by PL-G, CW, RF, VG, SK-S and KZ. All authors commented on manuscript drafts and gave final approval for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Calderón, A.P., Landaverde-Gonzalez, P., Wultsch, C. et al. Modelling jaguar gene flow in fragmented landscapes offers insights into functional population connectivity. Landsc Ecol 39, 12 (2024). https://doi.org/10.1007/s10980-024-01795-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10980-024-01795-2